Ответы на экзаменационные вопросы по химии 2 семестр:
реклама

Ответы на экзаменационные вопросы по химии 2 семестр:
1) Строение атома и его связь с периодическим законом
Квантовомеханическая модель строения атома: В соответствие с квантовомеханическими
представлениями невозможно точно определить энергию и положение электрона, поэтому в
квантовомеханической модели атома используют вероятностный подход для характеристики
положения электрона. Вероятность нахождения электрона в определенной области пространства
описывается волновой функции ψ, которая характеризует амплитуду волны, как функцию координат
электрона. В наиболее простом случае эта функция зависит от трех пространственных координат и
называет орбиталью. В соответствие с определением ψ, орбиталью называется область
простран6ства, в котором наиболее вероятно нахождение электрона. Так как электрон несет
отрицательный заряд, то его орбиталь представляет собой определенное распределение заряда, которое
получило название электронного облака.
Уравнение волны де Бройля: В 1924г де Бройль предположил, что корпускулярно-волновая теория
свойственна простейшим частицам.
h
8
- уравнение волны де Бройля. 10 см
mv
(приблизительный размер атома).
Принцип неопределенности Гейзенберга: «невозможно точно определить координату и импульс
частицы». px x h . Сведения о состоянии электрона в атоме носят вероятностный характер.
Представление об электронном облаке символизирует пределы, в котором существует электрон. Сумма
всех заряженных плотностей равна единичному отрицательному заряду электрона. Общая вероятность
равна 1. Решая уравнение Шрейдингера n ,l ,ml (r , , ) Rn ,l ( r )l ,ml ( ) m ,l ( ) (полная собственная
функция уравнения Шрейдингера с учетом всех условий для атома водорода, которая называется
атомной орбиталью). Rn ,l (r ) - радиальная составляющая; l ,ml ( ) m ,l ( ) - угловая составляющая.
Атомная орбиталь характеризуется тремя квантовыми числами.
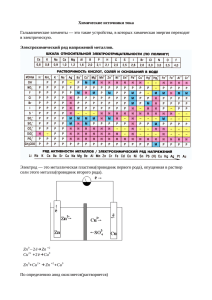
Квантовые характеристики электрона. 1) Главное квантовое число n характеризует энергию
электрона, чем меньше n - тем меньше энергия (принимает целые значения). 2) Орбитальное квантовое
число l характеризует форму электронного облака и величину орбитального момента колич. Движения
электронов. Определяется формулой l=n-1. 3) Магнитное квантовое число m лежит в интервале от –l до
l и проходит все целочисленные значения, включая ноль. Описывает различную ориентацию
электронных облаков в пространстве. 4) Спиновое квантовое число s можно упрощенно трактовать как
вращение электрона вокруг собственной оси – принимает значения -0.5 и 0.5.
Запрет Паули: «в атоме не может быть двух электронов с одинаковым набором всех четырех
квантовых чисел». У двух электронов могут быть попарно одинаковыми n,l,m-квантовые числа, но они
обязаны различаться по значениям «s». Наибольшее число электронов Z в оболочке с главным
квантовым числом «n»: Z
l n 1
2(2l 1) 2[1 3 5 ...(2n 1) n
2
. На уровне K-2 электрона, на L-
l 0
8, на M-18, на N-32.
Правило Гунда: «суммарный спин электронов данного подуровня должен быть максимальным», т.е.
электроны на подуровне занимают максимальное число свободных квантовых состояний.
Правило Клечковского: «Увеличение энергии и соответственно заполнение орбиталей происходит в
порядке возрастания суммы квантовых чисел n+l, а при равной сумме – в порядке возрастания числа n».
Соответственно этому правилу подоболочки выстраиваются в следующий ряд:
1s 2s 3s 3 p 3d 4 p 5s 4d 5 p 6s 4 f 6 p 7 s .
Порядок заполнения электронами энергетических уровней и подуровней:
C т.з. поступления электрона на тот или иной п/уровень все элементы в системе элементов могут
подразделяться на 4 семейства. K S-семейству. отн. гл. п/гр. 1, 2 гр., а также Н и Не. К Р-семейству отн.
элементы 3-8 группы (эл. поб. п/групп). d-все трех. элементы. f-28 элементов, лантаниды и актиниды.
П.С. отражает порядок заполнения электронами квантовых слоев атомов различных элементов.
Причины периодичности повторяемости свойств различных элементов в том, что в ряду элементов,
расположеннчх в порядке возрастания зарядов их ядер возникает периодически повторяемый процесс
застройки новых электронных оболочек. Чем меньше энергия ионизации, тем ярче проявляются Ме свва элементов. Однако по мере возрастания электронов на внешней оболочке атом с трудом теряет
электроны и более охотно их приобретает. Если к нейтральному атому присоединить электрон, то атом
превратится в положительно заряженный ион. Чем больше энергия сродства, тем ярче выраженны
неметаллические св-ва. Сумма Еионизации с Есродства к электрону носит название
электроотрицательности элемента.(Li-1, Na-0.9, K-0.8, Al-1.5, Sc-1.8, o-3.5, F-4) Чем больше ЭО, тем
легче атомы превращаются в отрицательно заряженные ионы. Т.о. основной характеристикой
элементов в системе элементов Менделеева Еи, Еср. к .эл. и ЭО.
1s 2 2s 2 2 p6 3s 2 3 p6 4s 2 3d 10 4 p6 5s 2 4d 10 5 p6 6s 2 5d 1 4 f 14 5d 210 6 p6 7s 2 6d 1 5 f 14 6d 210 7 p6 .
Энергия ионизации, энергия сродства, электроотрицательность, атомные радиусы: Э.и. – энергия,
которую необходимо затратить, чтобы оторвать электрон от нейтрального атома и удалить его на
бесконечно большое расстояние (эВ). Атом превращается в + ион. Потенциал ионизации – напряжение,
которое необходимо приложить, чтобы оторвать электрон от атома. Существует несколько
ионизирующих потенциалов (1-ый = энергии связи, 2-ой > энергии связи). Наиболее важный – 1 (Li 1 –
5,39 B; 2 – 75,62 B; 3 – 122,4 B). Скачкообразный характер потенциалов ионизации указывает на то, что
электроны вокруг ядра расположены слоями. Чем больше э.и. тем более выражены неметаллические
свойства элемента. Энергия сродства к электрону – энергетический эффект присоединения электрона к
атому (атом превращается в – ион). Чем больше э.с.э. тем ярче неметаллические свойства.
Электроотрицательность – количественная характеристика способности атома в молекуле притягивать
к себе электроны. Сумма энергии сродства к электрону и энергии ионизации. Чем больше
электроотрицательность, тем легче его атомы превращаются в – ион.
2) Химическая связь и межмолекулярное взаимодействие
Основные характеристики химической связи: энергия, длина, силовая постоянная, степень
ионности и дипольный момент, характеристичность. При возникновении химической связи
выделяется энергия. Чем больше энергии выделится, тем прочнее связь. Потенциальная энергия
образования многоатомных объектов всегда меньше суммарной потенциальной энергии атомов,
вступающих в связь.
Длинна связи – кратчайшее межъядерное расстояние в невозбуждённой молекуле 1-2 А, 1А=10-8см).
Энергия связи – кол-во энергии выделившейся при её возникновении. Силовая постоянная связи k
k , где - частота валентных колебаний. Степень ионности –
связана с валентными колебаниями,
характер распределения электронной плотности в пространстве между двумя эл. центрами. Дипольный
момент связи св. характеризует молекулу как систему частиц. св.
RAB el , мол. i .
i
Характеристичность 2-ух центровой связи – близость свойств конкретной химической связи в разных
соединениях.
Различные виды связи:
Ионная (электровалентность). Преобладают электростатические взаимодействия, возникает между
элементами, которые значительно различаются по величинам электроотрицательности.
Атомная (ковалентная, неполярная и полярная). Элементы с одинаковыми или близкими значениями
электроотрицательности. Имеет место смещение электронов и возникновение общих электронных пар.
Донорно-акцепторная (координационная). Разновидность ковалентной связи (различают валентнонасыщенную). Различие – в ковалентной участники связи равноправны, в донорно-акцепторной – один
участник – пару, др. – орбиту.
Водородная связь – дополнительная связь, осуществляемая атомами водорода, ковалентно
соединёнными в молекуле с атомами электроотрицательного элемента.
5)Металлическая связь. “Коллектив” катионов взаимодействует с “коллективом” электронов.
Диполный момент молекулы. Два заряда +е –е на расстоянии l друг от друга образуют диполь,
основной характеристикой которого является дипольный момент el , где l – плечо диполя, напряженность электростатического поля, создаваемого диполем.
Ковалентная связь. К.с. возникает между элементами с одинаковым или близким значением энергии
сродства к электрону. Валентность атомов в соединениях с ковалентной связью определяется по числу
электронных пар. Для определения относительных зарядов в атомах с ковалентной связью надо
мысленно связь разорвать и руководствоваться следующим правилом: при разрыве связи в пределах
периода электрон смещается от левее стоящего элемента к правее стоящего, а в пределах главной
подгруппы от ниже стоящему к выше.
Метод валентных связей. В основе м.в.с. лежит 3 положения:
- Химическую связь образуют 2 электрона с противоположно направленными спинами. Имеет место
взаимное перекрытие электронных орбиталей, при этом в пространстве между атомами возникает
повышенная плотность электронного облака и к этой области притягиваются ядра атомов и оставшиеся
электроны.
- Химическая связь имеет ориентацию в направлении, обеспечивающем максимально возможное
перекрытие орбиталей.
- Чем больше взаимное перекрытие электронных орбиталей, тем прочнее связь.
Валентность в МВС, типы связей(сигма, пи, дельта). Валентность в МВС – количественная мера
способности атома к образовыванию химической связи (способность присоединять и замещать
определенное количество других атомов). -связь – связь, образованная перекрыванием АО по линии,
соединяющей ядра взаимодействующих атомов. Сигма-связь обычно охватывает два атома и не
простирается за их пределы, поэтому является локализованной двухцентровой связью. Связь,
образованная перекрыванием АО по обе стороны линии, соединяющей ядра атомов (боковые
перекрывания), называется -связью. Связь, образованная перекрыванием d-орбиталей всеми
четырьмя лепестками, называется -связью.
Концепция гибридизации валентных орбиталей Полинга.
BeF2 Be* 2s12p1
BF3 B* 2s12p2
CF4 C* 2s12p3
Каждая связь должна отличаться своими характеристиками. Гибридизация относится к орбиталям и не
зависит от их заселения. В изолированном атоме существуют s, p, d ,f орбитали. Перекрывание
электронных облаков при образовании ковалентных связей возможно только при определенной их
взаимной ориентации в пространстве – отсюда направленность ковалентных связей, приводящая к той
или иной стереохимии молекул, т.е. к определенной их форме. При этом область перекрывания
располагается определенным образом по отношению к взаимодействующим атомам.
1)AA, BB. Этот тип молекул характерен для Н2 ( -связь), галогенов, и соединений Н с галогенами.
Молекулы водорода имеют линейную структуру. Химическая связь действует по кратчайшему
расстоянию, связывая атомы -связью.
2)А2В. Этот тип характерен для соединений, образованных элементами главной подгруппы 6-ой
группы: Н2О, Н2S. ( Н2О – угол –1050, Н2S – 920 35’) Максимальная плотность возникает при темп.=4
градуса цельсия. (линейная структура – угол)
3)А3В. Элементы главной подгруппы 3 группы(N,P,As,Sb). NH3.(трехгранная пирамида)
4)А4В Главная подгруппа 4 группа(Si,C,Ge,Sn). СН4 – электроны последнего уровня углерода – s2p2.
После возбуждения - sp3.Гибридизация электронных облаков. s2p2 sp3 q4 Гибридизация требует
затрат энергии, но эта энергия компенсируется при протекании реакции образования связи. Все связи
энергетически равноценны. (тетраэдр)
5)АВ3 – характерно для соединений гл. подгруппы 3-ей группы.(B,Al,Ga,In,Tl) BCl3. s2p sp2 q3.
(равносторонний треугольник – А в центре)
6)АВ2 –для некоторых соединений, образованными элементами главной подгруппы 2 группы. ВеCl2
(линейная структура, угол – 1800). sp sp q2 d2 sp3 q6 .
Строение молекул CH4, H2O, NH3, CO2 с точки зрения гибридизации. В случае ковалентной связи
взаимное перекрытие электронных облаков в пространстве определяет форму молекулы.
1)А2В. Этот тип характерен для соединений, образованных элементами главной подгруппы 6-ой
группы: Н2О, Н2S.(см.рис) ( Н2О – угол –1050, Н2S – 920 35’) Максимальная плотность возникает при
темп.=4 градуса цельсия.
2)А3В. Элементы главной подгруппы 3 группы(N,P,As,Sb). NH3 (см.рис).
3)А4В Главная подгруппа 4 группа(Si,C,Ge,Sn). СН4 – электроны последнего уровня углерода – s2p2.
После возбуждения - sp3. (см.рис).Гибридизация электронных облаков. s2p2 sp3 q4 Гибридизация
требует затрат энергии, но эта энергия компенсируется при протекании реакции образования связи. Все
связи энергетически равноценны.
4)АВ2 –для некоторых соединений, образованными элементами главной подгруппы 2 группы. ВеCl2
(линейная структура, угол – 1800). sp sp q2 d2 sp3 q6 (см.рис)
Ионная связь, как предельный случай ковалентной полярной связи. Ионная связь – такая связь,
при которой преобладают электростатические взаимодействия между атомами. И.с. возникает между
элементами, которые значительно отличаются по величинам электроотрицательности. (NaCl: Na – e ->
Na+ ; Cl + e -> Cl- ; Na+ + Cl- -> NaCl
Ионы натрия и хлора сближаются, пока силы притяжения не компенсируются силами отталкивания
одноимённых зарядов электронных оболочек. Ионы занимают в пространстве строго определённое
место. Если взаимодействуют тв. Na и г. Cl, то образуется твердокристаллическая решётка NaCl. Если
взаимодействие происходит в растворе – ионы Na и Cl – в сольватированном виде (в сл. воды – в
гидротированном). Вокруг ионов образуется оболочка растворителя. При кристаллизации из раствора
образуется кристаллическая решётка. Валентность атомов в соединениях определяется по числу
потерянных или приобретённых электронов. Координационное число показывает сколько ионов
противоположного знака в растворе ион данного знака (к.ч. Na=6, к.ч. Cl=6). Понятие молекулы не
применимо в случае ионной связи. Весь кристалл представляет собой одну гигантскую молекулу.
Соединений с чисто ионной связью не существует (имеет место определённая ковалентность).
Электростатическое взаимодействие очень мощное. Это объясняет высокую прочность соединений с
ионной связью, высокую температуру плавления и кипения. Соединения с ионной связью – проводники
второго рода. Ионная связь характеризуется: 1)наименьшими энергиями; 2) Определяется на
расстоянии, значительно превышающим размеры частиц, что полностью исключает перекрытие АО; 3)
Проявляется в любом агрегатном состоянии; 4) Имеют электрическую природу.
Металлическая связь. “Коллектив” катионов взаимодействует с “коллективом” электронов.
Межмолекулярное взаимодействие. Между валентнонасыщенными и в сумме электронейтральными
молекулами вещества в различных агрегатных состояниях действуют силы притяжения и отталкивания,
имеющие электростатическую природу. Относительная интенсивность этих сил во многом определяет
физико-химические свойства вещества. Например, проявление сил отталкивания объясняет малую
сжимаемость жидкостей и твердых тел. Силы притяжения лежат в основе таких явлений, как
сжимаемость газов, адсорбция и т.д. Силы отталкивания есть результат взаимного отталкивания
одноименно заряженных электронных оболочек. Силы отталкивания проявляются лишь на очень
n
маленьких расстояниях и быстро убывают с увеличением расстояния. Eотт Ar , n=12, r расстояние между молекулами. A и n - константы, характеризующие конкретное вещество. Часто силы
межмолекулярного притяжения называются силы Ван-Дер-Ваальсову. Различают
a) ориентационные: они проявляются в случае, если молекула- ярко выраженный диполь. Такие
молекулы стремятся расположиться упорядоченно: (+-)(+-)(+-) или {(+-) | (-+) | (+-)} (последнее –
столбик) Eор
2 4 N A
. Минус указывает на уменьшение E в системе.
3RTr 6
б) индукционные: имеют место в том случае, если одна из молекул полярна, а вторая – неполярна, но
легко поляризуема. Под действием электростатического поля, создаваемого полярной молекулой,
неполярная молекула деформируется, на ней наводится (индуцируется) диполь, что и обеспечивает
взаимное притяжение. Eинд
2 2 d
r6
в) дисперсионное. Если молекулы не полярны, возникает так называемые мгновенные диполи В случае
многоатомных молекул в эл.оболочке в одних местах сгущение, а в других - разряжение электронов : на
какой-то момент времени молекула - диполь. Дисперсионные силы суммируются. Это преобладающий
вид взаимодействия. Eдисп
3d 2T
.
4r 6
Ориентационное взаимодействие преобладает, если молекулы - яркие диполи. В общем виде:
Eприт Br m , m=6.
Eсумм Eотт Eприт Ar n Br m . Эти силы действуют на расстояниях 3-5 A (10^-8 см.)
E ~ 0.4*10^-4 кДж/Моль.
Водородная связь. Наблюдается при взаимодействии атома водорода с атомами сильно
электроотрицательных элементов – F, O, N, реже Cl и S. Природа водородной связи до конца не
выяснена: с одной стороны имеет место проявление сил межмолекулярного взаимодействия, но
характер этих связей – электростатический. В молекуле атомы водорода практически лишены
электронных оболочек, поскольку общие электронные пары смещены от атомов водорода к атому
кислорода. Атом водорода – протон – получает возможность притягиваться к электронной оболочке
атома кислорода соседней молекулы воды – возникают ассоциаты, отвечающие формуле ( H 2O) x , где x
– зависит от температуры. Пунктиром показана водородная свзяь.
Комплексные соединения: внутренняя и внешняя сферы, комплексообразователь и лиганды,
координационное число, координационная емкость. Донорно-акцепторная связь является
разновидностью ковалентной связи. Соединения первого порядка или валентно насыщенные
соединения. H2O, AgCl, NH3, HCl, KJ, HgJ2, CuSO4, FeCl3 и т.д. Они могут давать соединения высшего
порядка или комплексные соединения. 1)NH3+ HCl [NH4]Cl, 2)AgCl+2NH3=[Ag(NH3)2]Cl,
3)CuSO4+4NH3= [Cu(NH3)4]SO4, 4)2KJ+HgJ2= K2[HgJ4], 5)FeCl3+6H2O [Fe(H2O)6]Cl3. Комплексные
соединения- соединения, которые образуются без возникновения новых электронных пар, а за счет
проявления дон.-акц. связи. BF3+NH3 BF3NH3, B-акцептор, N-донор. Рассмотрим [Cu(NH3)4]SO4
(тетра-амимн-купрум-сульфат). В квадратных скобках заключена внутренняя сфера комплексного
соединения, за ними – внешняя сфера. Во внутреннюю сферу входят центральный ионкомплексообразователь (в данном случае – Cu2+) и лиганды, в данном случае молекулы NH3. В роли
центрального иона-комплексообразователя могут выступать положительно или отрицательно
заряженные ионы, а также нейтральные атомы. В роли лигандов выступают ионы; полярные молекулы;
неполярные, но легко поляризуемые молекулы. Координационное число (в данном случае 4),
показывающее, сколько лигандов может быть соединено с центральным ионом, принимает значения: 1,
2, 3, 4, 5, 6, 7, 8, 9, 12. Наиболее часто фигурируют значения 2 (линейная структура), 4 (тетраэдр или
квадрат), 6 (октаэдр); 12 возможно только для незаряженных лиганд. С точки зрения МВС: рассмотрим
NH3+ HCl [NH4]Cl:
3) Первое начало термодинамики.
Предмет и задачи химической термодинамики. Термодинамика - научная дисциплина, изучающая
переходы энергии из одной формы в другую, одних частей системы в другие, различные
энергетические эффекты физических и химических процессов, возможность, направление и пределы
самопроизвольного протекания процессов (т.е. условий установления хим. равновесия). Т.д. базируется
на двух законах-началах + тепловая теорема Нернста. Начала - это постулаты , т.е. они математически
недоказуемы. Предметом термодинамики является термодинамическое рассмотрение вопросов в
областях химии и физической химии . Справедливость законов т.д. основывается на том, что ни одно из
следствий из них не противоречит опыту.
Понятия: система, фаза, термодинамическое состояние, термодинамические свойства (параметры
и функции). Система - это тело или группа тел, находящихся в взаимодействии, которые мы мысленно
выделяем из окружающей среды. Системы бываю гомогенными (смесь газов, раствор) и гетерогенные
(вода со льдом, раствор с осадком) . В гомогенной системе между частями системы нет поверхностей
раздела , а в гетерогенной - есть. Если система не обменивается с окружающей средой веществом и
энергией, то она называется изолированной. Если отсутствует массообмен, а энергообмен присутствует,
то это закрытая система, иначе - открытая. Фаза - совокупность всех гомогенных частей системы,
одинаковых по составу и всем физ\хим свойствам, не зависящим от количества вещества. Фазы
отделены друг от друга поверхностями раздела, на которых все свойства фазы резко скачком меняются.
Термодинамическое состояние системы задается набором параметров p, V, T; f(p, V, T)=0. pV RT .
Функции и параметры: 1) Интенсивные: имеют определенное значение в каждой точке системы и не
зависят от размеров и массы системы. ( P, T ,Vm ,Vm ,U m , H m , Sm )2) Экстенсивные: ( U , S , H , V , m ).
Функции состояния: внутренняя энергия и энтальпия. Функции состояния – зависят только от
состояния системы. 2 1 ,
d 0 . U характеризует общий запас энергии системы,
включая энергию поступательного и вращательного движения молекул, энергию вращения
электронных ядер, нуклонов, и т.д., но без учета кинетической энергии в целом и потенциальной
энергии системы. Энтальпия (H=U+PV) (теплосодержание системы).
Процессы: теплота и работа. В отличие от U и H понятия Q и A относятся не к системе, а к
процессам. Теплота и работа проявляются только при протекании процессов, при изменениях
состояния, являясь лишь формами передачи энергии. Работа есть упорядоченная форма передачи
энергии от системы, совершающей работу, к системе, над которой она совершается; Работа,
совершаемая системой всегда связана с действием против внешних сил. Теплота же является
неупорядоченной формой обмена энергией между системами в следствии хаотического (теплового)
движения частиц. Работа может быть направлена на пополнение запаса любого вида энергии
(электрической \ магнитной \ итд); теплота же без преобразования ее в работу, может пополнять только
запас внутренней энергии. Итак, энергия может передаваться от одной системы к другой в форме
теплоты или в форме работы.
Работа различных процессов:
а) изобарический: A m =pdV , A m =
V2
V1
pdV p (V
2
V1 ) p V . Поскольку для 1 моля pV RT ,
A R(T2 T1 ) RT .
V dV
V
RT
RT ln( 2 ) . При постоянстве
, A m =pdV , Am 2
V
V1 V
V1
p V
p
p
температуры p1V1=p2 V2 , т.е. 1 2 , следовательно, Am 2 RTdp RT ln( 1 ) .
p2 V1
p1
p2
б) изотермический: pV RT , p
в) адиабатический (одновременно меняются и температура и давление): работа расширения
производится за счет убыли внутренней энергии, т.к. система не получает теплоты извне; газ при
расширении охлаждается. Для идеального газа внутренняя энергия является функцией только
температуры U=f(T); следовательно U равно произведению теплоемкости газа Cv на T : Ам=Cv(T1-T2).
г) изохорический: dV
V=0, Ам=0
Теплоты различных процессов:
Рассмотрим систему, представляющую собой идеальный газ. Если вся работа данной системы сводится
лишь к работе расширения, то, очевидно, A ' 0 и Q U
V2
V1
pdV .
изобарический - p=const. Q dU pdV A ', A ' 0 (по условию);
Q dU dpV d (U pV ) dH , поскольку H=U+pV. то., Q p dH , a Qp H H 2 H1 .
H= U+p V; pV=nRT => p V= nRT => H= U+ nRT (*)
изохорический - V=const , QV dU pdV , pdV 0 , QV dU , QV U U 2 U1 . Используя
уравнение (*) получаем Q p QV RT .
изотермический, T=const. Q dU pdV , dU 0 , поскольку внутр. энергия идеального газа является
функцией только температуры. Q pdV , Q A RT ln(
V2
p
) RT ln( 1 ) , т.е. при изотермическом
V1
p2
расширении идеального газа вся поглощаемая системой теплота затрачивается на совершение работы.
При протекании кругового процесса изменение функций состояния=0. Т.е. dU=0, U2-U1=0, Q=A.
Первое начало термодинамики. Первый закон термодинамики есть по сути закон сохранения энергии.
Первый закон термодинамики устанавливает связь между количеством теплоты полученной или
выделенной в процессе, количеством произведенной или полученной работы и изменением внутренней
энергии системы. Первый закон термодинамики имеет несколько формулировок:
-если в каком либо процессе энергия одного вида исчезает , то вместо нее появляется энергия в другой
форме в кол-ве строго эквивалентном исчезнувшему виду.
-Различные формы энергии переходят друг в друга в строго эквивалентных всегда одинаковых
соотношениях.
-в любой изолированной системе общий запас энергии сохраняется постоянным.
-работа - одна из форм перехода энергии => невозможно создание вечного двигателя 1 рода, который
давал бы возможность получать работу, не затрачивая на это соответствующее кол-во энергии.
-математическое выражение 1 закона т.д. U=Q-A. В любом процессе изменение запаса внутренней
энергии U=U2-U1 какой-либо системы складывается из теплоты Q, сообщенной системе минус кол-во
работы А, совершенной системой. Для процессов, связанных с бесконечно малыми
изменениями: dU Q A, A A ' pdV , где A ' - так называемая «полезная» работа , т.е
элементарная работа преодоления всех сил, кроме внешнего давления ; pdV - работа против сил
внешнего давления. I начало т.д. в развернутом виде: dU Q pdV A ' . В интегральном виде:
Q U
V2
V1
pdV A ' (второе слагаемое - работа расширения).
Теплоемкость: истинная и средняя, изобарная и изохорная. Понятие теплоемкости системы можно
приближенно определить как кол-во теплоты которое нужно сообщить системе при нагревании ее на 1
градус. Это определение является недостаточно строгим. В хим. т.д. различают понятия истинной
теплоемкости и средней теплоемкости. В общем случае истинная теплоемкость определяется как
предел отношения бесконечно малого кол-ва теплоты к тому изменению температуры, которое этим
изменением теплоты вызывается: C lim
T 0
Q
T
Для любого вещества и любого агрегатного состояния
справедливо:
CV (
U
H
)V ; C p (
) . Средняя теплоемкость системы в интервале температур от Т1до Т2
T
T p
определяется уравнением:
Ccp
Q
Q
2
3
. C p a bT cT dT ... (где a,b,c,d... - константы, характерные для данного
T2 T1 T
вещества.). [C]=Дж/(Моль*К)
Связь между истинной и средней теплоемкостью:
Ccp
1 T2
CdT Cср=1/(T2-T1) * (T2/T1)
T2 T1 T1
cdT (где Cср - сред. Тепл. На интервале температур
T1-T2; C- истинная тепл.)
Стандартное состояние вещества. Давление p=101325 Па, при котором твердые и жидкие вещества
находятся в наиболее устойчивом агрегатном состоянии, а газы обладают свойствами идеальных газов.
Т=298 К – стандартная температура.
0
Стандартная энтальпия образования вещества. H 298 - стандартная энтальпия образования одного
моля сложного вещества. H 298 - тепловой эффект образования одного моля сложного вещества из
простых веществ, находящихся в более устойчивых агрегатных состояниях при стандартных условиях.
0
H Q p
Закон Гесса и следствия из него. 1836г - «Тепловой эффект реакции не зависит от пути протекания, а
зависит от начальных и конечных состояний системы». Теплота, выделяющаяся или поглощающаяся
при протекании реакций – тепловой эффект. Условия: 1)p=const или V=const 2)A’=0 3)Температура
исходных веществ должна быть равна температуре продуктов реакции. Следствия закона Гесса:
1)Закон Лавуазье-Лапласа: «тепловой эффект прямой реакции равен по значению и противоположен по
знаку тепловому эффекту обратной реакции».
2)Тепловой эффект реакции, протекающей при стандартных условиях, равен сумме стандартных
энтальпий образования продуктов реакции за вычетом стандартных энтальпий образования исходных
веществ, с учетом стехиометрических коэффициентов.
Зависимость теплового эффекта реакции от температуры (закон Кирхгоффа). Термохимические
расчеты. Приближения Улиха. H H 2 H1 , p const
U U 2 U1 ,V const (
H
T
p C p
U
U
U
)V ( 2 )V ( 1 )V CV CV CV . температурный
T
T
T
2
1
коэффициент теплового эффекта процесса равен изменению теплоемкости, происходящему в
результате процесса.
Если эти 2 дифференциальных уравнения проинтегрировать в пределах T1-T2 , получим:
T2
T2
T1
T1
UT2 UT1 CV dT ; H T2 H T1 C p dT . Чтобы подсчитать тепловой эффект при T2 , мы
должны знать теплоемкость в интервале T1-T2.
При подсчете теплового коэффициента возможны допущения:
а) если температурный интервал T1-T2 невелик, можно в уравнение теплоемкости Cp=a+bT+cT2+...
ограничиться только первым слагаемым => Cp =const, тогда HT2= HT1+ Cp*(T2-T1). Как правило,
за T1 берут стандартную температуру, т.е. H T H 298
0
0
T2
C p dT . Иногда случается, что нет
298
данных по теплоемкости при T=298, тогда используют второе (грубое) допущение : HT0=298.
4) Второе начало термодинамики. Расчеты равновесий.
Второе начало термодинамики. 1) По Клаузиусу: теплота не может сама по себе переходить от более
холодного тела к более теплому. 2) Невозможен процесс, единственным результатом которого было бы
превращение теплоты в работу. 3) Невозможен вечный двигатель второго рода: который периодически
превращает тепло среды в работу (при постоянной температуре).
Понятие об энтропии. dS
Q
T
- энтропия. Энтропия – функция состояния, характеризующая меру
неупорядоченности (хаотичности) системы. [ S ]
Дж
. S k ln , где - термодинамическая
моль * К
вероятность реализации данного макросостояния.
Q dU A ' pdV ; TdS dU A ' pdV - объединяет первое и второе начало термодинамики.
dS
Q
T
для необратимых процессов
Связь первого и второго начала термодинамики. Неравенство Клаузиуса. Q dU A ' pdV ;
TdS dU A ' pdV - объединяет первое и второе начало термодинамики.
Q
dS
для необратимых процессов.
T
TdS dU A ' pdV - для обратимых и необратимых процессов.
A ' 0, TdS dU pdV - неравенство Клаузиуса.
Условия самопроизвольного протекания процессов в изолированных и неизолированных
системах. Запишем второе начало термодинамики для любых процессов dS
Q
T
. Пусть система
изолирована, т.е. Q 0 , тогда dS 0 - критерий направленности процесса в изолированной системе.
Все процессы идут до состояния химического равновесия: S 0, S равнов S max .
Изобарно-изотермический и изохорно-изотермический потенциалы. 1) FV ,T - свободная энергия
Гельмгольца (изохорно-изотермический Потенциал); 2) GP ,T - свободная энергия Гиббса.
F U TS , переписываем это уравнение в виде: pdV Am' d (U TS ) , где Am – максимальная
полезная работа. Имеем: pdV Am dF ; если V=const, Am 0 , то 0 dF F 0 . В
неизолированной системе процесс идет в сторону уменьшения свободной энергии Гельмгольца, до
'
'
состояния химического равновесия, т.е. Fравн Fmin . Если работа совершается, то она равна убыли
изохорно-изотермического потенциала ( Am dF , Am F ).
'
'
F pV G - свободная энергия Гиббса. Am' dG , если Am' 0 0 dG, G 0 . Вывод:
GP ,T 0 - процесс идет в обратном направлении, и соответственно если больше нуля – то наоборот.
Для расчетов: при переходе системы из состояния 1 в состояние 2:
G F pV U TS pV G H TS
Максимальная полезная работа. Внутренняя энергия представляет собой полную энергию системы.
Однако второе начало термодинамики запрещает превратить всю внутреннюю энергию в работу.
Можно показать, что максимальная полная работа (как над средой, так и над внешними телами),
которая может быть получена от системы в изотермическом процессе, равна убыли свободной энергии
f
Гельмгольца в этом процессе: Wmax A , где А – свободная энергия Гельмгольца. В этом смысле A
представляет собой свободную энергию, допускающую преобразование в работу. Оставшаяся часть
внутренней энергии может быть названа связанной. В некоторых приложениях приходится различать
полную и полезную работу. Последняя представляет собой работу системы над внешними телами,
исключая среду, в которую она погружена. Максимальная полезная работа системы равна
u
Wmax
G , где G – энергия Гиббса. В этом смысле энергия Гиббса также является свободной.
Am' G, Am' F (изобарно-изотрем. и изохорно-изотрем. соответственно)
H Q p , U QV , тогда подставляя в уравнения Гиббса-Гельмгольца
имеем: Am Q p T (
'
Am'
A'
) p , Am' QV T ( m )V .
T
T
Уравнения Гиббса-Гельмгольца.
G H TS ; F U TS , G H T S ; F U T S .
(
C
G
H
S
H
G
S
)p (
)p S T( )p , (
)p Cp , T ( )p p , (
) S , подставляем в
T
T
T
T
T p
T
T
первоначальные уравнения и получаем:
G H T(
G
F
G
F
)p; F U T (
) p , G H T (
) p ; F U T (
) - последние два
T
T
T
T p
уравнения – уравнения Гиббса-Гельмгольца.
Константа равновесия гомогенных и гетерогенных реакций. Обратимые реакции текут до
состояния химического равновесия. Признаки, характеризующие состояние химического равновесия: 1)
Скорость прямого процесса равна скорости обратного; 2) Неизменность равновесного состояния при
сохранении внешних условий; 3) Динамичность (способность равновесия восстанавливаться при
небольших смещениях внешних условий) 4) G p ,T 0 ( FV ,T 0 ).
После наступления химического равновесия концентрации исходных веществ и продуктов реакции
становятся неизменными.
Закон действия масс: отношение произведения равновесной концентрации продуктов реакции к
произведению равновесной концентрации исходных веществ в степенях, равным стехиометрическим
CDd CEe ...
, где Кс – константа равновесия.
C AaCBb ...
2CO( Г )
Константа равновесия гетерогенной реакции: CO2( Г ) C( ГРАФ )
коэффициентам, постоянна. K c
Kp
p 2 (CO( Г ) )
p(CO2( Г ) )
.
Различные способы выражения константы равновесия. p i Ni p , где Ni – мольная доля i-ого
компонента, p – общее давление, pi – парциальное давление i-ого компонента. pV nRT ,
тогда: K p K C ( RT )
n
n
C,
V
K N Pn . Если n 0 , то K p KC K N . Формулы для выражений
констант равновесий через мольные доли (KN), через активности (Ka), через концентрации (KC), через
парциальные давления (Kp) соответственно:
N Dd N Ee ...
aDd aEe ...
CDd CEe ...
pDd pEe ...
.
,
K
,
K
,
K
a
c
p
N Aa N Bb ...
a Aa aBb ...
C Aa CBb ...
p Aa pBb ...
KN
Принцип подвижного равновесия (принцип Ле-Шателье). Влияние температуры, давления и
концентрации на смещение равновесия. Если на равновесную систему оказываются внешние
воздействия, то в ней пойдут процессы, понижающие это воздействие.
а) Влияние температуры: повышение температуры смещает равновесие в сторону процесса идущего с
поглощением тепла (эндотермический процесс); понижение температуры; понижение температуры
приводит к противоположному результату.
б) Влияние давления: рост давления смещает равновесие в сторону меньшего числа молей, т.е. в сторону
падения давления. Уменьшение давления смещает равновесие в сторону увеличения числа молей
(учитываются только моли газов).
в) Влияние концентрации: 2SO2 O2
2SO3 . При увеличении концентрации SO2 или O2
равновесие смещается в сторону реакции, потребляющей эти вещества, т.е. вправо. При увеличении
концентрации SO3 равновесие смещается влево.
Изотерма химической реакции. Стандартное изменение свободной энергии. A+2B=3D. Пусть эта
реакция далека от равновесия. PA0 ; PB0 ; PD0 – исходные парциальные давления. G=0 - равновесие.
G<0 - равновесие смещено вправо. G>0 - равновесие смещено влево.
Изотерма хим. реакции позволяет зная PA0; PB0; PD0 подсчитатьG и определить в каком направлении
будет протекать реакция при задании исходных значений порционного давления.
G RT ln K p RT ln
( PD0 )3
( PA0 )( PB0 ) 2
при PA0=PB0=PD0=1 атм. - стандартное состояние: G RT ln K p - стандартное уравнение изотермы
химической реакции.
Для всех элементов в стандартном состоянии G0298 = условно приняты = 0!
Изобара и изохора химической реакции.
Изобара
уравнение изобары хим. р-и характеризует собой зависимость константы равновесия Kp от
температуры.
d ln K p
dT
H 0
. Знак производной определяется знаком теплового эффекта реакции. Если реакция
RT 2
эндотермическая, то Q такой реакции берется с (+) - производная положительна, и наоборот.
Изохора
d ln KC H 0
.
dT
RT 2
Методы экспериментального определения и методы расчета термодинамических функций и
констант равновесия. Из экспериментальных методов для газовых реакций отметим так называемый
метод “закаливания”. Сущность этого метода заключается в определении состава равновесной газовой
смеси при постоянной температуре. Газовая смесь выдерживается в замкнутом сосуде до достижения
равновесия при температуре, достаточно высокой для того, чтобы реакция протекала с заметной
скоростью. Далее равновесная газовая смесь резко охлаждается (“закаливается”), что даёт возможность
прямо проанализировать состав смеси на газоанализаторе. Установлено, что в “закалённой” смеси
равновесные концентрации компонентов–участников реакции сохраняют свои значения при комнатной
температуре в течение времени, необходимого для выполнения анализа. Зная равновесные
концентрации участников реакции, можно определить Кс.
Метод гальванического элемента p=1 атм. T=298 К.
Am' nFE , Am' Gp0,T , Gp0,T RT ln K p K p .
Энтропийный метод
Дана реакция: aA bB
1) H
0
298
2) S 298
0
dD eE , при этом p, T=const, T<>298 К.
T
3) H H
0
T
0
298
C dT , где
p
C p a bT ...
298
T
4) ST S 298
0
0
298
C p
T
dT
5) GT H T T ST
0
0
0
6) GT RT ln K p K p
0
7) GT RT (ln
pDd pEe
ln K p )
p Aa pBb
5) Кинетика химических процессов.
Истинная скорость реакции – скорость реакции в данный момент времени. Можно определить
истинную скорость химической реакции как lim ( с/ t) при t 0).
Истинная скорость реакции с точки зрения математики выражается через тангенс угла наклона
касательной к кривой зависимости концентрации реагентов от времени (рис.):
.
ист = tg
Средняя скорость - Рассмотрим в общем виде скорость реакции, протекающей по уравнению
А+В=С+D
(I)
По мере расходования вещества А скорость реакции уменьшается.
Отсюда следует, что скорость реакции может быть определена лишь для некоторого промежутка
времени. Так как концентрация вещества А в момент времени t1 измеряется величиной с1, а в момент t2
- величиной c2, то за промежуток времени ∆t = t2 - t1 изменение концентрации вещества составит ∆с =
с2 - с1, откуда определится средняя скорость реакции (
):
Знак минус ставится потому, что, несмотря на убывание концентрации вещества А и, следовательно, на
отрицательное значение разности с2 - с1, скорость реакции может быть только положительной
величиной.
Средняя скорость химической реакции определяется следующим образом (см. рис.):
.
ср = tg
Закон действующих масс – При постоянной температуре скорость хим. Реакции пропорциональна
произведению концентраций реагирующих веществ. Закон справедлив для наиболее простых по
кинетике гомогенных реакций, полностью выполняется для реакций с участием идеальных газов или
бесконечно разбавленных растворов.
A+B=D, v=k[A][B] k- константа скорости реакции
Физический смысл константы скорости – Константа скорости числено равна скорости реакции для
того случая когда концентрации каждого из реагирующего веществ равны единице.
Связь Константы равновесия с константами скоростей прямой и обратной реакций – константа
k/k
равновесия равна
Механизм химической реакции - Механизм химических реакций рассматривается как
сложная совокупность элементарных химических процессов с участием молекул, атомов,
свободных радикалов, ионов, возбуждённых частиц.(или последовательность протекания
простейших (элементарных) стадий, образования короткоживущих промежуточных
частиц (интермедиатов) реакции).
Одностадийные реакции. Реакции протекающие в одну стадию(например реакция
диссоциации) протекает через образование активированного комплекса, порядок реакции
совпадает с молекулярностью
Сложные реакции. Реакции протекающие последовательно через несколько стадий, или
параллельно. Большенство реакций многостадийно.
Колебательные реакции. Переодический процесс, характеризующийся колебаниями
концентраций некоторых промежуточных соединений и соответственно скоростей
превращения.
Цепные реакции. Идут в три стадии инициирование, рост и обрыв цепи. Цепные реакции
начинаются со стадии инициирования(зарождения цепи), т.е. образования активных частиц –
свободных радикалов(представляют собой осколки молекул имеющие не спаренные
электроны). Инициирование происходит в результате воздействия на систему светом,
излучением высокой энергии, теплом и т.д. также при экзотермических хим. реакций. В стадии
роста цепи происходит взаимодействия радикалов с молекулами образуются продукты реакции
и новые радикалы. В третьей стадии в результате взаимодействия радикалов на стенках
сосудов образуются нейтральные малекулы.
Разветвленные цепные реакции если входе цепной реакции вместо одного рождаются два
или более радикалов, то число радикалов растёт и происходит разветвление цепи. Вследствие
увеличения числа радикалов скорость реакции возрастает лавинообразно и может завершиться
взрывом.
Фотохимические процессы(неуверен что это вообще надо) Процессы протекающим под
влиянием света. Число молекул прореагировавших при поглощении одного кванта света,
называется квантовым выходом фотохимической реакции.
Молекулярность и порядок реакции – С точки зрения числа частиц, участвующих в реакции,
они классифицируются по признаку молекулярности и по признаку порядка. Молекулярность
определяется по числу частиц, одновременное соударение которых приводит к хим взаимодействию:
одномолекулярная -dc/dt=k*c, двухмолекулярная –dc/dt=k*c2, трехмолекулярная -dc/dt=k*c3.
Порядок реакции равен сумме показателей степеней у концентраций в уравнении, выражающем
зависимость скорости реакции от концентраций реагирующих веществ.
Зависимость скорости реакции от температуры - Согласно правилу Вант-Гоффа: повышение
t
температуры на каждые 10 градусов увеличивает скорость реакции в 2-4 раза.
vt 10
10 Основная
vt
причина: с ростом Т в систему поступает дополнительная энергия и число активных молекул резко
возрастает.
Правило Вант-Гоффа – При повышении температуры на каждые 10 градусов константа
скорости гомогенной элементарной реакции увеличивается в два — четыре раза.
Следует из формулы
v v0
T2 T1
10
d ln( Kp) H O
Уравнение Аррениуса:
K1/K2=Kp, H=E1-E2 => dlnK1/dT-dlnK2/dT=(E1dT
RT 2
E2)/(R*T^2) => dlnK1/dT=E1/(R*T2)+C, dlnK2/dT=E2/(R*T^2)+C. Допущения Аррениуса: H=const,
E1/=f(т), E2/=f(т), C=0 => проинтегрируем => lnK=-E/(R*T)+B, A=-E/R => lnK=A/T+B(вроде как
аналитическаое уравнение Аррениуса), А и В характерны для данной реакции.
ln(k)=ln(k0)-E/RT- эмперическое уравнение.
Активационные характеристики реакции
Активированный комплекс – Группировка частиц находящихся во взаимодействии в
результате которого происходит перераспределение связи между ними. В актив. Комплексе
старые связи еще не разорваны но и новые только наметились и еще не образовались. При
распаде комплекса образуются либо продукты реакции либо исходные вещ-ва
Энергия активации – Энергия необходимая для перехода вещ-ва в состояние
активированного комплекса
Энтропия активации – кароче хуй знает что это, вот что нашол я, но это бред=\ энтропиия
системы при переходе от реагентов к Активированному комплексу. Как правило,
определяется в осн. статистич. суммами:
; где
единице объема статистич. суммы Актив. Компл. и реагентов соотв
Графический и аналитический расчеты энергии активации:
и F-отнесенные к
ln(k)
ln(k0)
- Экспериментальные точки. tg ( )
E
R
k0 - экстрополяционное значение константы скорости,
отвечает придельному(гипотетическому) случаю, когда
все молекулы активны, т.е. реакционноспособными.
ln(k1)=ln(k0)-E/RT1
ln( k1 ) ln( k 2 )
ln(k2)=ln(k0)-E/RT2
E 1 1
R T1 T2
k2
k
2.303R lg 2
k1
k1
E
Энергия активации зависит от темеературы
1 1
1 1
T1 T2
T1 T2
R ln
Гомогенный и гетерогенный катализ – Изменение скорости реакции под действием катализаторов катализ. Положительный (просто катализ) и антикатализ. При катализе скорость реакции
увеличивается, при антикатализе - уменьшается. В-ва., увеличивающие скорость реакции катализаторы, уменьшающие - ингибиторы. Как правило катализатор при реакции не расходуется,
ингибитор расходуется. Автокатализ - в роли катализатора - продукты реакции. Прим.
FeO+H2=Fe+H2O
Использование катализатора не отражается на величине изменения свободной энергии системы. Нельзя
увеичить выход продуктов реакции. К сожалению катализаторам присуща определенная
избирательность. Катализатор ищут методом подбора. Реакция разложения: 1) 2) 3) 4) 5) примеров.
Различают гомогенный и гетерогенный катализ. А(г)+В(г)+Кат(г) - гомогенный - 1 фаза.
Ускорительное действие катализатора пропорционально его общему количеству.
Гетерогенный катализ. А(г)+В(г)+Кат(т) в различных фазах (часто газ и тв. в-во)
Ускорение катализа определяется состоянием поверхности катализатора.
Гомогенный катализ. Действие катализатора пропорционально его общему количеству. А+В=АВ (в
газовой фазе), Е, не большая скорость. 1) А+Кат=АКат, Е1 ; 2) АКат+В=АВ+Кат, Е2; Е1<<Е, Е2<<Е
Уменьшение энергии активации. Снижение потенциального барьера. ; NH3=1/2*N2+3/2*H2
E~=300кДж/моль
W E'~=160 кДж/моль 2so2+o2=2so3, NO (Суммарная реакция + еще две)
Катализатор не расходуется, происходит только чисто механический унос.
Гетерогенный катализ. При Г. катализе реагирующие в-ва и катализатор находятся в разных фазах.
А(г)+В(г)+Кат(т) При Г. кат. нужно учитывать адсорбционную способность катализатора, поверхность,
наличие активных центров, их геометрический рисунок, расстояние между активными центрами,
соотношение между активными молекулами и многое другое. Иногда порошкообразный гетерогенный
кат. вызывает мгновенное ускорение реакции. (пример: реакция разложения h2o2 на воду и кислород с
катализатором MnO2) .Электронно-химическая теория Рогинского. АВ+CD=AC+BD
Молекулы адсорбируются на поверхности кат. Ослабевают внутримолекулярные связи, т.е. молекула
перешла в возбужденное состояние. Теория мультиплетов (Баландин) Доказано, что работает не вся
поверхность кат., а только ее активные центры. Каталитические яды - As,P, C2H2, O2, цианиды...
Небольшое количество кат. ядов отравляет огромные поверхности катализатора. Согласно теории
мультиплетов, катализируемая молекула должна расположиться определенным образом на
поверхности кат. вблизи активных центров. (см. рисунки+ формулы) В некоторых случаях требуется
определенное расположение молекул(ы) между 4-6 центрами. СУТЬ КАТАЛИЗА - снижение энергии
активации.
Антикатализ (торможение коррозии) (вып. ингибиторами) Механизм действия: ингибитор
нейтрализует действие (+)кат., если реакция протекает по цепной реакции, то разрывает цепи.
Ингибитор может адсобрироваться на поверхности, препятствуя взаимодействию веществ.
Скорость гетерогенных реакций гетерогенными называются процессы, происходящие на
поверхности раздела соприкасающихся фаз(горение топлива, окисление металлов кислородом воздуха,
растворение газов и твердых тел в жидкостях и т.д. Скорость гетерогенных процессов зависит от
размеров и состояния поверхности раздела фаз. Гетерогенные процессы многостадийны. Кроме
основного процесса протекающего на поверхности раздела фаз, обязательны стадии подвода к этой
поверхности исходных веществ и отвода от неё продуктов реакции. Эти стадии протекают
последовательно, поэтому скорость суммарного процесса определяется скоростью наиболее медленной
стадии.
o Если определяющей стадией является химическая реакция на поверхности раздела фаз, то
гетерогенный процесс описывается законами хим. кинетики (протекает в кинетической
области)
o Если, как чаще бывает, наиболее медленно совершается подвод и отвод соответствующих
веществ то гетерогенный процесс описывается законами диффузии( протекает в
диффузионной области)
Температура сильнее влияет на скорость хим. реакции, нежели на коэффициент диффузии.
При низких температурах гетерогенные процессы протекают в кинетической области, при высоких в
диффузионной.
Скорость диффузии Д
dm
dc
DS
минус означает что диффузия направлена в сторону
d
dx
E
понижения концентрации. D B e RT Е- Энергия активации диффузионного процесса.
Скорость диффузии и хим. реакции одинаковы k c (c0 c) где с – концентрация на поверхности
c0
c0
1
1
-диффузное «сопротивление» системы;
- хим.
c0
1 1
k
k
k
«сопротивление» если k>> то скорость процесса контролируется диффузией и
c0 в противном случае процесс протекает в кинетической области: k c0
раздела фаз c
6) Растворы электролитов и неэлектролитов:
Растворы - гомогенные системы переменного состава, находящиеся в состоянии химического
равновесия.
Растворы - дисперсные системы (Взвеси, истинные, коллоидные)
Взвеси - гетерогенные системы (примеры: туман, суспензии, эмульсия). Не стабильны во времени.
Истинные растворы - однородные (гомогенные, или однофазные) (воздух, вода).
Коллоидные растворы - микрогетерогенные системы (между взвесями и истинными растворами)
Растворитель – вещество, которое находится в избытке и не меняет агрегатного состояния в растворе.
Способы выражения количественного состава растворов:
Концентрация - основная характеристика состава раствора.
1. Процентное выражение концентрации: C (%)
2. Молярная концентрация: CM
раствора.
m1
*100
m2
m1 ì î ëü
[
] -сколько молей растворенного вещества содержится в 1 л
M 1V
ë
3. Моляльная концентрация: Cm
растворителя.
4.Нормальная концентрация: Ñí
m1 *1000 ì î ëü
[
] -количество вещества, растворенного в 1000 г
M 1 * m2 1000 ã
m1 ã ýêâ
[
] -количество эквивалентного вещества в литре
Ý1 * V
ë
раствора.
5. Титр-число граммов растворенного вещества в 1 мл раствора.
6. мольная доля: N i
ni
ni
i
По количеству растворенного вещества и по характеру установившегося равновесия между
растворенным веществом и раствором растворы: насыщенные, ненасыщенные и перенасыщенные.
Растворимость-концентрация вещества в его насыщенном растворе.
Предельная растворимость - сколько граммов вещества растворяется при данной температуре в 100 г
растворителя.
1. Хорошо растворимые - в 100 г растворителя растворяется больше 10 г вещества.
2 .Мало растворимые – в 100 г растворителя 1-10 г в-ва.
3. Плохо растворимые – в 100 г растворителя 0,1-1 г в-ва.
4. Нерастворимые- в 100 г растворителя меньше 0,1 г в-ва.
Физические и химические процессы при растворении:
Растворам присущи признаки как хим. соединений и признаки механической смеси. Признаки хим.
соединения - однородность р-ра, наличие энергетич. эффектов при растворении (поглощение/выделение
теплоты), изменение объема при растворении (объем или больше или меньше суммарного объема
растворимого и растворителя) - перестройка на молекулярном уровне. Признаки мех. смеси - широкий
диапазон изменения состава р-ра (не характерен для хим. соединения), возможность обнаружения в в-вах
р-ра свойства отдельных его составляющих компонентов.
Теории растворов: физическая (Вант-Гофф): согласно физической теории р-ров р-ритель является
индифферентной средой в которой равномерно размешаны молекулы растворенного в-ва.
Взаимодействия между молекулами нет. (Оправдывается для разбавленных р-ров не электролитов).
Химическая (Менделеев):согласно теории Менделеева молекулы р-рителя и р-ренного в-ва образуют
связи. Молекулы сольватов (гидратов) образуют смеси.
3 вида взаимодействия:
1. Электростатическое взаимодействие
2. Дипольное взаимодействие
3. Донорно-акцепторное взаимодействие
Сольватация- взаимодействие растворенного вещества с растворителем.
Теплота растворения в-ва: Теплота, выделяющаяся или поглощающаяся, 1 моля вещества при данной
температуре и бесконечном разбавлении.
Бесконечное разбавление- раствор, в котором частицы вещества не взаимодействуют между собой, и
дальнейшее растворение не приводит к изменению внутренней энергии раствора.
Í
p
H êðèñò . Í
ñî ëüâ.
Í
äèô ô óçèè
Растворимость твердых веществ в жидкостях зависит от природы твердых тел, природы растворителя
и температуры. Происходит с эндоэффектом.
С повышением температуры растворимость, как правило, увеличивается. Давление мало влияет на
растворимость твердых тел в жидкостях.
Растворимость жидкостей в жидкостях:
Обычно увеличивается с повышением температуры и почти не зависит от давления. Оно проходит с
эндоэффектом.
Критическая температура-температура, при которой возникает неограниченное растворение жидкости в
жидкости. Неограниченная растворимость- жидкости смешиваются друг с другом в любых пропорциях.
Закон распределения: Если в систему из двух несмешивающихся жидкостей внести тритий компонент,
то он распределится в первых двух согласно коэффициенту распределения. K
C1
C2
Растворимость газов в жидкостях: растворимость газов в жидкостях с повышением температуры
уменьшается. При кипячении жидкости происходит практически полное удаление из нее растворенных
газов. Растворимость газов в жидкостях зависит от давления, под которым он находится над жидкостью.
Происходит с экзоэффектом.
Закон Генри: при растворении газа в жидкости общий объем системы уменьшается. При постоянной
температуре растворимость газа пропорциональна его давлению:
C Ã * ð , где р- давление, Г- постоянная Генри, С- концентрация.
Закон Дальтона: при рассмотрении смеси газов общее давление равно сумме всех парциальных
давлений: p
pi . При растворении смеси газов растворимость каждого из них пропорциональна его
i
парциальному давлению: Ci à * pi .
Законы Рауля: 1. Давление пара растворителя над раствором пропорционально мольной доле
0
растворителя в растворе. P1 P1 * N1
Либо: Относительное понижение давления насыщенного пара над раствором равно мольной доле
растворенного вещества. P1 P *(1 N 2 ) ;
0
1
0
1
0
1
P
N2
P
2. Повышение температуры кипения и понижение температуры замерзания пропорциональны числу
частиц растворенного вещества и не зависит от его природы.
Определение молекулярных масс растворенных веществ (эбулиоскопия и криоскопия)
Tk K ýÑm , где K ý - эбулиоскопическая константа
T K êð Ñm , где K êð - криоскопическая константа
ç
Константы зависят от природы растворителя.
Эбулиоскопия и криоскопия - методы определения молекулярных масс либо по повышению температуры
кипения, либо по понижению температуры замерзания.
Ñm
m1 *1000
m *1000
m *1000
, отсюда Tk K ý 1
; M x Ký 1
M x * m2
M x * m2
Tk * m2
Раствор электролитов: Электролиты-вещества, растворы или расплавы, проводящие электрический ток.
(проводники 2-го рода).
Характеризуются наличием ионной связи. Электролиты не подчиняются законам Рауля, так как
температура кипения и замерзания зависит не только от аналитической концентрации, но и от степени
диссоциации. Степень диссоциации различна для разных электролитов и характеризует молярную
электропроводимость.
Для одного и того же раствора вещества степень диссоциации растет при уменьшении концентрации
раствора.
Электролитическая диссоциация:
Электролиты - в-ва, которые в р-ре или расплаве полностью или частично состоят из ионов, заряженных
частиц. Электролиты 2 рода. При прохождении эл тока через электролиты происходит электролиз. Р-ры
электролитов не подчиняющиеся законам Рауля. Р-ры э-литов кипят при температуре большей, и
замерзают при температуре меньшей, чем р-ры не электролитов той же молекулярной концентрации.
Причину этого объяснил С.А. Аррениус. Теория электролитической диссоциации.
Согласно Аррениусу электролиты при растворении или плавлении распадаются на ионы. При этом
каждый ион – самостоятельная частица. В результате диссоциации моляльная концентрация
увеличивается, отсюда отклонение от закона Рауля. Степень полярности характеризуется величиной
диэлектрической проницаемости р-рителя “Е” Чем больше Е тем ярче выражена гидратация. (ряд
примеров)
По способностям к диссоциации электролиты делятся на сильные и слабые:
Сильные- практически полностью диссоциируют на ионы, процесс необратим.
Слабые- обратимый процесс, так как в растворах слабых электролитов есть как и ионы так и
недиссоциированные молекулы.
Степень диссоциации:- та доля растворенного электролита, которая распалась на ионы.
Удельная электропроводимость- величина обратная удельному электросопротивлению.
1
[Î ì 1ñì
1
] , т. е. это электропроводимость объема раствора, заключенного в кубический сосуд с
2
ребром 1 см, противоположные стенки которого – электроды площадью 1ñì .
Эквивалентная электропроводимость - электропроводимость такого объема раствора, в котором
содержится 1 г-эквивалент растворенного вещества.
1
* [Î ì
Ñ
1
* ñì 2 * ã ýêâ1 ]
Константа диссоциации в одном и том же растворителе при постоянной температуре – величина
постоянная, она свойственна данному электролиту.
[ A ]n [ B ]m
Kä
[ An Bm ]
Степень диссоциации характеризует состояние электролита в растворе только данной концентрации и
меняется с ее изменением.
Kä
- закон разведения Освальда.
C
>0.7 – сильные э-литы
<0.3 – слабые э-литы
=0.3-0.7 – средние э-литы
1. Сильные – почти все соли, кроме CdCl2, HgCl2, Fe(CNS)3, Pb(CH3COO)2
Также минеральные кислоты, включая H2SO4, HNO3, HCl, HBr, HY, HClO4
Гидроксиды щелочных и щелочноземельных металлов.
2 Слабые – почти все органические кислоты, некоторые минеральные к-ты, включая H2CO3, H2S, HCN,
H2SiO3, H3AsO3, H3AsO4, HF, основания NH4OH, H2O
3. Средние э-литы H3PO4, H2C2O4, Mg(OH)2 и другие.
Сильные электролиты в растворах диссоциируют практически полностью. Однако из-за
электростатического притяжения разноименно заряженными ионами сильный электролит ведет себя так,
как если бы он диссоциировал не полностью. Это и называется кажущийся степенью диссоциации.
Если условно взять какой-либо ион за центр и окружить его сферой произвольного радиусы, то его в
большей степени будут окружать ионы противоположного знака. Это окружение- ионная атмосфера.
Активность учитывает все виды взаимодействия между ионами. Активность определяется как величина,
подстановка которой вместо концентрации в термодинамические уравнения, действительные для
идеальных систем, делает их применимыми к реальным системам.
a * c , где а- активность, с – концентрация, а -коэффициент активности.
Произведение растворимости: в насыщенном растворе трудно-растворимого электролита произведение
концентраций (активностей) его ионов при данной температуре есть величина постоянная, называемая
произведением растворимости.
Если р-р сильно разбавлен, то вместо активности можно брать концентрацию.
Ï Ð [ A ]n *[ B ]m или Ï Ð An * Bm
Условия образования осадка:
[ A ]n *[ B ]m >ПР
Условия растворения осадка:
[ A ]n *[ B ]m <ПР
Электролитическая диссоциация воды. Ионное произведение воды. Водородный показатель.
Удельная электроотрицательность дистиллированной воды составляет:
=5.48*10-8 Ом-1*см-1
H20H++OH- - 57.54кДж/моль (термохимическое правило знаков т.е. теплота поглощается)
Kд=[H+][OH-]/[H20]=1.86*10-18 при t=220
Вода-смесь молекул воды, катионов водорода и анионов гидроксила, в знаменателе – концентрация
недиссоциированных молекул воды в воде.
KД[H2O]=[H+][OH-]=KВ – ионное произведение воды.
Полагаем, что концентрация недиссоциированных мол-л воды в воде равна общей мольной концет-ии
воды.
[H2O]=1000/18.016=55.56 моль/л
КВ=[H+][OH-]=1.8*10-16*55.56=10-14 при t=220С
Поскольку процесс диссоц-ии воды – пр-сс эндотермический, то с ростом температуры дис-ция воды
будет усиливаться,. а значит ионное произведение воды будет увеличиваться.
КВ=а*10-14
t0C
a
KB
0
0.13
0.13*10-14
22
1.0
10-14
25
1.008
…
50
5.66
…
100
74.0
…
[H+]=[OH-] – условие нейтральности воды.
[H+]=КВ=1.008*10-14=1.004*10-7 г-ион/л
t=250C - [H+]=10-7 г-ион/л (приблизительно)
Мы можем определить :
=[H+]/[H20]=1.004*10-7/55.56=1.8*10-9 при t=250C
нейтральный раствор:[H+]=10-7 г-ион/л
кислый раствор: [H+]>10-7 г-ион/л
щелочной раствор: [H+]<10-7 г-ион/л
Допустим : [H+]=10-4; КВ=[H+][OH-]=10-14
Отсюда находим [OH-]=КВ/[H+]=10-10
10-4>10-10[H+]>[OH=], раствор имеет кислую реакцию. Т.к. неудобно иметь дело с отрицательными
степенями договорились степень кислотности растворов оценивать при помощи водородного покпзателя
pH=-lg[H+]
нейтральный раствор pH=7
кислый раствор pH<7
щелочной рас-р pH>7
Сок лимона-2.1, черный кофе-5, дождевая вода-6.5, кровь здоровогочел-ка-7.4.
Индикаторы- это слабые органич-ие кислотыили основания, к-ые меняют окраску при изменении
велечины pH.
НиндН++инд - (лакмус, фенолфталеин)
индОНинд++ОНрН для лакмуса=3ед(58) или 2ед(68) (красный окрассиний окрас)
Введем каплю лакмуса в раствор кислоты. Равновесие реакции смещается влево или вправо(кислая –
щелочная среда)
Фенолфталеин – кислота. Молекулы – бесцветные,а анион (инд-) емеет малиновый цвет). Если введем в
рас-р щелочи, то за счет связывания катионов Н, получается малиновый цвет, в кислом рас-ре окрас
бесцветный.
Наиме
кислота
интервал изменен
нован
или
изменен
ие
ие
основание
ия рН
окраски
Инд
метил основание
3.1-4.4
красный
-желтый
оранж
метил основание
4.2-6.3
красный
-рот
-желтый
лакму
кислота
5(6)-8
красный
с
-синий
фкислота
8.3-9.8
бесцветфтале
малинов
ин
ый
тимол
кислота
9.8-10.5 бесцветфтале
синий
ин
Универсальный Инд-смесь целого ряда инд-оров.
Гидролиз солей.
-разложение солей под действием воды.
С точки зрения теории электрической диссоциации, кислота – вещ-во, кот-е в водном раст-ре отщепляет
только 1-н вид катионов – Н+. НnAnH++An-.Основание – такое вещ-во,кот-е в водном р-ре отщепляет
только 1-н вид анионов – анионы гидроксила. Me(OH)p Mep++pOH- - нейтрализация. Наряду с
кислотами и основ-ми сущ-ют амфолиты – вещ-ва, кот-е в зависимости от величины РН могут дисс-ть
или по схеме кис-ты или по схеме основ-я. Zn(OH)2, Cr(OH)3.. Zn(OH)2Zn2+ +2OH-H2ZnO2
2H++ZnO22- Гидролиз – реакция обратная р-ции нейтрализации. Если ра-ряем соль в воде, то соль
диссоц-т на ионы (катион и анион), но одновременно вода диссоц-т на катион водорода и ОН-. Ионы соли
могут связываться с Н+ и ОН-. Если степень связанности Н+ с ионами соли будет >, чем ОН-, то р-р соли
перейдет в щелочную реакцию, если будут > связаны ОН-, то в кислую р-ю.
Примеры гидролиза.
1) соль слабой к-ты и сильного основ-я.
CH3COONaCH3COO-+Na+
H20 Н++ OH_
CH3COONa+ H20 CH3COOH+NaOH
Раствор соли, образов-й слабой кисло-й и силь-м основанием имеет РН>7
Константа гидролиза КГ – это константа равновесия реакции гидролиза.
Кв
КГ = Кв/КД[CH3COOH]
КГ = [CH3COOH] [OH-]/[CH3COO-] [H20]
КГ = [CH3COOH] [OH] [H+]/[CH3COO-] [H+]
+
КД CH3COOH =[CH3COOH ] [H ]/[CH3COOH]
2) соль слабой 2-х – основной кислоты и сильного основания.
1 ступень: Na2CO3+H20=NaHCO3+NaOH
2 ступень: NaHCO3 +H20= NaOH+H2CO3
{ионный вид} РН>7
{ионный вид} PH>7
3) cоль сильной к-ты и слабого основания
КГ = Кв/КД[NH4OH]
NH4Cl+H20=HCL+NH4OH
PH>7
4)соль сильной к-ты и слаб многоатомного основ-я
Al2CO3AlOHCl2Al(OH)2ClAl(0H)3
5) соль сильной к-ты и силь. основ-я
KNO3+H2O=KOH+HNO3 Гидролизу не подвергается.Р-р такой соли имеет нейтральную р-цию. РН=7
6) соль слаб к-ты и слаб основ-я
CH3COONH4+H20=CH3COOH+NH4OH
КГ = Кв/КД CH3COOHКД NH4OH Если к-та сильнее основания , то р-р имеет кислую р-цию, если основ-е
сильнее, то щелоч-ю реакц-ю
7 ) соль очень слаб к-ты и очень слаб основ-я Al2S3+6H20=2Al(OH)3+3H2S2. Соль подвергается
полному гидролитическому разложению.
Степень гидролиза h=число гидролизов молей соли / общ число растворен-х молей соли *100%. h
увеличив-я с ростом t и с ростом разбавления. h при t т.к. Кв с ростом
SbCl3+2H20Sb(OH)2Cl+2HCl
РН>7 Р-ры многих солей во избежание гидролиза можно
хранить в подкисленном или в подщелочном виде.
7) Физико-химический анализ
Физико-химический анализ, его сущность: Наиболее полная характеристика системы может быть
получена с помощью ф.-х. анализа, основателем которого был академик Н.С. Курнаков. Сущность ф.-х.
анализа заключается в том, что на основании экспериментальных данных строят диаграммы состояния
системы, выражающие зависимость какого-либо физического свойства системы (температуры
плавления, твердости, электропроводимости, плотности, удельного объема и т.д.) в виде функции ее
химического состава. На основании вида полученной диаграммы представляется возможным сделать
целый ряд выводов относительно физических и химических свойств системы и ее строения.
Для металлических систем особое значение имеет построение диаграмм плавкости. Геометрические
особенности диаграммы позволяют наглядно определить появляющиеся в этой системе новые фазы,
границы устойчивости границы устойчивости отдельных фаз, условия их совместного существования,
характер химических превращений в данной системе и т.д.
Понятия: фаза, независимые компоненты, степени свободы, вариативность химического
равновесия:
Фаза: гомогенная часть гетерогенной системы, обладает поверхностью раздела и определяется
набором физических свойств.
Независимые компоненты (компоненты системы) – индивидуальные вещества, наименьшее число
которых необходимо и достаточно для образования всех фаз данной системы. Под индивидуальным
веществом понимаются как простые вещества, так и химические соединения.
Степени свободы – независимые переменные (условия), которые можно произвольно менять, е
нарушая состояние системы (числа и характера фаз)
Вариативность химического равновесия:
Правило фаз Гиббса: позволяет определить число фаз в системе при равновесии и изменение этого
числа при изменении какого-либо параметра. Для системы, состоящей из «К» компонентов и имеющей
«Ф» фаз, число степеней свободы «С»: С=К-Ф+2. Если давление или температура =const, то С=К-Ф+1.
Диаграмма состояния унарной системы-воды: Диаграмма состояния – это диаграмма, указывающая,
в каких фазовых состояниях находится система в зависимости от условий: температуры (Т), давления
(Р) и состава.
P
В качестве однокомпонентной системы возьмем воду. Поскольку
К=1, состав как переменная отпадает, и фактически имеем Р-Т
I
диаграмму. С=К-Ф+2=3-Ф; максимальное число фаз, находящихся в
A
C I D
равновесии, равно 3. I- поле СОВ – область существования твердой
фазы-льда; II-СОА – жидкая фаза – вода; III-АОВ – водяной пар.
I
О
Линия ОА – сосуществование двух фаз: ж – г, т.е.равновесие
«испарение – конденсация». Линия ОВ – равновесие «сублимация –
B
III
конденсация», т – г. Линия ОС – «плавление – кристаллизация», т – ж.
Точка «О» отвечает равновесному сосуществованию 3 фаз, где С=3T
Ф=0, т.е. это точка нонвариантного равновесия, положение ее строго
определено: Р=4,6 мм рт.ст., Т=+0,01°С.
В тоске D: С=3-1=2, т.е. оставаясь на поле можно менять 2 переменные.
Кривые охлаждения и нагревания: Диаграммы плавкости строят, исходя обычно из кривых
охлаждения, которые изображают в координатах температура –
время. На рис.1 система I в интервале t1-t2 не испытывает никаких
фазовых превращений, температура меняется монотонно. Кривая II
отвечает охлаждению воды от 150°С (пар при атмосферном
давлении) до 0°С и ниже. При 100°С наблюдатеся горизонтальный
участок, отвечающий процессу конденсации пара, температура
поддерживается неизменной, пока не исчезнет весь пар.
Постоянство температуры обеспечивается за счет выделения в
процессе конденсации пара теплоты испарения воды. От 100
до 0°С наблюдается монотонный ход кривой охлаждения,
остывает жидкая вода. При 0°С наблюдается второй
горизонтальный участок, отвечающий фазовому переходу –
процессу кристаллизации. Температура поддерживается
постоянной за счет выделения теплоты плавления.
Рассм.кривые охлаждения чистого металла I и сплава с
эвтектикой (рис.2). На кривой охлаждения I чистого метала А
наблюдается одна площадка вс, отвечающая процессу
кристаллизации металла. При tпл. В равновесии находится
жидкая и твердая фазы одинакового состава. Образование твердой фазы сопровождается
выделением теплоты плавления. Кривая II отвечает охлаждению сплава А-В. От «а» до «а’»
температура меняется монотонно, остывает жидкая фаза. При достижении точки а’ из жидкой фазы
начинают выпадать кристаллы А, при этом жидкая фаза начинает обогащаться компонентом В. Таким
образом, в интервале «а’-в» в равновесии находятся жидкая и твердая фазы расславленного состава,
площадки на кривой охлаждения не наблюдается. За счет выделения при кристаллизации А теплоты
плавления этого компонента темп охлаждения сплава замедляется, и на кривой охлаждения
наблюдается перелом. Выпадение кристаллов А продолжается до тех пор, пока жидкая фаза не
достигнет так называемого «эвтектического состава». Основной особенностью эвтектического сплава
является то, что он кристаллизируется подобно чистому компоненту – на кривой охлаждения
наблюдается площадка. Из жидкой фазы одновременно выпадают кристаллы А и В и, таким образом,
при tЕ в равновесии находятся (отрезок «вс») жидкая и твердая фазы одинакового состава. Отрезок
«cd» отвечает охлаждению твердого сплава.
Остановки в падении температуры могут наблюдаться и на кривой охлаждения уже твердого металла,
указывая на переходы металла из одной аллотропной формы в другую.
Принцип построение диаграмм плавкости бинарных систем:
Т
для построение необходимо знать Р,Т и состав: Х1+Х2=1 (Х –
мольная доля компонента, %) Т.к. для бинарных систем Х1=1-Х2,
то состав таких систем изображается в виде отрезка.
М
А
В
В конденсированных системах Р=const, величина постоянная и не оказывает существенного влияния на
положение равновесия. С=3-Ф (К=2).
Таким образом можно изобразить состояние бинарной системы на плоскости. (Т-х. диаграмма). Т –
температура фазовых превращений.
Здесь любая точка отражает состояние системы, т.е. фигуративная.
Построение диаграммы осуществляется путем изучения кривых охлаждения и нагревания (т.е.
изменения Т со временем) – исслед. их перегибы, переломы, площадки…
Правила построения:
Принцип непрерывности и соответствия Курнакова: принцип непрерывности устанавливает, что
при непрерывном изменении давления, температуры, концентраций свойства отдельных фаз системы
изменяются так же непрерывно. Свойства всей системы в целом изменяются непрерывно лишь до тех
пор пока не изменится число или характер ее фаз. При появлении новых или исчезновении имеющихся
фаз свойства системы в целом меняются скачком.
По принципу соответствия каждой совокупности фаз, находящихся в равновесии в данной системе,
отвечает на диаграмме определенный геометрический образ. В двухкомпонентной системе одной фазе
на диаграмме соответствует участок плоскости, кристаллизации твердой фазы – кривая начала
кристаллизации, равновесию между тремя фазами – точка пересечения кривых и т.д.
Диаграмма плавкости бинарной системы с эвтектикой: Для бинарных систем с эвтектикой,
характерна полная взаимная растворимость компонентов в жидком состоянии и нерастворимость в
твердом (Pb-Ag, Cd-Bi, Tl-Au). По правилу рычага можно определить соотношение по массе жидкой и
твёрдой фаз сплава определённого состава при определённой температуре, Мж/Мтв=хy/yz. Чем ближе
сплав к эвтектическому состоянию, тем больше доля жидкой фазы при данной температуре.
Эвтектический состав кристаллизуется подобно чистому компоненту. Линия отделяющая чистую
жидкую фазу – линия ликвидуса.
Правило рычага: (для определ.относит.количеств. сосуществующих равновесных фаз) массы
сосуществующих равновесных фаз соотносятся друг с другом как отрезки на осях, отсекаемые их
общей ординатой.
Диаграмма плавкости бинарной системы с неограниченными твердыми растворами: Для
бинарных систем с неограниченными твердыми растворами, характерны полная взаимная
растворимость компонентов как в жидком так и в твердом состоянии (Cu-Ni, Bi-Sb, Ag-Au). Первые
порции твердой фазы обогащены более тугоплавкими компонентами, для не чистого вещества
отсутствуют площадки на кривых охлаждения. Ликвация: более твердоплавкие сплавы в слитке –
снаружи, а более легкоплавкие – внутри. Для устранения ликвации проводят отжиг – нагрев слитка до
температуры солидуса в течение 10 часов, за это время диффузия выравнивает состав слитка по
сечению. Правило рычага: Мж/Мтв=zу/хy. Верхняя линия – ликвидуса, нижняя – солидуса.
Диаграмма плавкости бинарной системы с ограниченными твердыми растворами: Для бинарных
систем с ограниченными твердыми растворами – с эвтектикой и с перитектикой, характерна полная
взаимная растворимость компонентов в жидком состоянии и ограниченная в твердом. Если с
эвтектикой (Cu-Ag, Pb-Sn), то точка нонвариантного равновесия лежит ниже температур плавления
чистых компонентов, если с перитектикой (Hg-Cd), то точка лежит между температурами плавления
чистых компонентов. Перитектическое превращение – результат химического взаимодействия ранее
выпавшей твердой фазы с жидкой фазой определенного состава, в результате образуется новая твердая
фаза.
Диаграмма плавкости бинарной системы с устойчивым и неустойчивым химическим
соединением: (с дистектикой) Устойчивое, плавящееся без разложения химическое соединение АmВn
(с так называемой конгруэнтной температурой плавления) проявляется на диаграмме плавкости в виде
максимума. Чем круче максимум, тем устойчивее химическое соединение. Состоит как бы из двух
простейших эвтектических диаграмм, где рольодного из компонентов играет устойчивое химическое
соединение АmВn.
8) Электрохимия
Скачок потенциала на границе "металл-раствор".
Поверхность электрода заряжается отрицательно за счёт оставшихся на нём электронов, вследствие
чего перешедшие в раствор гидратированные катионы не могут отойти от катиона и остаются вблизи
него, создавая в совокупности с электронами так называемый двойной электрический слой.
Пограничный слой жидкости заряжается положительно, поверхность металла –отрицательно,
образуется своего рода конденсатор, состоящий из двух противоположно заряженных обкладок.
Возникает скачок электрического потенциала- электродный потенциал.
Равновесное значение скачка потенциалов на границе раздела электрод/раствор определяется исключительно
особенностями электродной реакции и не зависит от природы электрода и адсорбции на нём поверхностноактивных веществ. Эту абсолютную разность потенциалов между точками, находящимися в двух разных фазах,
нельзя измерить экспериментально или рассчитать теоретически. Практическое значение имеют относительные
электродные потенциалы, обычно называемые просто электродные потенциалы, представляющие собой разность
электродных потенциалов рассматриваемого электрода и электрода сравнения — чаще всего нормального
водородного электрода, электродный потенциал которого условно принимается равным нулю.
Равновесный электродный потенциал.
Это потенциал, устанавливающийся в условиях равновесия электродной реакции. Электродные потенциалы
отвечают обратимому протеканию процессов или, что тоже самое, состоянию электрохимического равновесия на
электродах. Для его нахождения, как правило, находят разность потенциалов измеряемого электрода, потенциал
которого условно принимают равным нулю.
Водородный электрод.
Водородный электрод (ВЭ) представляет собой пластинку или проволоку из металла, хорошо поглощающего
газообразный водород (обычно используют платину или палладий), насыщенную (абсорбированную) водородом
(при атмосферном давлении) и погруженную в раствор, содержащий ионы водорода. Потенциал платины зависит
от концентрации ионов [Н+] в растворе. Электрод является эталоном, относительно которого ведется отсчет
электродного потенциала определяемой химической реакции. При давлении водорода 1 атм., концентрации
протонов в растворе 1 моль/л и температуре 298 К потенциал ВЭ принимают равным О В. При сборке
гальванического элемента из ВЭ и определяемого электрода, на поверхности платины обратимо протекает реакция:
2Н++ 2е- =Н2
то есть, происходит либо восстановление водорода, либо его окисление - это зависит от потенциала реакции,
протекающей на определяемом электроде. Измеряя ЭДС гальванического электрода при стандартных условиях
определяют стандартный электродный потенциал определяемой химической реакции.
Потенциал ВЭ воспроизводится с очень высокой точностью, поэтому он-то и принят как эталон при создании
шкалы электродных потенциалов!
ВЭ применяют для измерения стандартного электродного потенциала электрохимической реакции, для
измерения концентрации (активности) водородных ионов, а так же любых других ионов. Применяют ВЭ так же
для определения произведения растворимости, для определения констант скорости некоторых электрохимических
реакций.
Концентрация растворённого в платине водорода пропорциональна его парциальному давлению [Н2]=kр(Н2),
где к - постоянная при данной температуре.
Потенциал ВЭ
ф=фO+0,059lg[Н+ ]-0,0301lg(р(Н2)), но обычно парциальное давление водорода поддерживается равным
атмосферному, а следовательно 0,0301g(p(H2)=0, и т.к. фO) принят равным нулю, то ф=0,059lg[Н+], или, учитывая
что lg[H+]=- рН => ф= - 0,059рН.
Стандартные электродные потенциалы.
Потенциал электрода в растворе, в котором ионы, определяющие электродный процесс, имеют активность, равную
единице. Величины стандартного электродного потенциала измеряются относительно стандартного (нормального)
водородного электрода, потенциал которого условно принимается равным нулю и выражается в вольтах. Ряд
стандартных электродных потенциалов позволяет решать вопрос о направлении самопроизвольного протекания
ox-red реакций. Определяющим фактором служит знак изменения энергии Гиббса реакции. В гальваническом
элементе реакция может самопроизвольно протекать в таком направлении, при котором электрохимическая
система с более высоким значением электродного потенциала выступает в качестве окислителя, т. е.
восстанавливается. 1) Если ох и red расположены далеко друг от друга в ряду (ЭП), то направление реакции
практически полностью определяется их взаимным расположением в этом ряду. (пр. цинк будет вытеснять медь из
водного раствора её соли при любой практически осуществляемой концентрации этого раствора, ибо фZn=--0763, а
фСu=+0,337). 2) Если ох и red расположены близко друг к другу в ряду (ЭП), то при решении вопроса о
направлении самопроизвольного протекания реакции надо учитывать влияние на электродные потенциалы также и
концентраций соответствующих веществ.
Ряд напряжений Бекетова.
Если из всего ряда стандартных (ЭП) выделить только те, электродные процессы которые отвечают Mn+ + ne-=M то
получим ряд напряжения металлов. Последовательность расположения металлов в порядке возрастания значений из
стандартного потенциала (за ноль принят потенциал водорода). Ряд напряжений некоторых металлов: К, Са, Mg,
Al, Zn, Сr(III), Fe(II), Н2, Си, Ag, Hg, Au.
Каждый металл вытесняет из растворов солей металлы, стоящие справа от него; металлы, которые
расположены левее H2 вытесняют его из кислот. При этом чем дальше расположен металл в ряду
напряжений, тем более сильным окислителем в водном растворе являются его ионы, и наоборот, чем
ближе металл к началу ряда, тем более сильные восстановительные свойства проявляет простое
вещество - металл.
Активные металл вытесняют водород не только из воды, но и из любого водного раствора, поэтому
взаимное вытеснение металлов из растворов их солей практически происходит лишь в случае
металлов, расположенных в ряду после магния. Вытеснение металлов из их соединений другими
металлами впервые подробно изучал Бекетов Н.Н. Он расположил металлы по их химической
активности в "вьггеснительный ряд", являющийся прототипом ряда напряжений металлов
При сравнении металлов в ряду напряжений за меру химической активности принимается работа
превращения металла, находящегося в твёрдом состоянии, в гидратированные в водном растворе. Эта
работа - сумма трёх слагаемых: энергии атомизации - (характеризует прочность кристаллической
решетки данного металла) превращения кристалла металла в изолированные атомы, энергии ионизации
свободных атомов металла (отрыва от них валентных электронов - определяется положением в (ПС)) и
энергии гидратации образующихся ионов (зависит от электронной структуры иона, его заряда и
радиуса).
Гальванические элементы.
(хим.Е->электр. Е)
Это химический источник электрического тока, названный в честь Луиджи Гальвани. Принцип
действия гальванического элемента основан на взаимодействии двух различных металлов через
электролит, приводящем к возникновению в замкнутой цепи электрического тока, за счёт
самопроизвольной ox-red реакции. Такая система делает возможным пространственное разделение oxred реакции: ох - протекает на одном металле, a red на другом. Таким образом, электроны передаются
от восстановителя к окислителю по внешней цепи. ЭДС гальванического элемента зависит от
материала электродов и состава электролита.
Электрод - система из двух контактирующих фаз.
Медно-цинковый гальванический элемент. Это элемент Яхоби-Даниэля.
Состоит из медной пластины, погруженной в раствор сульфата меди (медный электрод), и цинковой
пластины, погруженной в раствор сульфата цинка (цинковый электрод). Оба раствора соприкасаются
между собой через перегородку из пористого материал а( чтобы не смешивались). При работе элемента
(когда цепь замкнута), цинк окисляется: на поверхности его соприкосновения с раствором атомы цинка
превращаются в ионы и, в процессе гидратации, переходят в раствор. Высвобождающиеся при этом
электроны движутся по внешней цепи к медному электроду. Полуреакции (электрохимическое ур-е)
Zn=Zn2++2e- Cu2+ + 2e-=Cu. Суммарное ур-е получается при сложении уравнений обоих полуреакций:
Zn + CuSO4 > Сu + ZnSO4 ИЛИ Zn + Cu2+ > Zn2+ + Сu. (анод (Zn)- ох; катод (Сu) - red).
Процессы на электродах.
Электрод - система из двух контактирующих фаз. (анод - ох; катод - red).
Ox-Red реакции складывается из полуреакций окисления и восстановления. Каждая реакция протекает
на соответствующем электроде. В связи стем, что реакцию разделяют на полуреакции, ЭДС тоже
принято представлять в виде разности двух величин, каждая из которых отвечает одной полуреакции
(электродные потенциалы). Е = ф+ -ф-. Потенциалы зависят от: 1) природы в-в - участков электродного
процесса. 2) соотношения между ко1ще1пу>Ш1иями(акт1шностей) этих веществ. 3) температуры
системы. Эта зависимость выражается уравнением: ф=фO+(2,ЗRТ)/(zF)*lg([Ох]/[Red]), где: фO стандартный (ЭП) данного процесса (типа const, учитывает влияние природы в-в); R - универс. газ.
const; Т - абсолютная температура; z - число электронов; участвующих в процессе; F - const Фарадея;
[Ох] и [Red] - произведения концентраций веществ, участвующих в процессе в окисленной и
восстановленной формах. (ф=фO при lgl-О… При "стандартной концентрации")
Зависимость электродных потенциалов от концентрации электролита: ф=фО+(0,059/n)*1g[Меn+], (если
условия отличаются от стандартных) где: [Men+] - концентрация ионов металла в электролите, моль/л.,
n- заряд иона металла.
+ см. водородный электрод.
ЭДС и напряжение разряда
ЭДС гальванического элемента зависит от материала электродов и состава электролита.
ЭДС - разность электродных равновесных значений электродных потенциалов, измеренных в
отсутствии тока во внешней цепи.
+ см. процессы на электродах
Термодинамика гальванического элемента: зависимость ЭДС элемента от природы
реагирующих веществ, температуры и концентрации (активности).
ЭДС элемента связана с энергией Гиббса - GT zFE , где: z - заряд ионы в-ва; F - consi Фарадея; Е
- ЭДС. Изменение энтальпии при работе гальванического элемента: H T GT TnF (
dE
)
dT
dE
dT
Изменение энтропии реакции, при работе гальванического элемента: S T nF
Зависимость ЭДС от концентрации электролита: E=EO+(2,3RT)/(zF )*lg([Meln+]/[Me2n+]) где [Meln+]
концентрация ионов металла, имеющего более электроотрицательное значение потенциала моль/л
[Me2n+]-концентрация ионов металла, положительного электрода гальванического электрода, моль/л.
Формула Нернста для электродного потенциала.
ф =фО + ((RT/(nF)) ln([Ox]*[A]p/[Red]*[B]m), где: [Ох] и [Red] — начальные концентрации
окислительной и соответственно восстановительной формы вещества, для которых рассчитывается
окисшггельно-восстш1овитеяьнып потенциал; [А] и [В] - начальные концентрации других веществ,
участвующих в электродной реакции (обычно вода, ионы Н+ или ОН-); тир - стехиометрические
коэффициенты в уравнении электродной реакции.
или ф=фO+(0,059/n)*lg[Men+];
или еще E=EO-((RT)(nF))*ln((Cdd*Cee)/ (Caa*Cbb))
Расчет ЭДС элемента.
1)Е=ф+-фф=фO+(0,059/n)*lg[Men+]
ф=фO + ((RT)/(nF)) ln([Ox]*[A]p/[Red]*[B]m) см. Формула Нернста
2)E=EO-((RT)(nF))*ln((Cdd*Cee)/ (Caa*Cbb))
Ox-Red процессы.
Ox-Red реакции - реакции, в результате которых изменяется степень окисления элементов реакции. Ох
- (отдача электронов) - сопровождается повышением степени окисления (эл-ты с высшей степенью
окисления). Red - (присоединение электронов) - сопровождается понижением степени окисления (эл-ты
с низшей степенью окисления).
Вещество, в состав которого входит окисляющийся элемент - восстановитель, а в-во содержащее
восстанавливающийся элемент - окислитель.
Число электронов, отдаваемых молекулами (атомами, ионами) восстановителя, равно числу
электронов, присоединяемых молекулами (атомами, ионами) окислителя.
Пример: HI+H2SO4->I2+H2S+H2O; йод окисляется, сера восстанавливается ; 2I-=I2+2е - (окисление);
SO42-=>H2S;
SO42-+10H+=>H2S+4H2O; SO42-+10H++8e=H2S+4H2O - (восстановление); татем уравниваем складывая
уравнения Ох и Red.
Red: Свободные металлы, соединения Fe(2),Pb(2), Ci(2), Cu(l); H2, С.
Ох: Соединения металлов, в котором у металла макс, степень окисления, равная номеру группы. НеМе
могут быть как Ох, так и Red.
Ox-Red двойственность - когда элемент находится в промежуточной степени окисления, а
следовательно может проявлять свойства как окислителя, так и восстановителя, (пр. HN02 может быть
сильным ох и red).
Ионно электронный метод уравнивания Ox-Red реакций.
Это метод уравнивания окислительно-восстановительных реакций, в котором число отданных и
полученных электронов подсчитывают по зарядам реальных ионов, которые имеются в растворе.
В сложных случаях электронно-ионный баланс помогает определить, что с чем реагирует и какие
продукты при этом получаются. И позволяет определить коэффициенты уравнения реакции. Особенно
эффективен метод электронно-ионных полуреакций в тех случаях, когда трудно или вообще
невозможно определить степени окисления атомов в веществах -реагентах и/или продуктах реакции.
I) Записываем левую часть уравнения предполагаемой реакции; 2) рассматриваем (и записываем, если
надо) формулы частиц, присутствующих в растворе; 3) Очень хочется определить, какой из ионов
здесь будет выступать в роли окислителя, а какой - в роли восстановителя. Выяснить это можно по
степеням окисления, если повезет, 4) После того, как мы определили функции реагентов, надо
выяснить, какие продукты будут получаться, а то мало ли что; 5) Далее следует составить уравнения
полуреакции; 6)А вот тут-то надо достичь равенства числа отданных и принятых в реакции электронов.
Для этого, тихонечко умножаем первую полуреакцию на коэффициенты, которые нужно подобрать
чуток пошевелив мозжечком. 7) Теперь по уравнениям полуреакций составить ионное уравнение всего
процесса. Складывая два уравнения полуреакций (после умножения их на нужные для баланса
электронов коэффициенты); 8) По ионному уравнению составляем молекулярное уравнение. 9)
Радуемся полученному результату, но, все-же полезно помнить, что в щелочной среде не может
получиться кислота, а в кислотной среде не может получиться щелочь.
Расчет ЭДС и определение направленности Ox-Red реакций.
ЭДС:
см. Стандартные электродные потенциалы, см. Расчет ЭДС элемента.
см. Термодинамика гальванического элемента: зависимость ЭДС элемента от природы реагирующих
веществ, температуры и концентрации (активности).
см. Формула Нернста для электродного потенциала
Направление ox-red реакции:
Рассмотрим реакцию хлорида железа (Ш) с хлоридом олова (П), которая протекает по схеме:
2FeC13 + SnCI2 = 2FeC12 + SnCl4. Для предсказания возможности протекания данной реакции
необходимо рассчитать величину изобарно-изотермического потенциала ΔG. Реакция протекает в
сторону уменьшения свободной энергии Гиббса (ΔG < 0). Однако для окислительновосстановительных реакций критерием направленности процесса могут служить величины
окислительно-восстановительных потенциалов. Представим реакцию в виде: Fe3+ + е = Fe2+; Sn2+ + 2e =
Sn4+. Каждая из этих реакций характеризуется своим потенциалом (0,771 и 0,1 S3 В соотв.). Так как
перемещение электрона возможно от электрода с меньшим потенциалом к электроду с большим
потенциалом, так и ОВР
протекает от меньшего значения окислительно-востсановительного потенциала к большему. На
электроде с меньшем значением ф идет процесс окисления, а на электроде с большим значением ф восстановления. Следовательно, в стандартных условиях катионы Fe3+ могут окислить катионы Sn2+,
так как значение меньше. С увеличением концентрации катионов Sn4+ величина окислительновосстановитсльного потенциала увеличивается, а с увеличением концентрации Fе2+ величина
уменьшается. ОВР возможна до установления в системе pane потенциалов. Таким образом, разность
ΔФ (т. е. ЭДС) при стандартньк условиях может служить критерием возможности протекания
процесса. ОВР протекает в направлении от меньшего окислительно-восстановительного потенциала к
большему до состояния равновесия. В этом же направлении изменяется (уменьшается) свободная
энергия Гиббса, которая связана с изменением потенциала уравнением: Δ = -nFE, где n - число
электронов, F -число Фарадея.
Типы электродов.
1-го рода - в электрохимии - электрод, потенциал которого определяется активностью катиона,
участвующего в установлении электрохимического равновесия на поверхности электрода.
(металл (или неметалл), погруженный в электролит, содержащий ионы этого же элемента. Металл Э.
является восстановленной формой в-ва, а его окисленной формой - простые или комплексные ионы
этого же металла).
2-го рода - в электрохимии - электрод потенциал которого определяется активностью аниона,
участвующего в установлении электрохимического равновесия на поверхности электрода
(относятся многие электроды сравнения, напр. каломельный, хлорсеребряный, оксидно-ртутный).
3-го рода - в электрохимии - система из металла, контактирующего с двумя растворимыми солями,
находящимися в растворе электролита, имеющего общий катион с более растворимой солью.
(В результате электрохимической р-ции на Э. менее растворимая соль превращается в более
растворимую, а потенциал Э. определяется термодгашогческой активностью катионов более
растворимой соли).
Электролиз.
Это физико-химическое явление, состоящее в выделении на электродах составных частей
растворенных веществ или других веществ, являющихся результатом вторичных реакций на
электродах, которое возникает при прохождении электрического тока через раствор электролита. Это
Ox-Red процесс. Упорядоченное движение ионов в проводящих жидкостях происходит в
электрическом поле, которое создается электродами — проводниками, соединёнными с полюсами
источника электрической энергии. Анодом называется положительный электрод, катодом —
отрицательный. Положительные ионы —катионы—(ионы металлов, водородные ионы, ионы аммония
и др.)—движутся к катоду, отрицательные ионы — анионы — ионы кислотных остатков и
гидроксилытой группы—движутся к аноду, (знаки катод/анод - противоположны, не такие как в
гальваническом эл-те).
Процессы, идущие при электролизе противоположны процессом идущим в гальваническом эл-те. При
электролизе хим. реакция осуществляется за счет энергии электрического тока подведенной извне, в то
время как при работе ГЭ энергия самопроизвольно протекающей в нем хим. реакции превращается в
электрическую.
Какие именно электрохим. процессы будут протекать у электродов при электролизе будет зависеть от
относительных значений электродных потенциалов соответствующих электрохим. систем.
Характер катодного процесса при электролизе водных растворов определяется положением
соответствующего металла в ряду напряжений, а также рН р-ра, концентрация ионов Me и т.д. Катод
восстанавливается, что приводит к выделению элементов в свободном состоянии. Нужно учитывать
величину потенциала процесса восстановления ионов водорода. Он зависит от концентрации ионов
водорода, (при рН=7 ф=-0,41) Поэтому, если катионом электролита является металл, электродный
потенциал которого значительно положительнее -0,41, то из нейтрального раствора такого электролита
на катоде будет выделятся металл. А если катионом электролита является металл, имеющий потенциал
более отрицательный чем -0,41 металл восстанавливаться не будет, а произойдет выделение водорода.
Ну а если потенциал металла близок к -0,41 (металл средней части ряда Zn, Сr, Fe, Cd, Ni) то в
зависимости от концентрации раствора и условий электролиза возможно как восстановление металла,
так и выделение водорода; нередко наблюдается совместное выделение металла и водорода.
Электрохимическое выделение водорода из кислотных растворов происходит в следствие разряда
ионов водорода. В случае нейтральных или щелочных сред оно является результатом
электрохимического восстановления воды: 2Н2O+2ел(-)=H2+2OНАнод может окислятся в ходе электролиза. Активный анод - окисляется, число конкурирующих
окислительных процессов возрастает до трех: электрохимическое окисление воды с выделением
кислорода, разряд анионы(т.е. его окисление), и электрохимическое окисление металла анода (анодное
растворение металла) и из этих процессов будет идти тот, который наиболее энергетически выгоден.
Инертный анод - (платина уголь, графит) при электролизе водных растворов щелочей,
кислородсодержащих кислот и их солей, фтороводорода и фторидов происходит электрохимическое
окисление воды с выделением кислорода. В зависимости от рН раствора этот процесс протекает по рал
гаму. В щелочной среде: 40Нл(-)=02+2Н20+4е(-), в кислотной или нейтральной: 2Н2О = О2+4Н++4е.
Кислородсодержащие анионы или неспособны окислятся или их окисление происходит при очень
высоких потенциалах. При электролизе водных растворов бескислородных кислот и их солей (кроме
HF и фторидов) у анода разряжаются анионы.
Эффективность электролиза оценивают рядом факторов, к которым относятся: сила тока, напряжение,
плотность тока, КПД источника тока, выход по току, выход по веществу, коэффициент полезного
действия электроэнергии (выход по энергии), расход электроэнергии на единицу полученного
продукта.
Сипа тока или нагрузка на электролизер характеризуют его производительность. Чем выше сила
тока, пропускаемого через электролизер, тем больше продукта можно получить при эксплуатации
данного электролизера. Наблюдается тенденция к созданию мощных электролизеров, рассчитанных в
некоторых случаях на десятки и сотни тысяч Ампер (производство хлора, алюминия и т. д.)
Напряжение разложения вещества.
Протекание электрического тока через электрическую ванну приводит к сдвигу потенциалов от их
равновесных значений, отвечающему определенной плотности тока, т. е. к поляризации электродов.
Каждому электролиту- свойственно определенное минимальное напряжение, которое необходимо
приложить к раствору, чтобы начался электролиз. Такое необходимое напряжение получило название
«напряжение разложения». В результате химической поляризации электродов возникает
гальванический элемент, электродвижущая сила которого равна Е(пол) = - = 1,36 - 0,000 = 1,36 В и
имеет направление противоположное внешней ЭДС. Возникновение обратной ЭДС при электролизе,
называемой ЭДС поляризации, составляет сущность явления поляризации при электролизе. Поэтому
электролиз возможен при условии компенсации ЭДС поляризации внешним напряжением. Часто
реально напряжение разложения оказывается много больше ЭДС поляризации, так как необходимо
скомпенсировать еще и перенапряжение на катоде и аноде, вызванные замедленностью реакции
разряда. Таким образом, в общем случае напряжение, при котором будет происходить электролиз
(напряжение разложения), складывается из ЭДС поляризации (Е(иол) = фк - фА), анодного и катодного
перенапряжения (нюА и нюk), омического падения напряжения на электролите (ΔUэл). Напряжение
разложения обусловлено природой реагирующего вещества, а поэтому не может быть изменено.
Перенапряжение.
ЭДС работающего элемента всегда меньше той расчетной величины, которая отвечает обратимой
электрохимической реакции. Причиной этого является поляризация.
Поляризацией называется явление изменения потенциала от своего равновесного значения при
протекании тока. Если в отсутствии тока система находится в равновесии, то мерой поляризации
служит перенапряжение:
з = фР – ф(i)
где фР - равновесный потенциал, ф(i) - потенциал при протекание тока.
Изменение потенциала, обусловленное замедленностью электрохимических стадий реакций называется электрохимической поляризацией и характеризуется перенапряжением перехода. Эта
стадия играет главную роль в электрохимической кинетике, т.к. на перенос заряда непосредственно
влияет потенциал электрода. Связь между перенапряжением перехода и плотностью тока выражается
уравнением Тафеня:
з = а + в lgi
Константа «в» зависит от природы окислителя и восстановителя, температуры, «а» - от природы
окислителя и восстановителя, состава раствора, температуры, материала электрода, «i» - плотность
тока.
Законы электролиза.
Количественная характеристика электролиза выражается двумя законами Фарадея:
1) Масса вещества, выделяющегося на электродах, прямо пропорциональна количеству прошедшего
через электролит электричества m = KэQ = КэI/t, где ш - масса выделившегося вещества, Q количество электричества (Кл) I - сила тока (A), t - время (с), Кэ - электрохимический эквивалент
вещества, выделившегося на электродах при протекании тока силой в 1 А в течение 1 сек (или
количеством электричества в I Кл).
2) При электролизе различных электролитов равные количества электричества выделяют на электродах
массы веществ, пропорциональные молярным массам их эквивалентов. Кэ = Мэ/F; га =(M3/F)*I*t, где F
- число Фарадея, Мэ - молярная масса эквивалента Если I / t = F = 96500 Кл, то га = Мэ. Для
химического превращения молярной массы эквивалента любого вещества необходимо пропустить
через электролит количество электричества, равное числу Фарадея (96500 Кл). На законах Фарадея
основан один из точных способов измерения количества электричества, прошедшего в системе. Для
этого служат приборы, называемые кулонометрами. Вследствие параллельных побочных процессов
масса вещества, получаемого при электролизе, оказывается часто меньше той, которая соответствует
количеству прошедшего электричества. Отношение массы реально выделенного вещества на электроде
к теоретической и умноженное на 100% называют выходом по току:
Зэ= Мэкс./Мтеор.
Аккумуляторы..
Вторичным источником тока являются аккумуляторы. Работоспособность разряженного аккумулятора
можно восстановить, зарядив его, т.е. пропустив через него в обратном направлении ток от внешнего
источника (электролиз). При зарядке аккумулятор работает как электролизер, а при разрядке - как
гальванический элемент. Процессы заряда аккумуляторов осуществляются многократно.
Свинцово-кислотный аккумулятор.
Свинцовый аккумулятор представляет собой систему свинцовых перфорировал перфорированных
пластин, заполненных губчатым свинцом и являющихся катодом, а положительным электродом
служит оксид свинца РbO2, впрессованный в свинцовую решетку. В качестве электролита
используется 30%-ный раствор серной кислоты. Схема аккумулятора:
Pb | H2S04 | PbO2
При погружении пластины в серную кислоту на их поверхности образуется труднорастворимая сольсульфат свинца PbSО4. В этом состоянии электроды имеют одинаковый химический состав и
окислительно-восстановительное взаимодействие невозможно, аккумулятор разряжен. Поэтому
предварительно проводят зарядку аккумулятора, пропуская через него постояшгый электрический ток
от внешнего источника Процессы, протекающие при зарядке подобны ггроцессам при электролизе.
На катоде (-) происходит процесс восстановления: Рb2+ + 2е = РЬ0, ф= -0,36 В. Этой реакции разряда
соответствует электрохимическая реакция: PbS04 + 2Н+ + 2е=-> Pb0 + H2S04. На аноде (+) ионы РЬ2+
окисляются: Рb2+ - 2е => Pb+4); PbSO4 - 2e + SO42- => Pb(S04)2. Образующаяся соль подвергается
гидролизу: Pb(S04)2 + 4Н20 => Pb(0H)4 + H2S04 с последующим разложением РЬ(ОН)4 = РbO2 +
2Н20, т.е. конечным результатом является образование оксида свинца (IV) - РbO2. Таким образом,
после зарядки один электрод аккумулятора представляет собой губчатый металлический свинец, а
другой - оксид свинца (IV). Общее химическое уравнение процесса зарядки: PbSO4 + 2Н20 ->Рb+РbO2
+ 2H2S04. При работе аккумулятора (разрядке) процессы на электродах протекают в обратном
направлении. Окисление на аноде: Рb - 2е + S042-> PbS04. Восстановление на катоде: РЮ2 + 4Н+ +
S042- + 2е=> PbS04 + 2Н20. Суммарная реакция: РЬ02 + Pb + 2H2S04 (разрядка->; зарядка<-) 2PbS04 +
2Н20.В процессе зарядки концентрация кислоты увеличивается, а в процессе разрядки наоборот,
уменьшается. Относительная плотность серной кислоты указывает насколько разряжен аккумулятор.
ЭДС свинцового аккумулятора достигает 2,1 В. Напряжение при заряде выше ЭДС и растет в течение
заряда. В конце напряжение достигает значения, достаточного для электролиза воды, тогда начинается
выделение водорода и кислорода, поэтому выделение пузырьков газа (кипение) служит признаком
окончания заряда аккумулятора. При разрядке аккумулятора его ЭДС и напряжение падают. Если
напряжение упадет ниже 1,7 В, на электродах начинается образование пленки PbS04 особой
кристаллической структуры (так называемое сульфатирование), которая изолирует электроды от
электролита. Вследствие этого недопустимо снижение напряжения до ] ,7 В. Свинцовый аккумулятор
обладает высоким КПД ( 80%), высокой ЭДС, простотой и невысокой ценой. Недостатки -небольшая
удельная энергия 20-30 Вт*ч/кг и малый срок службы (от 2 до 5 лет).
Коррозия металлов.
Коррозией называется процесс самопроизвольного разрушения металлов под влиянием внешней
среды. В процессе коррозии происходит переход из металлического состояния в ионное. Коррозия самопроизвольный процесс, сопровождается уменьшением свободной энергии (ΔG<0), увеличением
энтропии системы и oопределенным энергетическим эффектом. В некоторых случаях коррозия
поражает всю поверхность, в других - часть ее. Характер разрушения зависит от свойств металла и
условий протекания процесса. Возможны различные виды коррозийных разрушений равномерное,
пятнами, точечное, питтинг, межкристаллитное, растрескивающее, селективное. Наиболее опасными
являются питтинг и межкристаллитное разрушения.
+ см. все далее.
Классификация процессов коррозии.
По принципу их протекания все коррозийные процессы принято подразделять на: химические и
электрохимические Химическая и электрохимическая коррозия.
Химическая коррозия. Обусловлена взаимодействием металла с сухими газами или жидкостями, не
проводящими электрического тока. Химической коррозии подвергаются детали и узлы машин,
работающих в атмосфере кислорода, галогенов, при высоких температурах (турбинные, ракетные
двигатели, оборудование химических производств). Механизм реакции сравнительно прост. Продукты
реакции образуются на тех участках металлической поверхности, которые вступили в реакцию. Так, на
железе при 250-З00оС появляется видимая пленка оксидов, при б00оС и выше поверхность металла
покрывается слоем окалины, состоящей из оксидов железа различной степени окисления (FeO, Fe304,
Fe203). Окалина не может защитить металл от дальнейшего окисления, так как содержит трещины и
поры, которые позволяют проникать кислороду к металлу. При повышении температуры свыше 800оС
скорость окисления железа резко увеличивается. Образующиеся на металле оксидные пленки часто
препятствуют дальнейшему окислению (образуется так называемая «защитная» пленка, которая
препятствует проникновению к металлу как газов, так и жидкостей). Защитными свойствами обладает
только та пленка, которая может покрывать сплошь весь металл. Расчеты показывают, что это
возможно, если объем оксида металла больше объема самого металла, пошедшего на образование этого
оксида: VOKC/VM> 1, Для щелочных
И для щелочно -земельных металлов это условие не соблюдается (Vokc/Vme<1 ). Такие пленки
защитнными свойствами не обладают и эти металла ввиду своей химической активности принадлежат
к числу коррозионно нестойких. На алюминии и хроме образуются хорошие защитные пленки,
благодаря чему металлы в атмосферных условиях коррозийно стойки (хотя химически активны).
Толщина таких пленок крайне мала, например, на алюминии она достигает только 100 Е. Такие пленки
состоят всего из нескольких слоев молекул и могут быть обнаружены только специальными
оптическими методами. Если металл, покрытый пленкой, продолжает коррозировать, то это означает,
что имеет место диффузия атомов кислорода сквозь пленку к металлу, а атомов металла - в обратном
направлении. Диффузия металла и кислорода в слое твердого защитного оксида может происходить по
одному из двух возможных механизмов: а) движение ионов в междоузельном пространстве
кристаллической решетки; б) движение ионов по пустым узлам решетки Первый механизм имеет место
при образовании пленок ZnO, CdO, BeO, А1203; второй - Cu20, FeO, NiO, СоО, Zr02, Ti02. диффузия
катионов в защитной пленке.
сопровождается одновременным перемещением в том же направлении эквивалентного числа
электронов в междоузлиях при первом механизме и по «электронным дефектам» (катионом с более
высокой валентностью) при втором механизме. При повышении температуры окисление металлов на
воздухе происходит более интенсивно (увеличивается скорость диффузии).
Электрохимическая коррозия. Разрушение металла при соприкосновении с электролитом с
возникновением в системе электрического тока называется электрохимической коррозией.
В атмосферных условиях роль электролита играет водная пленка на металлической поверхности, в
которых часто растворены электропроводящие примеси. Электродами обычно являются сам металл и
примеси в нем содержащиеся. В качестве примера рассмотрим действие серной кислоты на железо,
содержащего примесь меди. При таком контакте возникает множество микроскопических
гальванических элементов. (-) Fe | H2S04 | Cu(+), ФFe = - 0,44 В., ФCu = 0,34 В. Более активный
металл-железо - окисляется, посылая электроны атомам меди и переходит в раствор в виде ионов Fe2+,
а ионы водорода разряжаются (восстанавливаются) на меди. Помимо этого железо может растворяться
и при непосредственном взаимодействием с кислотой (Fe + 2Н+ = Fe2+ + Н2), но опыт показывает, что
скорость этой реакции мала по сравнению со скоростью растворения железа как анода гальванопары.
Механизм коррозии в кислой и нетралы юй среде различен. Рассмотрим коррозию технического железа
на воздухе, когда оно покрыто влажной пленкой или находится в растворе электролитов с небольшой
концентрацией гидроксид ионов. В качестве анода здесь служит основной металл -железо, катодом,
являются примеси содержащиеся в металле, например, зерна графита:
Анод: Fe - 2e= Fe2+
Катод: 2Н2О + О2 + 4е = 4ОН-. Ионы ОН:(-) соединяются с перешедпшми в раствор ионами Fe2+. Fe2+ +
2ОН- = Fe(OH)2 , 4Fe(OH)2 + О2 + 2Н2О = 4Fe(OH)3, Fe(ОH)3 = Ре(ржавчина. одновременно =OH
и=О) + Н2О. Согласно теории электрохимической коррозии при соприкосновении металла с
электролитом на его поверхности возникает множество микрогальванических элементов. При этом
анодами являются частицы металлов, катодами - загрязнения, примеси и участки металла, имеющие
более положительный потенциал. Механическая обработка изменяет электродный потенциал, поэтому
соприкосновение двух участков металла деформированного и недеформированного достаточно для
появления разности потенциалов. Коррозировать будет деформированый участок. Разность
потенциалов возникает и там где соприкасаются обнаженный и покрытый пленкой участки.
Поскольку ионы и молекулы, связывающие электроны в катодном процессе, называют
деполяризаторами, то часто говорят о водородной деполяризации в кислой и кислородной
деполяризации в нейтральной и слабощелочной средах. Поскольку электрохимическая коррозия
обусловлена деятельностью гальванических элементов, можно сделать вывод, что факторы
способствующие деятельности гальванических элементов усиливают коррозию. Скорость коррозии
тем больше, чем дальше отстоят в ряду напряжений металлы, из которых образовалась гальваническая
пара. Скорость коррозии возрастает с ростом температуры и с увеличением концентрации окислителя в
растворе. Нередко продукты коррозии оказываются малорастворимыми в данной среде и своим
присутствием защищают металл от дальнейшего разрушения. Например, железо в щелочных
растворах, свинец в разбавленной серной кислоте коррозионно стойки вследствие образования пленок
Fe(OH)2, PbSО4.
Большое значение в процессах коррозии имеют поляризация электродов, образование пленок на
металлах, перенапряжение водорода. Если бы в коррозионных процессах не происходило поляризации
электродов, то процессы коррозии протекали бы с такой скоростью, что железо и ряд других металлов
потеряли бы свое значение в технике.
Пассивация металлов.
Сравнительно легко пассивирующие металлы, такие, как титан, никель, нержавеющая сталь, можно
защитить от коррозии, присоединяя их к положительному полюсу генератора (так называемая анодная
защита). Анодную плотность тока доводят до величины, которая приводит к полной пассивации, в
результате чего металл перестает растворяться. Этот метод применяют для защиты конструкции от
коррозии в сильно агрессивных средах.
Защитные плёнки на поверхности металлов.
Вообще-то это оксид: хМе + уО -> МехОу. Наличие пассивных пленок, образующихся в атмосфере на
поверхности таких металлов, как алюминий, титан, хром, никель, значительно повышает их
коррозионную стойкость. Защитная способность этих пленок зависит от плотности и электронной
проводимости. Пассивные пленки наносят искусственно на такие металлы, как алюминий, железо
воронение железа), медь, магний. Такие искусственно созданные пленки имеют значительно большую
толщину и обладают большей механической и противокоррозийной стойкостью. Широко используют
жаростойкое легирование, т.е. введение в состав сплава компонентов, повышающих жаростойкость
Например для повышения коррозионной стойкости стали в нее вводят легирующие элементы - хром,
никель, титан, вольфрам присутствие которых обеспечивает возникновение на поверхности сплошных
пассивных пленок
Фактор Пиллинга - Бэдвордса у разных металлов имеет разные значения и если сплошные и
устойчивые оксидные слои образуя при значениях = 1,2-1,6, то при больших значениях пленки
получаются не сплошные, легко отделяющиеся от поверхности металла (железная окалина) в
результате возникающих внутренних напряжений
Гальванокоррозия при контакте двух металлов, при наличии примесей в металле, при контакте металла
стокопроводящими неметаллическими примесями, при неодинаковой аэрации, при неодинаковой
механической обработке, при неодинаковой концентрации электролита.
Основные методы в борьбе с коррозией: нанесение защтных покрытий, электрохимическая
защита, обработка коррозийной среды.
Широко применяются следующие методы защиты металлических конструкций от коррозии: 1)3ащита
покрытия; 2) Антикоррозионное легирование металла; 3) Электрохимическая защита; 4) Обработка
коррозионной среды; 5) Разработка и производство новых металлических конструкционных
материалов повышенной коррозионной устойчивости. Выбор одного из методов защиты основывается
не только на технических соображениях, но и на экономических расчетах. Наиболее дешевым и
распространенным методом являются лакокрасочные покрытия. Наличие пассивных пленок,
образующихся в атмосфере на поверхности таких металлов, как алюминий, титан, хром, никель,
значительно повышает их коррозионную стойкость. Защитная способность этих пленок зависит от
плотности и электронной проводимости. Пассивные пленки наносят искусственно на такие металлы,
как алюминий, железо воронение железа), медь, магний. Такие искусственно созданные пленки имеют
значительно большую толщину и обладают большей механической и противокоррозийной стойкостью.
Широко используют жаростойкое легирование, т.е. введение в состав сплава компонентов,
повышающих жаростойкость. Например, для повышения коррозионной стойкости стали в нее вводят
легирующие элементы - хром, никель, титан, вольфрам, присутствие которых обеспечивает
возникновение на поверхности сплошных пассивных пленок. Для защиты от коррозии используют
металлические покрытия. По характеру защитного действия различают анодные и катодные покрытия.
К анодным покрытиям относятся такие покрытия, в которых покрывающий металл обладает в данной
среде более отр1щательным электродным потенциалом (оцинкованное железо). К катодным относятся
покрытия из менее активных металлов (луженое железо, никелевое покрытие железа). Пока слой,
покрывающий основной металл, полностью изолирует его от воздействия окружающей среды,
принципиального различия между этими двумя видами покрытий нет. При нарушении целостности
покрытия создаются совершенно различные условия. В порах цинкового покрытия на железе при
образовании микрогальваноэлемента цинк является анодом, а железо - катодом. Цинк растворяется в
электролите, а железо не будет разрушаться до тех пор, пока сохраняется цинковое покрытие.
Никелевое покрытие на железе - катодное. При образовании микрогальваноэлемента железо выполняет
роль анода и разрушается. Для зашиты от коррозии в коррозионную среду вводят небольшое
количество добавок, или замедлителей коррозии (ингибиторов), которые в зависимости от среды
разделяют на парофазные (или летучие) и жидкофазные. В качестве жидкофазных ингибиторов для
нейтральных растворов применяют в основном неорганические ингибиторы анионного типа (нитриты,
хроматы, фосфаты). Их тормозящее действие сводится к образованию или оксидных пленок, или
пленок труднорастворимых соединений, в результате которого потенциал сдвигается в область более
положительных значений. Ингибиторами кислотной коррозии, как правило, являются органические
соединения, которые, абсорбируясь на поверхности металла, тормозят как анодный, так и катодный
процесс. В агрессивных средах (морская вода, почва) применяют электрохимический метод зашиты. В
промышленности часто применяют так называемую протекторную защиту. К стальной конструкции
корпус судна, трубопровод) присоединяют пластины циника или цинка с алюминием. При этом
образуется макрогальванический элемент, где роль анода выполняет цинк, а защищаемая конструкция
становится катодом, коррозия конструкции вследствие сдвига потенциала уменьшается или вообще
прекращается. В других методах, называемых катодной защитой, аналогичный результат достигается
присоединением защищаемого металла к отрицательному полюсу внешнего источника. Сравнительно
легкопассивирующие металлы, такие, как титан, никель, нержавеющая сталь, можно защитить от
коррозии, присоединяя их к положительному полюсу генератора (так называемая анодная защита).
Анодную плотность тока доводят до величины, которая приводит к полной пассивации, в результате
чего металл перестает растворяться. Этот метод применяют для защиты конструкции от коррозии в
сильно агрессивных средах.