ПАТОФИЗИОЛОГИЯ
УЧЕБНИК
ПЯТОЕ ИЗДАНИЕ
Том 2
И З Д А Т Е Л Ь С К А Я ГР У П П А
«ГЭОТАР-Медиа
■ ^ ч - -7* V
■
1
••• V *.
Ч- ч ' V .• ч ; ч- *.
................
Н
■ „11Л
•Ж •
1 I
тШ < А 1
Ж о уш
П.Ф. Литвицкий
ПАТОФИЗИОЛОГИЯ
УЧЕБНИК
В 2 ТОМАХ
ПЯТОЕ ИЗДАНИЕ,
ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
М и н и с т е р с т в о о б р а з о в а н и я и н а у к и РФ
Р е к о м е н д о в а н о ГБОУ ВПО « П е р в ы й М о с к о в с к и й г о с у д а р с т в е н н ы й м е д и ц и н с к и й
у н и в е р с и т е т и м е н и И . М . С е ч е н о в а » в к а ч е с т в е у ч е б н и к а дл я с т у д е н т о в у ч р е ж д е н и й
в ы с ш е г о п р о ф е с с и о н а л ь н о г о о б р а з о в а н и я , о б у ч а ю щ и х с я по с п е ц и а л ь н о с т я м
0 6 0 1 0 1 . 6 5 « Л е ч е б н о е д е л о » , 0 6 0 1 0 3 . 6 5 « П е д и а т р и я » по д и с ц и п л и н е
«П атоф изиология, клиническая патофизиология»,
по с п е ц и а л ь н о с т и 0 6 0 1 0 5 .6 5 « М е д и к о -п р о ф и л а к т и ч е с к о е дело»
по д и с ц и п л и н е « П а т о ф и з и о л о г и я »
Р е г и с т р а ц и о н н ы й н о м е р р е ц е н з и и 7 о т 2 4 я н в а р я 2 0 1 2 г.
ФГАУ « Ф е д е р а л ь н ы й и н с т и т у т р а з в и т и я о б р а з о в а н и я »
Москва
ИЗД АТЕ Л ЬСКАЯ ГРУППА
«ГЭОТАР-Медиа»
2015
П.Ф. Литвицкий
ПАТОФИЗИОЛОГИЯ
Том 2
!УОТ АКАО
?н
рг
■ Г
ч» СА2‘ -
Москва
И ЗД АТЕ Л ЬСКАЯ ГРУППА
«ГЭОТАР-Медиа»
2015
;
'
УДК 616-092(075.8)
Б Б К 52.52я73
Л64
Рецензент:
д-р мед. наук, академик МАН ВШ, зав. кафедрой патофизиологии ме­
дицинского факультета Российского университета дружбы народов,
проф. В.А. Фролов.
Литвицкий,П. Ф.
Л64
Патофизиология : учебник : в 2 т. / П. Ф. Литвицкий. — 5-е изд.,
перераб. и доп. — М .: ГЭОТАР-Медиа, 2015. — Т. 2. — 792 с . : ил.
18ВЫ 978-5-9704-3176-4 (общ.)
18ВК 978-5-9704-3177-1 (т. 2)
Учебник подготовлен с учетом требований ФГОС и современных программ
по патофизиологии и клинической патофизиологии для медицинских вузов.
В нем изложены материалы, характеризующие предмет, цели и методы патофи­
зиологии; основные понятия нозологии, общей этиологии и патогенеза; совре­
менные представления о типовых патологических процессах, формах патологии
органов и физиологических систем, о наиболее распространенных клинических
синдромах и болезнях, принципах их диагностики, лечения и профилактики.
Учебник является информационным компонентом учебно-методического
комплекса, в который также входят: учебно-методическое пособие «Патофизиоло­
гия. Задачи и тестовые задания». — М.: ГЭОТАР-Медиа, 2013 (основная дидакти­
ческая часть учебного комплекса); учебно-методическое пособие «Патофизиоло­
гия. Руководство к занятиям». — М.: ГЭОТАР-Медиа, 2010; РаШорНумо^ёу. СопС18е 1есШге8,
сигисо-ра1орЬу$ю1о§юа1 кНиайопа апс! сНтсо-1аЬога1огу са$е$. —
М.: ГЭОТАР-Медиа, 2014; набор цветных настенных учебных таблиц.
Материалы учебника необходимы и достаточны для формирования основ
компетентного врачебного мышления, умения эффективно решать профессио­
нальные врачебные задачи.
УДК 616-092(075.8)
Б Б К 52.52я73
Права на данное издание принадлежат ООО Издательская группа «ГЭОТАР-Медиа».
Воспроизведение и распространение в каком бы то ни было виде части или целого изда­
ния не могут быть осуществлены без письменного разрешения ООО Издательская
группа «ГЭОТАР-Медиа».
1 8 1 ^ 978-5-9704-3176-4 (общ.)
1 8 1 ^ 978-5-9704-3177-1 (т. 2)
©Литвицкий П.Ф., 2012
© ООО Издательская группа «ГЭОТАР-Медиа», 2015
© ООО Издательская группа «ГЭОТАР-Медиа»,
оформление, 2015
ОГЛАВЛЕНИЕ
С писок сокращ ений и условных о б о зн а ч е н и й ....................................................13
Глава 22. Типовые формы патологии системы к р о в и ........................................... 18
Наруш ения общего объема крови и гем а то к р и т а......................................... 18
Наруш ения объема к р о в и ................................................................................18
Н ор м о вол ем и и ...............................................................................................19
Г иперволем ии.................................................................................................20
Гиповолем ии................................................................................................... 21
К ровоп отеря......................................................................................................... 23
Этиология к р овоп о тери ............................................................................. 23
М еханизм развития острых постгеморрагических
с о с т о я н и й ....................................................................................................... 24
А даптивные механизмы при постгеморрагическом
с о с т о я н и и ....................................................................................................... 26
Виды кровоп отери ........................................................................................ 28
П ринципы и методы лечения кровопотери.........................................28
Патофизиология системы эритроцитов............................................................ 29
Э р и т р о ц и т о зы ..................................................................................................... 30
Виды эритроц и тозов.................................................................................... 30
А нем ии.................................................................................................................... 34
Общие лабораторные признаки ан ем и и ............................................... 34
Виды а н е м и й ....................................................................................................... 35
М еханизмы развития а н е м и й ...................................................................37
П остгеморрагические ан ем ии ...................................................................37
Гемолитические ан ем и и ............................................................................. 38
Д изэритропоэтические а н е м и и .............................................................. 42
А немия вследствие наруш ения синтеза глобиновых Д Н К .............43
А немии, развиваю щ иеся при наруш ениях обмена ж елеза.............45
А немии, развиваю щ иеся вследствие наруш ения
синтеза гл о б и н о в .......................................................................................... 50
П атофизиология системы л ей коц и тов .............................................................. 52
Типовые изменения количества лейкоцитов в единице
объема крови: лейкопении и л ей к о ц и т о зы ............................................... 52
Л ей коп ен и и ..................................................................................................... 53
Л ей коц и тозы ................................................................................................... 55
Патофизиология тром боцитов..............................................................................62
Т р о м б о ц и то зы ..................................................................................................... 62
Абсолютные тромбоцитозы ....................................................................... 62
Относительные тром боц итозы .................................................................63
Значение тр о м б о ц и то зо в ........................................................................... 63
Т ром боцитопении.............................................................................................. 63
П ричины тром б оц итоп ен и й .....................................................................63
М еханизм развития тром боцитопений................................................. 64
П роявления тром боцитопений.................................................................64
Лечение больных с тр о м б о ц и то п ен и ям и ............................................. 65
Т ром б оц и топ ати и ...............................................................................................66
Виды тр о м б оц и топ ати й ..............................................................................66
Патогенез тром боцитопатий.....................................................................67
П роявления тром боцитопатий.................................................................68
Лечение при тром боцитопатиях.............................................................. 68
Наруш ения системы гемостаза....................................................................... 69
Типовые формы патологии системы гемостаза...................................69
Гемобластозы ..............................................................................................................83
Общая характеристика л ей к о зо в ...................................................................84
Этиология лейкозов...................................................................................... 84
Патогенез л е й к о зо в ...................................................................................... 84
Л имфомы и другие гем о б л астозы .................................................................85
Опухолевый атипизм гемобластозов......................................................85
Лейкемоидные р е а к ц и и .................................................................................... 88
Л ей к о зы .................................................................................................................. 89
Острые л е й к о з ы .............................................................................................89
Х ронические л ей ко зы ..................................................................................94
Глава 23. Типовые формы патологии сердечно-сосудистой систем ы ............... 98
Недостаточность кр о вооб ращ ен и я..................................................................... 98
Ф акторы риска сердечно-сосудистой патологии .........................................100
Типовые формы патологии сердечно-сосудистой системы ..................... 100
К оронарная н ед остаточн ость....................................................................... 100
Обратимые наруш ения коронарного кровотока.............................. 101
Необратимые наруш ения коронарного кровотока..........................103
П ричины коронарной н едостаточности ............................................. 104
М еханизмы повреж дения сердца при коронарной
недостаточности.......................................................................................... 107
Э ф фекты постокклю зионной реперфузии м и о к а р д а ................... 114
П остиш емическая реперфузия м иокарда........................................... 114
Изменение основных показателей ф ункции сердца
при коронарной н ед остаточ н ости ........................................................ 115
А ритмии сер д ц а................................................................................................. 116
Виды аритмий, их этиология и п ат о г е н е з......................................... 117
Э к стр аси сто л и я.......................................................................................... 128
Н аруш ения в миокарде, предшествующие ар и тм и ям ................... 129
Электрофизиологические механизмы развития а р и т м и й ...........131
С ердечная н ед о стато ч н о сть ......................................................................... 133
П ричины сердечной н ед о стато ч н о сти ............................................... 133
Наиболее значимые факторы, вызывающие перегрузку сердца---- 134
Виды сердечной н ед остаточ н ости ........................................................ 134
М еханизмы экстренной ком пенсации сократительной
ф у н к ц и и ..........................................................................................................137
К леточно-м олекулярны е механизмы сердечной
недостаточности...........................................................................................139
П роявления сердечной недостаточности........................................... 142
Ф ормы сердечной недостаточности...................................................... 143
П ри н ци п ы норм ализации ф ун кц ии сердца
при его н ед о стато ч н о сти ......................................................................... 147
Н аруш ения системного артериального д а в л е н и я .......................................148
Т ерм инология...............................................................................................148
Артериальные гипертензии........................................................................... 149
К лассиф икация артериальной ги п ертен зи и .....................................149
Распространенность артериальных гипертензий............................ 150
Риск пораж ения сердца и со су д о в ........................................................ 150
О рганы-миш ени при артериальной г и п е р т е н з и и .......................... 151
Виды артериальной гипертензии по патогенезу.............................. 151
Этиология и патогенез артериальны х ги п ертен зи й ........................153
О слож нения артериальной ги п е р т е н зи и ........................................... 168
Основные нозологические формы артериальных
гипертензий................................................................................................... 169
П ринципы лечения больных артериальными
ги п ер тен зи я м и ............................................................................................. 175
Артериальные г и п о т е н з и и ........................................................................... 175
Виды артериальной ги п отен зи и ............................................................ 176
Этиология и патогенез артериальных гипотензий.......................... 176
Наруш ения регионарного к р о в о т о к а .............................................................. 180
Н аруш ения кровотока в сосудах среднего диам етра............................ 181
Артериальная ги п ер ем и я......................................................................... 181
Венозная г и п е р е м и я ..................................................................................185
И ш ем и я...................................................................................................................... 187
П ричины и ш е м и и ...................................................................................... 187
М еханизмы развития и ш е м и и .............................................................. 188
П роявления и ш ем и и ..................................................................................190
Последствия и ш е м и и ................................................................................190
С т а з .................................................................................................................. 192
Нарушение крово- и лим ф ообращ ения в сосудах
микроциркуляторного р у с л а ....................................................................... 194
С л а д ж ..............................................................................................................200
Глава 24. Типовые формы патологии системы внешнего д ы х а н и я................. 202
О ценка ф ун кц ии внешнего д ы х а н и я ................................................. 202
С пирометрические признаки дыхательной
недостаточности.......................................................................................... 205
Типовые формы патологии внеш него д ы х а н и я ........................................... 207
Наруш ения вентиляции альвеол л е г к и х ................................................. 208
А львеолярная г и п о в е н т и л я ц и я ............................................................ 208
А львеолярная ги п е р в е н т и л я ц и я .......................................................... 213
Расстройства кровообращ ения в л е г к и х ................................................. 214
Гипертензия в сосудах малого круга кровообращ ени я................. 214
Легочная ги пертен зия................................................................................214
Гипотензия в сосудах малого круга кровообращ ени я................... 215
Н аруш ения вентиляционно-перф узионного с о о т н о ш е н и я ............. 216
Н аруш ения дифф узии кислорода и углекислого газа
через аэрогематический б а р ь е р ...................................................................217
П ричины сниж ения дифф узионной сп особ н ости ..........................218
Д ыхательная недостаточность........................................................................... 219
П ричины дыхательной недостаточности........................................... 219
Формы дыхательной недостаточности......................................................220
Гипоксемическая (паренхиматозная, типа I)
форма дыхательной недостаточности..................................................221
Гиперкапническая (гиповентиляционная, типа II)
форма дыхательной недостаточности..................................................221
С меш анная форма дыхательной недостаточности..........................221
Респираторный д и стр есс-си н др ом ............................................................ 221
П ричины респираторного дистресс-синдрома
взрослы х......................................................................................................... 222
Патогенез респираторного дистресс-синдром а
взрослы х......................................................................................................... 222
П роявления респираторного дистресс-синдром а
взрослы х..........................................................................................................222
Глава 25. Типовые формы патологии системы пищ еварения............................ 224
Этиология типовых форм патологии
желудочно-киш ечного т р а к т а ..................................................................... 224
Ф ун кц ии БАВ в пищ еварительном т р а к т е .......................................226
Ф акторы риска расстройств п и щ е в а р е н и я .......................................227
Типовые формы патологии желудочно-киш ечного т р а к т а ..................... 228
Расстройства в к у с а .......................................................................................... 228
Агевзии и ги п о ге в зи и ................................................................................228
Гипергевзия...................................................................................................229
Наруш ение адекватности вкусовых о щ у щ е н и й .............................. 229
Н аруш ения а п п е т и т а ...................................................................................... 229
А норексия и г и п о р е к с и я ......................................................................... 229
Гиперрексия и б у л и м и я ........................................................................... 230
П а р а р е к с и я ...................................................................................................230
Расстройства пищ еварения в полости р т а .............................. .................230
Н аруш ения с а л и в а ц и и ............................................................................. 230
Наруш ения переж евывания п и щ и ........................................................231
Расстройства гл о т а н и я ............................................................................. 232
Д исф ункции п и щ е в о д а ..................................................................................232
Д исф ункция пищ евода на уровне его верхнего
сф инктера и т е л а ........................................................................................ 233
Д исф ункция пищ евода на уровне его ниж ней части
и ниж него с ф и н к т е р а ................................................................................233
Наруш ения пищ еварения в ж елудке.......................................................... 234
Расстройства секреторной ф ункции ж елудка.................................. 234
Н аруш ения моторики ж елудка.............................................................. 236
Расстройства всасывания в ж елу д ке....................................................239
Наруш ение барьерной и защ итной ф ун кц ии слизистой
оболочки ж е л у д к а ...................................................................................... 239
Расстройства пищ еварения в к и ш е ч н и к е ................................................240
Н аруш ения переваривающ ей ф ун кц ии к и ш е ч н и к а ......................241
Расстройства всасывательной ф ун кц ии ки ш е ч н и к а......................241
Наруш ение моторной ф ун кц ии к и ш е ч н и к а .....................................242
Н аруш ения барьерно-защ итной ф ункции к и ш е ч н и к а ............... 243
П атологические синдром ы системы п ищ еварен и я.....................................243
Я звенная болезнь желудка и двенадцатиперстной к и ш к и ............... 244
Разновидности язвенной б о л е з н и ........................................................ 244
Этиология язвенной болезни желудка и двенадцатиперстной
к и ш к и ..............................................................................................................245
Ф акторы риска язвенной б о л е з н и ........................................................ 245
Патогенез язвенной б о л е зн и ...................................................................247
Основные проявления язвенной болезни ж е л у д к а ........................249
Основные проявления язвенной болезни двенадцатиперстной
к и ш к и ..............................................................................................................249
С индром наруш енного в с а с ы в а н и я .......................................................... 249
П ричины синдрома м альаб со р б ц и и ....................................................249
П роявления синдрома м альабсорбции............................................... 249
Энтеропатии (э н т е р и т ы )......................................................................... 250
Патогенез энтеропатий............................................................................. 251
Проявления и механизмы развития э н те р о п а ти й ..........................252
К олиты .................................................................................................................. 253
Х ронический к о л и т.................................................................................... 253
Синдром раздраж енной к и ш к и ............................................................ 254
Н еспецифический язвенны й к о л и т......................................................255
Глава 26. Типовые формы патологии печени.......................................................... 257
Ф ун кц ии п е ч е н и ...............................................................................................257
Участие в обмене в е щ е с т в ....................................................................... 258
Недостаточность ф ункций п е ч е н и ............................................................ 258
Виды печеночной недостаточности......................................................258
П ричины печеночной недостаточности............................................. 259
Общие звенья патогенеза печеночной недостаточности............... 262
П роявления печеночной недостаточности.........................................263
П еченочная к о м а ........................................................................................ 264
Ж ел ту х а................................................................................................................ 265
М етаболизм б и л и р у б и н а......................................................................... 265
Виды ж елтух и ...............................................................................................267
Внепеченочные желтухи........................................................................... 271
Глава 27. Типовые формы нарушений экскреторной функции п о ч е к .............275
П ричины патологии п о ч е к ........................................................................... 275
Общие механизмы возникновения и развития
почечной п а т о л о г и и ........................................................................................ 277
Нарушение клубочковой ф и л ь т р а ц и и ............................................... 277
Н аруш ения канальцевой реабсорбц и и ............................................... 278
Н аруш ения с е к р е ц и и ................................................................................278
Виды почечной п а т о л о г и и ..................................................................................278
Виды почечной патологии по происхождению .......................................279
С индромы , развиваю щ иеся при пораж ении п о ч е к .............................. 280
Характеристика отдельных форм патологии п о ч е к ........................280
Н е ф р и т ы ....................................................................................................... 280
П и ел о н еф р и ты .............................................................................................284
Н еф ротический си н д р о м ......................................................................... 285
Почечная н ед остаточ н ость........................................................................... 289
Острая почечная недостаточность........................................................ 289
Патогенез острой почечной недостаточности...................................291
Х роническая почечная недостаточность..................................................292
П ричины хронической почечной недостаточности........................292
Патогенез хронической почечной н ед остаточ н ости ......................293
У р ем и я.................................................................................................................. 293
П ричины у р е м и и ........................................................................................ 293
Патогенез урем ии........................................................................................ 293
Н е ф р о л и т и а з ..................................................................................................... 294
М еханизмы ф орм ирования к о н к р е м ен т о в .......................................294
Проявления почечной п ат о л о ги и .....................................................................295
Изменения параметров м о ч и ....................................................................... 295
Изменения объема и состава крови ............................................................ 296
Общие нефрогенные си н дром ы ...................................................................297
П ринципы лечения почечной п а т о л о г и и ......................................................297
Глава 28. Типовые формы патологии эндокринной си стем ы ............................ 299
Э ндокринны е ж елезы ...................................................................................... 299
Э ндокринны е клетки органов и тк а н е й ....................................................299
Г о р м о н .................................................................................................................. 300
Рецепторы гормонов и вторые п о ср ед н и к и .......................................300
Варианты воздействия гормонов на к л е т к и -м и ш е н и ................... 300
М еханизмы нейроэндокринной р е г у л я ц и и ........................................... 301
Регуляторные контуры нейроэндокринной р е г у л я ц и и ............... 301
Общая этиология и общ ий патогенез эндокринны х р асстрой ств.........301
Общие звенья патогенеза эндокринны х р ас стр о й ств ................... 301
Н аруш ения ф ун кц ий гипоталамо-гипоф изарной с и с т е м ы ................... 304
Типовые формы патологии ги п о ф и за........................................................ 305
Типовые формы патологии аден оги поф и за.......................................305
Отдельные формы патологии аден оги поф и за.................................. 306
Г ипопитуитаризм........................................................................................ 306
Г иперпитуитаризм...................................................................................... 309
Типовые формы патологии н е й р о ги п о ф и за .................................... 312
Наруш ения ф ун кц ий надпочечников........................................................ 314
Типовые формы патологии надпочечников.......................................315
Гиперальдостеронизм................................................................................316
Гиперкортизолизм ...................................................................................... 318
К ортико-генитальны й (адреногенитальны й) с и н д р о м ............... 320
Г и перкатехолам и нем и я........................................................................... 322
Н адпочечниковая недостаточность......................................................323
Наруш ения ф ункций щ итовидной ж елезы ............................................. 327
Типовые формы патологии щ итовидной ж елезы ............................ 328
Г и пертиреозы ...............................................................................................328
Г и поти реозы .................................................................................................334
Наруш ения ф ункций паращ итовидны х ж ел ез.......................................343
Гиперпаратиреоидные с о с т о я н и я ........................................................ 343
Гипопаратиреоидные с о с т о я н и я .......................................................... 347
Наруш ения эндокринной ф ункции половых ж е л е з ............................ 350
Типовые формы патологии в результате эндокринопатий
половых ж елез...............................................................................................352
Наруш ения половой диф ф еренцировки............................................. 352
Э ндокриногенные расстройства полового развития
и половой ф ункции у лиц генетически женского п о л а ................. 352
Э ндокриногенны е наруш ения полового развития
и половой ф ун кц ии у лиц генетически мужского пола................. 358
Глава 29. Типовые формы патологии нервной системы.......................................361
Общая этиология расстройств нервной д ея те л ь н о сти ........................361
П ричины повреж дения нервной с и с т е м ы .........................................361
Общий патогенез расстройств нервной д е я т е л ь н о с т и ........................363
Повреждение нейронов..............................................................................363
М еханизмы расстройств интегративной деятельности
нервной с и с т е м ы ........................................................................................ 367
Типовые формы наруш ений деятельности нервной систем ы ................. 368
Патологическое ослабление нервных в л и я н и й .....................................369
Патологическое усиление нервных влияни й
на эф ф екторны е структуры ........................................................................... 370
Фазовые состояния в нервной системе......................................................371
Нейрогенные расстройства д в и ж е н и й ......................................................372
Виды нейрогенных расстройств д в и ж е н и й .......................................372
Системы регуляции д в и ж ен и й .............................................................. 373
Характеристика типовых форм расстройств д в и ж е н и я ............... 374
Г и п о к и н ези и .................................................................................................374
Г и перки незии ...............................................................................................377
Н аруш ения ч у встви тел ьн ости .....................................................................381
Типовые формы расстройств ч у в с тв и тел ь н о сти ............................ 381
Гипо- и а н е с т е з и и ...................................................................................... 382
Гиперестезии.................................................................................................383
Д изестези и ..................................................................................................... 384
Общие механизмы расстройств ч увстви тел ьн о сти ........................385
Б о л ь .................................................................................................................. 387
Н ейрогенные расстройства т р о ф и к и ........................................................ 393
М еханизмы нейротрофического к о н т р о л я .......................................393
Н аруш ения высшей нервной деятельн ости ............................................. 395
Экспериментальные н е в р о зы .................................................................397
Э тиология неврозов....................................................................................400
Л и тер ату р а...................................................................................................................... 406
П ри лож ен и я.................................................................................................................... 407
С правочник те р м и н о в .......................................................................................... 407
А вторский с п р а в о ч н и к ........................................................................................ 745
Словарь у д а р е н и й ................................................................................................... 761
Л абораторные п оказател и .................................................................................... 761
П редметны й у к а з а т е л ь ...............................................................................................773
СПИСОК СОКРАЩЕНИЙ И УСЛОВНЫХ ОБОЗНАЧЕНИЙ
* — торговое название ЛС
10 — ЛС не зарегистрировано в РФ
* или # — с последующим кодом из шести цифр (символы * или # ука­
зывают на наличие аллелей, разных фенотипов заболевания или же на то,
что в состав нозологической единицы включено несколько разных пораженных
генов) — менделевское наследование (по ЬИ р://ш \у\^псЫ .п1т.тЬ.§оу/О гш т/)
<-> — синоним
'.Я — аутосомное доминантное наследование
р — аутосомное рецессивное наследование
К — связанное с X-хромосомой наследование
[] — концентрация ионов
АВ — атриовентрикулярный
Аг — антиген, антигены
АД — артериальное давление
АДГ — антидиуретический гормон (вазопрессин)
АДФ — аденозиндифосфат
АКТГ — адренокортикотропный гормон
АЛТ — аланинаминотрансфераза
АНТ — адениннуклеотидтрансфераза
апоЛП — аполипопротеин
АПФ — ангиотензинпревращающий фермент
АСТ — аспартатаминотрансфераза
АТ — антитело, антитела
АТФ — аденозинтрифосфат
АТФаза — аденозинтрифосфатаза
БАВ — биологически активное вещество информационного характера
БЦ Ж (ВСО — ЬасШиз Са1те(1е — Сиепп) — вакцина Кальметта—Герена —
вакцинный штамм МусоЬас1епит Ьт'т пониженной вирулентности
в/в — внутривенно, внутривенный
в/м — внутримышечно, внутримышечный
ВЖ К — высшие жирные кислоты
ВНД — высшая нервная деятельность
ВОЗ — Всемирная организация здравоохранения
ВЗОМТ — воспалительные заболевания органов малого таза
ВПР — врожденный порок развития
Г-6-ФД — глюкозо-6-фосфатдегидрогеназа
ГАМК — у-аминомасляная кислота
ГБ — гипертоническая болезнь
ГМ К — гладкомышечная клетка
ГП К — глюкоза плазмы крови
Да — дальтон
ДВС — диссеминированное внутрисосудистое свертывание (крови)
ДН — дыхательная недостаточность
ДО — дыхательный объем легких
Дс — диффузионная способность
Дссо — диффузионная способность по окиси углерода
ЖДА — железодефицитная анемия
ЖЕЛ — жизненная емкость легких
Ж К Т — желудочно-кишечный тракт
И БН — система иммунобиологического надзора
И БС — ишемическая болезнь сердца
ИВЛ — искусственная вентиляция легких
ИЗСД — инсулинзависимый сахарный диабет
ИЛ — интерлейкин, интерлейкины
ИНСД — инсулиннезависимый сахарный диабет
инфБ — инфекционная болезнь
инф П — инфекционный процесс
И Ф Н — интерферон, интерфероны
кДа — килодальтон
КОС — кислотно-основное состояние
КТ — кетоновые тела
КФ — Классификация ферментов (<ЬПр: / / \у'л'\у.ехра8у.сЬ/8рго 1/е п 2уш е.
Ы т1>). КФ приведена по Е п гу те ТЧотепсЫиге (1ЧС-Ш ВМ В, Комитет
по номенклатуре Международного союза по биохимии и молекулярной био­
логии)
К Ф К — креатинфосфокиназа
К Щ Р — кислотно-щелочное равновесие
КЭАг — карциноэмбриональный Аг
ЛГ — лютеинизирующий гормон, лютропин
Л П — липопротеины
ЛПВП — липопротеины высокой плотности
ЛПЛаза — липопротеинлипаза
Л П Н П — липопротеины низкой плотности
Л П О Н П — липопротеины очень низкой плотности
Л П П П — липопротеины промежуточной плотности
Л П С — липополисахарид
ЛС — лекарственное средство
МВЛ — максимальная вентиляция легких
М К — молочная кислота
М КБ-10 — Международная классификация болезней, 10-й пересмотр
МОД — минутный объем дыхания
М О К — минутный объем кровообращения
М П — мембранный потенциал (покоя)
М СФ — микробоцидная система фагоцитов
НД — несахарный диабет
ННД — нефрогенный несахарный диабет
НПВС — нестероидные противовоспалительные средства
НЯК — неспецифический язвенный колит
ОЕЛ — общая емкость легких
ОЛЛ — острый лимфобластный лейкоз
ОМЛ — острый миелобластный лейкоз
ООЛ — остаточный объем легких
ОПН — острая почечная недостаточность
ОПСС — общее периферическое сопротивление сосудов
ОСН — острая сердечная недостаточность
ОФВ, — объем форсированного выдоха за 1 с
О Ц К — объем циркулирующей крови
п /к — подкожно, подкожный
ПКА — почечный канальцевый ацидоз
ПВ — протромбиновое время
ПГ — простагландин
ПД — потенциал действия
ПТГ — паратиреоидный гормон
РДС — респираторный дистресс-синдром
РОвыд — резервный объем выдоха
СД — сахарный диабет
сдп — сопротивление дыхательных путей
СЕ — субъединица
СКВ — системная красная волчанка
СМ Ж — спинномозговая жидкость, ликвор
СНАДГ — синдром неадекватной секреции АДГ
СНД — сердечная недостаточность диастолическая
СОД — супероксидцисмутаза
СОС25_75% — средняя объемная скорость выдоха
СОЭ — скорость оседания эритроцитов
ССС — сердечно-сосудистая система
СТГ — соматотропный гормон
Т3 — трийодтиронин
Т4 — тетрайодтиронин, тироксин
Т К — титруемая кислотность (суточной мочи)
ТТГ — тиреотропный гормон
УЗИ — ультразвуковое исследование
УФ — ультрафиолетовый
ФЖЕЛ — форсированная жизненная емкость легких
Ф Ж ЕЛ , — форсированная жизненная емкость легких за 1 с
Ф ИО — фактор некроза опухолей
ФОЕ — функциональная остаточная емкость легких
ФСГ — фолликулостимулирующий гормон, фоллитропин
ХГТ — хорионический гонадотропин
ХЛЛ — хронический лимфобластный лейкоз
ХМЛ — хронический миелобластный лейкоз
ХПН — хроническая почечная недостаточность
ХССН — хроническая систолическая сердечная недостаточность
цАМФ — циклический аденозинмонофосфат
цГМ Ф — циклический гуанозинмонофосфат
Ц Н С — центральная нервная система
ЧС С — частота сердечных сокращений (в минуту)
ЧТВ — частичное тромбопластиновое время
ЭАГ — эссенциальная артериальная гипертензия
ЭКГ — электрокардиограмма
ЮГА — юкстагломерулярный аппарат
В-клетки (В произносят как «бэ») — В-лимфоциты
В-ХЛЛ — В-клеточный лимфолейкоз
С1, С2, СЗ (произносят как «си») и т.д. — компоненты системы компле­
мента 1, 2, 3 и т.д.
Са2+ — катион(ы) кальция, ион(ы) кальция; ионизированный (свободный)
кальций
С Б (произносят как «си ди») — кластер дифференцировки (от англ. с1и$1ег
о / сИ//егепИаИоп), см. «Маркёр»
С1_ — анион(ы) хлора
Е О Р — эпидермальный фактор роста (от англ. ерШегта1 %гоц?1к /асЮг)
РаЬ — см. «Фрагмент»
Рс (произносят как «эф си») — см. «Фрагмент»
Н + — ион(ы) водорода
НЬ — гемоглобин
НЬСО — карбоксигемоглобин
Н Ь 0 2 — гемоглобин оксигенированный
НЬА (произносят как «эйч эль эй», от англ. Нитап 1еикосу(е аШщет) — см.
«Антиген», «МНС»
Ш — гематокрит
1САМ (от англ. 1п(егсе11и1аг АсИгеыоп М оксик, С Б54) — молекула межкле­
точной адгезии
1§ — иммуноглобулин(ы)
10 — англ. 1п1е11щепсе диоНеп1, см. «Коэффициент»
К + — катион(ы) калия
ЬРА (от ЬутрНосук РипсИоп-АззоаШей АпИ^еп) — связанный с функцией
лимфоцитов Аг (интегрин), рецептор 1САМ1 (С Б54)
МеШЪ — метгемоглобин
М Н С (произносят как «эм эйч си», от англ. та]ог НШосотраНЬИНу сотркх) —
главный комплекс гистосовместимости
Мг — кажущаяся молекулярная масса
ИА — N отта АпаЮтка (Анатомическая номенклатура)
№ + — катион(ы) натрия
1ЧСАМ (от англ. Иеига! Сей АсИгезюп М оксик) — молекула адгезии нервных
клеток
р — короткое плечо хромосомы (при номере хромосомы)
раС 0 2 — парциальное напряжение двуокиси углерода в артериальной
крови
РАР (от Р1а1е1е( АсИуаИп§ Рас1ог) — фактор активации тромбоцитов
рА0 2 — парциальное давление кислорода в альвеолярном воздухе
ра0 2 — парциальное напряжение кислорода в артериальной крови
р С 0 2 — парциальное давление двуокиси углерода
РЕСАМ1 (от англ. Р1а1е1е(-Епс1о1кеПа1 Се11 Адкезюп М оксик, С Б31) — моле­
кула адгезии тромбоцитов и эндотелия
р 0 2 — парциальное давление кислорода
руС 0 2 — парциальное напряжение двуокиси углерода в венозной крови
ру0 2 — парциальное напряжение кислорода в венозной крови
Я — длинное плечо хромосомы (при номере хромосомы)
§а0 2 — сатурация (насыщение) НЬ кислородом в артериальной крови
8У0 2 — сатурация (насыщение) НЬ кислородом в венозной крови
1: (х; хх) — транслокация между хромосомами (например, I (9; 22) — транс­
локация между хромосомами 9 и 22)
Т-клетки — Т-лимфоциты
У/<3 — вентиляционно-перфузионный
Уа0 2 — объемное содержание кислорода в артериальной крови
УСАМ1 (от англ. Уазси1аг Се11 АйИеа'юп М оксик) — молекула адгезии сосу­
дистых клеток
У1Р (01 англ. УазоасИуе 1пкзНпа1 Ро1урерМе) — вазоактивный интестиналь­
ный (кишечный) полипептид (недопустимо написание «ВИП»)
УЬА (от англ. Уегу Ьа(е АсйуаИоп рго(ет) — очень поздно активируемый
белок
Уу0 2 — объемное содержание кислорода в венозной крови
Г!,<
I
ОТ АКАОЕМ/У
н И М /.
Глава 22
ТИПОВЫЕ ФОРМЫ ПАТОЛОГИИ
СИСТЕМЫ КРОВИ
Кровь — внутренняя среда организма и одна из его интегрирующих
систем. В связи с этим различные отклонения в состоянии организма приво­
дят к изменениям в системе крови, и наоборот. Именно поэтому при оценке
состояния здоровья или нездоровья человека тщательно исследуют параме­
тры, характеризующие кровь (гематологические показатели).
Нарушения общего объема крови
и гематокрита
Общий объем крови составляет 6—8% массы тела. Так, у взрослых мужчин
общий объем крови равен в среднем 5 л. При этом 3 ,5 -4 л обычно циркули­
рует в сосудистом русле и полостях сердца (циркулирующая фракция крови),
а 1,5—2 л депонировано в сосудах органов брюшной полости, легких, под­
кожной клетчатки и других тканей (депонированная фракция).
Форменные элементы составляют 36—48% от общего объема крови.
Гематокрит (Ш , или гематокритное число) — отношение объема форменных
элементов крови к объему ее плазмы. В норме гематокрит равен у мужчин
0,41-0,50, у женщ ин — 0,36—0,44.
НАРУШЕНИЯ ОБЪЕМА КРОВИ
При различных патологических процессах, болезнях и болезненных состо­
яниях может изменяться как общий объем крови, так и соотношение между
ее форменными элементами и плазмой (Ш ). Выделяют три группы типовых
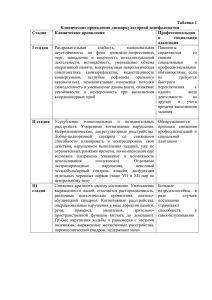
форм нарушений: нормоволемии, гиповолемии, гиперволемии (табл. 22.1).
Таблица 22.1. Типовые формы изменений общего объема и/или соотнош ения
форменных элементов и плазмы крови
Типовые формы
Нормоволемии:
— олигоцитемическая
— полицитемическая
Гиповолемии:
— нормоцитемическая (простая)
— олигоцитемическая
— полицитемическая
Ш (по сравнению
с нормой)
Снижен
Увеличен
Не изменен
Снижен
Увеличен
Окончание табл. 22.1
Типовые формы
Гиперволемии:
— нормоцитемическая (простая)
— олигоцитемическая
— полицитемическая
Ш (по сравнению
с нормой)
Не изменен
Снижен
Увеличен
Нормоволемии
Нормоволемии — состояния, характеризующиеся нормальным общим
объемом крови при сниженном или увеличенном Ш.
Выделяют олигоцитемические и полицитемические нормоволемии.
Олигоцитемическая нормоволемия
Олигоцитемическая нормоволемия — состояние с нормальным общим объ­
емом крови при уменьшении количества ее форменных элементов (главным
образом эритроцитов), что проявляется сниженным гематокритом.
Основные причины олигоцитемической нормоволемии:
— массированный гемолиз эритроцитов (например, при образовании антиэритроцитарных 1§; действии гемолитических веществ: змеиного яда,
соединений свинца, мышьяка, фенилгидразина и др.);
—длительное и значительное угнетение гемопоэза, главным образом эритропоэза (например, при апластических анемиях);
— острая большая кровопотеря. В этой ситуации общий объем крови срав­
нительно быстро нормализуется за счет транспорта жидкости из тка­
ней в сосудистое русло, а число форменных элементов крови остается
еще сниженным (олигоцитемия).
Проявления олигоцитемической нормоволемии:
—анемия (в связи с уменьшением количества эритроцитов и, соответ­
ственно, — гемоглобина) и, как следствие, гемическая гипоксия;
—тромбоцитопения (при кровопотере или реакциях иммунной аутоагрес­
сии в отношении тромбоцитов);
—снижение свертываемости крови, сочетающееся нередко с геморрагиче­
ским синдромом;
—лейкопения, обусловливающая снижение противоинфекционной рези­
стентности организма;
—уменьшение вязкости крови — наблюдается в условиях восстановления
объема жидкой части крови при значительном уменьшении количества
ее форменных элементов (например, на этапе гидремической компенса­
ции при острой кровопотере).
Полицитемическая нормоволемия
Полицитемическая нормоволемия — состояние, характеризующееся нор­
мальным общим объемом крови при увеличении количества ее форменных
элементов, что проявляется значением Ш выше нормы.
Наиболее частые причины полицитемической нормоволемии:
—инфузии пациентам фракций форменных элементов крови (эритроцитарной, лейкоцитарной или тромбоцитарной массы);
—хроническая гипоксия (вызывает эритроцитоз вследствие активации эритропоэза) и эритремии.
Проявления полицитемической нормоволемии:
—увеличение вязкости крови;
—развитие тромботического синдрома;
— нарушения микрогемоциркуляции (замедление тока крови в микрососу­
дах, стаз), которые обусловливают снижение транскапиллярного обмена
в тканях;
— повышенное артериальное давление (АД) в результате увеличения сердеч­
ного выброса в связи с повышенной вязкостью крови.
Гиперволемии
Гиперволемии — состояния, характеризующиеся увеличением общего
объема крови и обычно изменением Ш.
Выделяют нормоцитемическую, олигоцитемическую и полицитемическую
гиперволемии.
Нормоцитемическая гиперволемия
Нормоцитемическая гиперволемия (простая) — состояние, проявляющееся
эквивалентным увеличением объема форменных элементов и жидкой части
ОЦК. Ш при этом остается в диапазоне нормы.
Основные причины простой гиперволемии:
—переливание большого объема донорской крови;
—острые гипоксические состояния, сопровождающиеся выбросом крови из ее
депо, а также значительная физическая нагрузка, приводящая к гипоксии.
Олигоцитемическая гиперволемия
Олигоцитемическая гиперволемия (гидремия, гемодилюция) — состояние,
характеризующееся увеличением общего объема крови вследствие возраста­
ния объема ее жидкой части. Показатель Ш при этом ниже нормы.
Основные причины олигоцитемической гиперволемии:
—избыточное поступление в организм жидкости при патологической жажце
(например, у пациентов с сахарным диабетом — СД) или при введении
в сосудистое русло большого количества плазмозаменителей или плаз­
мы крови;
—замедленное выведение жидкости из организма в результате недостаточ­
ности экскреторной функции почек (например, при почечной недо­
статочности), гиперпродукции АДГ, значительной гиперосмоляльности
плазмы крови.
Полицитемическая гиперволемия
Полицитемическая гиперволемия — состояние, проявляющееся увеличени­
ем общего объема крови вследствие преимущественного повышения количе­
ства ее форменных элементов, в связи с чем Ш превышает верхнюю границу
нормы.
Основные причины полицитемической гиперволемии:
—эритроцитозы (группа патологических состояний, характеризующихся
увеличением количества эритроцитов независимо от количества лейко­
цитов и тромбоцитов);
— истинная полицитемия (ро1усу{кет1а уега, болезнь Вакеза) — хронический
лейкоз с поражением на уровне клетки-предшественницы миелопоэза
с характерной для опухоли неограниченной пролиферацией этого клона
клеток, сохранившей способность дифференцироваться по четырем рост­
кам (преимущественно по эритроцитарному). Эритремия сопровождается
значительным эритроцитозом и, как следствие, повышенным Н1;
—хроническая гипоксия любого типа (гемическая, дыхательная, циркуля­
торная, тканевая и др.).
Полицитемия отражает гиперрегенераторное состояние костного мозга, кото­
рое сопровождается повышенной пролиферацией клеток крови, главным образом
эритроцитов, и выходом их в сосудистое русло. Полицитемическая гиперволемия
выявляется при хронической недостаточности кровообращения, альвеолярной
гиповентиляции, снижении кислородной емкости крови и эффективности био­
логического окисления, при экзогенной (нормо- и гипобарической) гипоксии.
Проявления гиперволемии
Для гиперволемий характерно увеличение сердечного выброса и повы ­
ш ение АД:
—увеличение сердечного выброса в результате компенсаторной гипер­
функции сердца (в связи с увеличением объема и/и ли вязкости крови).
Однако при декомпенсации функции сердца и развитии его недостаточ­
ности сердечный выброс, как правило, снижается;
— повышение АД обусловлено, главным образом, увеличением сердечного
выброса, а также О Ц К и тонуса резистивных сосудов.
Для истинной полицитемии (эритремии) характерны:
—увеличение вязкости крови;
—повышенная агрегация и агглютинация форменных элементов крови;
—диссеминированное тромбообразование;
— расстройства микрогемоциркуляции.
Гиповолемии
Гиповолемии — состояния, характеризующиеся уменьшением а§негв
объема крови и, как правило, нарршеянем соотношения ее
элементов и плазмы (гематокрита).
Различают нормоцитемическую, олигоцитемическую и полицитемическую
гиповолемии.
Нормоцитемическая гиповолемия
Нормоцитемическая гиповолемия — состояние, проявляющееся уменьшени­
ем общего объема крови при сохранении Ш в пределах нормы.
Причины нормоцитемической гиповолемии:
— острая кровопотеря;
— шоковые состояния;
— вазодилатационный коллапс.
В двух последних случаях нормоцитемическая гиповолемия развивается
в результате депонирования большого объема крови в венозных (емкостных)
сосудах и значительного снижения в связи с этим ОЦК.
Проявления нормоцитемической гиповолемии определяются характером при­
чины, вызвавшей ее (кровопотеря, шок, коллапс), а также включением меха­
низмов компенсации, направленных на устранение острой гипоксии.
Олигоцитемическая гиповолемия
Олигоцитемическая гиповолемия — состояние, характеризующееся сниже­
нием общего объема крови (с преимущественным уменьшением количества
ее форменных элементов) и снижением, в связи с этим, гематокрита.
Наиболее частые причины олигоцитемической гиповолемии:
—состояния после острой кровопотери (на том ее этапе, когда транспорт
жидкости из тканей и выход депонированной крови в сосудистое русло
еще не устраняют гиповолемии, а поступление клеток крови из органов
гемопоэза — дефицита эритроцитов);
—эритропении в результате массированного гемолиза эритроцитов (напри­
мер, при ожогах большой поверхности тела, когда гемолиз сочетается
с потерей организмом жидкой части крови в связи с плазморрагией)
и подавления эритропоэза (например, при апластических или арегенераторных состояниях).
Проявления олигоцитемической гиповолемии:
—снижение кислородной емкости крови (в результате эритропении);
— гемическая гипоксия (проявляющаяся меньшим содержанием кислорода
в крови, ацидозом, уменьшением р 0 2 венозной крови и др.);
— расстройства органно-тканевого кровообращения и микрогемоциркуляции
различной степени, обусловленные, помимо прочих факторов, умень­
шением ОЦК.
Полицитемическая гиповолемия
Полицитемическая гиповолемия — состояние, при котором снижение обще­
го объема крови в организме обусловлено в основном уменьшением объема
плазмы; показатель № при этом выше нормы.
Наиболее частые причины полицитемической гиповолемии:
—повышенная потеря организмом жидкости — при повторной рвоте (напри­
мер, у беременных или в результате экзогенной интоксикации), длитель­
ной диарее (в частности, при нарушении мембранного пищеварения,
кишечных токсикоинфекциях), полиурии (в том числе при почечной
недостаточности), повышенном и длительном потоотделении (напри­
мер, в условиях жаркого климата или в горячих цехах на производстве),
обширных ожогах кожи (сопровождающиеся плазморрагией);
—недостаточное поступление жидкости в организм (водное «голодание»),
например, при отсутствии питьевой воды и/и ли невозможности питья
воды (например, в результате спазма мускулатуры при столбняке
или бешенстве).
Проявления полицитемической гиповолемии:
—нарушения микрогемоциркуляции в связи с гиповолемией и полицитемией;
— повышение вязкости крови, агрегация форменных элементов крови
в микрососудах органов и тканей и диссеминированный микротромбоз;
—признаки основной патологии, вызывающей полицитемическую гиповолемию, например шока, несахарного диабета (НД), почечной недоста­
точности, ожоговой болезни и др.
КРОВОПОТЕРЯ
Кровопотеря — состояние, характеризующееся утратой организмом части
крови. При этом развивается комплекс патогенных и адаптивных реакций
организма, совокупность которых называют состоянием после кровопо­
тери. Состояние после кровопотери проявляется расстройством жизне­
деятельности организма различной степени (в зависимости от величины
кровопотери и реактивности организма).
Кровопотеря является следствием кровотечения (геморрагии) — излияния крови
из кровеносных сосудов и/или полостей сердца во внешнюю среду (внешнее
кровотечение) или в полости организма (внутреннее, полостное кровотечение).
Наличие крови в полостях организма обозначают специальными терми­
нами.
• Гемоторакс — наличие крови в плевральной полости.
• Гемоперикардиум — кровь в полости перикарда.
• Гемоперитонеум — излияние крови в брюшную полость.
• Гемартроз — кровь в полости сустава.
Кровотечение следует отличать от кровоизлияния и гематомы.
• Кровоизлияние — очаговое или диффузное пропитывание тканей (напри­
мер, подкожной клетчатки, мышц) кровью.
• Гематома — локальное скопление крови в ткани.
При кровоизлиянии и гематоме из сосудистого русла выходит сравни­
тельно небольшой объем крови, и существенных расстройств системного
кровообращения не наблюдается. Развивающиеся в организме нарушения
определяются в основном ролью органа или ткани, в которые произошло
кровоизлияние или в котором сформировалась гематома (мозг, печень, почки,
мышцы, подкожная клетчатка).
Этиология кровопотери
Наиболее частые причины кровопотери
• Нарушение целостности стенок сосудов или сердца при механическом воз­
действии (например, разрез или разрыв стенки), гнойном расплавлении
стенки сосудов или разрушении ее растущей опухолью, разрыве стенок
желудочков или предсердий в зоне инфаркта миокарда или аневризмы.
• Значительное повышение проницаемости стенок сосудов, особенно микроциркуляторного русла. Наблюдается при лучевой болезни, экстрамедуллярных очагах кроветворения (например, у пациентов с лейкозами),
инфекционных процессах (например, сыпном тифе, сепсисе), тяжелом
гиповитаминозе С (цинга).
• Существенное снижение свертываемости крови. Это обстоятельство (осо­
бенно в сочетании с повышенной проницаемостью стенок микрососудов)
может привести к потере организмом значительного количества крови
(например, при маточных и желудочно-кишечных кровотечениях).
Условия, влияющие на течение и исходы кровопотери
• Особенности кровопотери:
—объем потерянной крови:
о выход из сосудистого русла до 20-25% ОЦК, как правило, не опасен
и компенсируется вследствие включения экстренных механизмов ком­
пенсации;
о потеря 25—35% О Ц К сопровождается значительными расстройствами
центральной, органотканевой циркуляции и микрогемоциркуляции;
о потеря 50% и более от общего объема крови (особенно быстрая) деталь­
на;
— скорость кровопотери: чем меньше скорость кровопотери, тем менее
выражены расстройства жизнедеятельности. Так, утрата даже половины
общего объема крови в течение нескольких дней (при маточном, желу­
дочном, геморроидальном и других видах кровопотери), как правило,
не приводит к смерти.
• Соотношения активности факторов свертывающей, противосвертывающей
и фибринолитической систем организма. Снижение активности или содер­
жания факторов свертывающей и/или повышение противосвертывающей
и фибринолитической систем, ведущее к меньшей свертываемости крови,
может обусловить увеличение скорости и объема кровопотери, что усугу­
бляет ее течение и последствия.
• Реактивность организма. Течение и последствия кровопотери в существен­
ной мере зависят от реактивности организма, определяемой:
—полом (женщины менее чувствительны к кровопотере), возрастом
(взрослые переносят кровопотерю легче, чем дети);
—текущим состоянием организма (при перегревании или охлаждении
последствия кровопотери тяжелее, чем при нормальной температуре;
в условиях глубокого наркоза расстройства жизнедеятельности более
выражены, чем в бодрствующем состоянии).
Механизм развития острых постгеморрагических
состояний
Механизм острых постгеморрагических состояний представлен на рис. 22.1.
На начальном этапе кровопотери в большей или меньшей мере снижается
ОЦК при сохранении нормального Ш, т.е. развивается нормоцитемическая
гиповолемия. В связи с этим уменьшается приток венозной крови к сердцу,
снижается его ударный и минутный выброс.
Кровопотеря
Уменьшение объема циркулирующей крови
Снижение притока венозной крови к сердцу!
______________________ ±______________________
Уменьшение ударного и минутного выброса крови сердцем |
Снижение артериального давления
__________________I ___________________
Уменьшение перфузионного давления в сосудах
___________ *___________
Нарушения микроциркуляции |
Расстройства жизнедеятельности организма |
Рис. 22.1. Основные звенья патогенеза постгеморрагических состояний
Снижение сердечного выброса крови приводит к падению АД и, как след­
ствие, перфузионного давления в сосудах органов и тканей. В результате умень­
шается транспорт кислорода и субстратов метаболизма из крови к клеткам,
а от последних — углекислого газа и продуктов обмена веществ. Развивается
капилляротрофическая недостаточность, интоксикация организма продуктами
нарушенного метаболизма, гипоксия, ацидоз.
Указанные изменения, в свою очередь, вызывают расстройства энерге­
тического обеспечения клеток и пластических процессов в них. Нарушается
функция органов и тканей, что нередко сопровождается их недостаточностью,
выраженной в большей или меньшей мере. Существенно расстраивается
жизнедеятельность организма в целом. Крайняя степень этих расстройств —
постгеморрагический шок.
Нарушение системной гемодинамики и меньш ая интенсивность био­
логического окисления в клетках обусловливают включение или активацию
адаптивных механизмов.
Адаптивные механизмы при постгеморрагическом
состоянии
Основные адаптивные механизмы компенсации кровопотери
• Активация свертывающей системы крови и процесса тромбообразования.
• Реакции сердечно-сосудистой компенсации кровопотери (гидремическая ком­
пенсация кровопотери):
—сужение просвета резистивных сосудов;
— выброс крови из депо;
— повышение сердечного выброса;
— поддержание О Ц К на максимально возможном уровне (за счет посту­
пления в сосуды жидкости из интерстиция, а также тока лимфы).
• Восстановление белкового состава крови (вследствие синтезов в печени):
реакция белковой компенсации кровопотери.
• Устранение дефицита форменных элементов крови вследствие активации
гемопоэза: клеточная, костномозговая компенсация.
• Активация механизмов экстренной и долговременной адаптации к гипоксии
(подробнее см. раздел «Адаптивные реакции организма при гипоксии»
в главе 16 «Патофизиология гипоксии», т. 1).
Стадии компенсации кровопотери
Названные выше механизмы активируются в разные сроки после крово­
потери.
В связи с этим выделяют следующие стадии развития процессов компенсации
кровопотери:
—сердечно-сосудистую;
—гидремическую;
— белково-синтетическую;
—костномозговую.
Следует помнить, что многие из названных процессов протекают в орга­
низме не строго последовательно (стадийно), а чаще параллельно, совпадая
во времени и, как правило, потенцируя друг друга. Это способствует более
быстрой и эффективной ликвидации последствий кровопотери.
Сердечно-сосудистая компенсация острой кровопотери
Заключается в стимуляции работы сердца и в изменениях тонуса и про­
света артериол.
Стимуляции работы сердца (в связи с активацией симпатико-адреналовой
системы) способствует:
—увеличению ЧС С и ударного выброса (как правило);
— возрастанию (в связи с вышеуказанными изменениями) интегрального
показателя функции сердца — сердечного выброса (однако при значи­
тельной кровопотере он может оставаться ниже необходимого).
Изменение тонуса и просвета артериол обеспечивает развитие феномена «цен­
трализации кровотока». Этот феномен характеризуется:
—расширением артериол мозга и сердца (в результате быстрого образования
факторов с сосудорасширяющим действием: аденозина, ПГ, кининов, N 0 ),
а также накоплением в клетках и интерстиции ионов Н +, выходом из клеток
ионов К +. Увеличением содержания в них Иа+ и некоторых других ионов.
Указанные и другие изменения способствуют снижению тонуса стенок
артериол и поддержанию оптимального кровоснабжения сердца и мозга;
— одновременным сужением просвета артериол подкожной клетчатки, кожи,
мышц, органов брюшной полости, почек и некоторых других тканей и орга­
нов, что уменьшает в них объем кровотока. Повышение тонуса артериол
в указанных органах и тканях обусловливает также выброс депониро­
ванной крови в сосудистое русло и увеличение ОЦК. В целом, на этапе
сердечно-сосудистой компенсации острой кровопотери еще сохраняется
нормоцитемическая гиповолемия.
Гидремическая компенсация острой кровопотери
В первые же минуты после кровопотери активируются механизмы, обе­
спечивающие усиление тока жидкости из тканей в сосудистое русло.
Пусковой фактор гидремической компенсации при острой кровопотере — сни­
жение ОЦК. Основную роль в механизме гидремической компенсации играют
вазопрессин (АДГ) и альдостерон, при этом происходят следующие процессы:
— гиповолемия стимулирует секрецию АДГ через барорецепторы каротид­
ной области;
—АДГ повышает активность образованного аквапорином-2 водного кана­
ла в собирательных трубочках, что усиливает реабсорбцию жидкости
из просвета собирательных трубочек в межклеточное пространство;
—под влиянием АДГ сужается просвет междольковых артерий и принося­
щих артериол нефронов, снижается клубочковая фильтрация и умень­
шается степень гиповолемии;
—АДГ снижает также кровоснабжение клеток юкстагломерулярного аппа­
рата, в связи с чем возрастает секреция ими ренина, образование под его
влиянием ангиотензина II, повышающего тонус стенок артериол, стиму­
лирующего высвобождение катехоламинов и секрецию альдостерона;
—повышение уровня альдостерона в крови стимулируется также реаб­
сорбцией № + в почечных канальцах почек, благодаря чему развивается
гиперосмия плазмы крови;
— гиперосмия крови включает осморефлекс — активацию осморецепторов
сосудистого русла и стимуляцию секреции АДГ нейронами гипоталамуса;
—альдостерон, кроме того, активирует реабсорбцию К а 1 из первичной
мочи в кровь, что стимулирует высвобождение АДГ, потенцирующего
ток жидкости в сосудистое русло и восстановление утраченного объема
жидкой части крови.
Одновременно с описанными выше изменениями активируется ток жид­
кости из клеток в межклеточное пространство (по градиенту осмотического
давления), в лимфатические капилляры и затем в кровь.
Таким образом, на этапе гидремической компенсации (на 2—3-и сутки
после кровопотери) наблюдается олигоцитемическая гипо- или нормоволе­
мия. Поступающая в сосудистое русло интерстициальная жидкость содержит
меньшее в сравнении с плазмой количество белка. Это стимулирует в орга­
низме синтетические процессы.
Белковая компенсация острой кровопотери
Гипопротеинемия устраняется в связи с активацией протеосинтеза в пече­
ни уже через несколько часов после кровотечения. Признаки повышенного
синтеза белков регистрируются еще в течение 1,5—3 нед и более в зависимости
от объема кровопотери и реактивности организма.
Помимо прочих белков, в печени синтезируются также прокоагулянты.
Это сочетается с активацией реакций гомеостаза. Последнее способствует
увеличению так называемого гемостатического потенциала, тромбированию
дефекта сосудистого русла и снижению интенсивности или прекращению
кровотечения.
Клеточная (костномозговая) компенсация острой кровопотери
Причины костномозговой реакции в ответ на кровопотерю:
—гипоксия (она носит смешанный характер и по существу является гемической, циркуляторной, дыхательной);
—физико-химические изменения в тканях и биологических жидкостях (уве­
личение содержания Н +, Ыа+, продуктов гидролиза АТФ и др.), стиму­
лирующие синтез факторов роста в гемопоэтических клетках костного
мозга, а также лимфоидной ткани. Наибольшее значение среди этих
веществ имеет эритропоэтин.
Виды кровопотери
В зависимости от поврежденного сосуда или отдела сердца, из которого
возникает кровотечение, от объема потерянной крови, времени, отделяющего
кровотечение от травмы сердца или сосудистой стенки, места кровоизлияния
выделяют следующие виды кровопотери.
• По виду поврежденного сосуда или камеры сердца:
— артериальная;
— венозная;
— капиллярная;
— смешанная.
• По объему потерянной крови:
—легкая (до 20-25% ОЦК);
— средняя (25-35% ОЦК);
—тяжелая (более 35—40% ОЦК).
• По времени, отделяющему кровотечение от травмы сердца или сосуда:
— первичная — кровотечение начинается сразу после травмы;
— вторичная — кровотечение отдалено по времени от момента травмы.
• По месту излияния крови:
— наружная — кровоизлияние во внешнюю среду;
— внутренняя — кровоизлияние в полости тела или в органы.
Принципы и методы лечения кровопотери
Лечение при кровопотере базируется на этиотропном, патогенетическом
и симптоматическом принципе.
Этиотропный принцип лечения при кровопотере
Для прекращения кровопотери (уменьшения ее степени) необходимо воз­
действовать на причину кровопотери: восстановить целостность стенки сосуда
или сердца, повысить свертываемость крови.
П атогенетический принцип лечения при кровопотере
Восстановление ОЦК достигается устранением или уменьшением степени
расстройств центрального и органотканевого кровообращения путем пере­
ливания крови, плазмы, плазмозаменителей, например декстрана (полиглюкин*), гемодеза* и др.
Нормализацию транскапиллярного обмена обеспечивают, уменьшая сте­
пень расстройств микроциркуляции вливанием плазмозаменителей, например
декстрана (реополиглюкин*), желатиноляА, изотонического раствора натрия
хлорида.
Устранение сдвигов или уменьшение степени нарушения водного, белкового
и ионного дисбаланса достигается (помимо восстановления ОЦК и нормали­
зации транскапиллярного обмена) введением растворов, которые содержат
белки и ионы в таком количестве и соотношении, чтобы устранить их дис­
баланс в организме.
Коррекцию КОС выполняют, восстанавливая ОЦК, уменьшая степень рас­
стройств микроциркуляции при введении буферных растворов и жидкости,
нормализуя функции органов, компенсирующих сдвиги КОС (почки, печень,
легкие и др.).
Симптоматический принцип лечения при кровопотере
Этот принцип включает мероприятия, которые направлены на нормали­
зацию функций органов и их систем, нарушенных в результате кровопотери
и гипоксии (ССС, дыхательная система, почки, печень и др.).
Патофизиология системы
эритроцитов
Эритроцит — безъядерная клетка диаметром 7—8 мкм (нормоцит).
Количество эритроцитов колеблется в диапазоне 3,9—4,9хЮ12/л у женщин
и 4,0—5,2хЮ12/л у мужчин. Более высокое содержание эритроцитов у муж­
чин обусловлено стимуляцией эритропоэза андрогенами. Продолжительность
жизни (время циркуляции в крови) эритроцитов равна 100-120 сут. Форма
эритроцита (двояковогнутый диск) создает наибольшую площадь поверх­
ности по отношению к объему, что обеспечивает максимальный газооб­
мен. Эритропоэз и другие направления гемопоэза рассмотрены в разделах
«Гемопоэз» и «Эритропоэз» в приложении «Справочник терминов».
К типовым формам изменений и патологии в системе эритроцитов отно­
сятся эритроцитозы, эритропении и анемии.
Э РИТР0Ц ИТ03Ы
Эритроцитозы (эритрсмии) — состояния, характеризующиеся увеличением
количества эритроцитов в единице объема крови выше нормы.
Виды эритроцитозов
Различают первичные и вторичные эритроцитозы (рис. 22.2).
Рис. 22.2. Виды эритроцитозов
К первичным эритроцитозам (самостоятельные формы болезни) относят
истинную полицитемию (болезнь Вакеза) и семейные (наследуемые) формы.
Среди вторичных эритроцитозов (симптомы других болезней, патологиче­
ских состояний или процессов) различают абсолютные (вследствие усиления
эритропоэза и/или поступления эритроцитов в сосудистое русло из костного
мозга) и относительные формы. Последние могут быть гемоконцентрационными (гиповолемическими) и перераспределительными.
Первичные эритроцитозы
Наиболее часто из первичных эритроцитозов встречается болезнь Вакеза
(см. статью «Полицитемия истинная» в приложении «Справочник терми­
нов»).
Патогенез болезни Вакеза
Ключевыми звеньями патогенеза первичного эритроцитоза при болезни
Вакеза считают:
—увеличение в гемопоэтической ткани количества злокачественных про­
лиферирующих клеток-предшественниц миелопоэза;
—усиление миелопролиферативного опухолевого процесса в гемопоэтиче­
ской ткани. Это отмечается не только в костном мозге, но также нередко
в селезенке и печени, колонизируемых клетками-предшественницами
миелопоэза.
О моноклональном характере миелопролиферации при болезни Вакеза
свидетельствуют факты обнаружения в эритроцитах, гранулоцитах и тромбо­
цитах одного и того же дефекта хромосом (аберрации, анеуплоидии и др.)
или дефектного фермента, кодируемого одним и тем же мутантным аллелем.
Проявления болезни Вакеза
Эритремия сопровождается существенными изменениями в костном мозге,
периферической крови, нарушениями функций ССС и других систем.
Гематологические проявления эритремии
• В костном мозге:
—интенсивная пролиферация опухолевого пула миелоидных клеток (в основ­
ном в проксимальных, нередко в дистальных отделах трубчатых костей,
а также в плоских костях, печени и селезенке);
—ускорение обмена железа (введение в кровь препаратов, содержащих 59Ре
и 52Ре, сопровождается большей скоростью процессов утилизации желе­
за тканью костного мозга и последующего выведения его);
—уменьшение массы эритропоэтической ткани костного мозга (постэритремический миелофиброз). В поздних стадиях эритремии это приводит
к развитию анемии и тромбоцитопении.
• В периферической крови:
— эритроцитоз, ретикулоцитоз, тромбоцитоз, нейтрофилия (с ядерным
сдвигом влево до метамиелоцитов и даже миелоцитов), эозинофилия
и базофилия, моноцитоз;
—гиперволемия (полицитемическая);
—увеличение содержания НЬ (обычно до 180—200 г/л);
— гипохромия эритроцитов (является результатом отставания синтеза НЬ
от темпов эритроидной пролиферации).
На заключительных этапах болезни, напротив, развиваются эритропе­
ния, тромбоцитопения и даже панцитопения: уменьшение количества всех
или многих клеток миелоидного ряда в связи с постэритремическим миелофиброзом (рис. 22.3).
Рис. 22.3. Основные гематологические проявления эритремии
Нарушения кровообращения при эритремии
Нарушения в системе кровообращения приведены на рис. 22.4.
И з м е н е н и я к р о в о о б р а щ е н и я при э р и т р е м и и
'
Артериальная
гипертензия
1Г
Расстройства
органотканевого
кровотока
’Г
1Г
Нарушения Тромботический
микро­
синдром
циркуляции
V
Геморрагический
синдром
Рис. 22.4. Изменения кровообращения при эритремии
При эритремии, как правило, выявляются следующие отклонения в систе­
ме кровообращения.
•Артериальная гипертензия (наблюдается почти у половины пациентов
с эритремией; сочетание эритроцитоза с артериальной гипертензией назы ­
вают синдромом Гайсбека). Причины артериальной гипертензии при эри­
тремии:
—увеличение сердечного выброса крови (как следствие гиперволемии;
при длительном течении эритремии сердечный выброс снижается
в связи с развитием сердечной недостаточности);
— повышение ОПСС, которое также является следствием гиперволемии;
— активация системы «ренин — ангиотензин-альдостерон — АДГ» (обу­
словлена нарушением кровообращения в почках, склерозированием
и тромбозом почечных артерий).
• Расстройства органотканевого кровотока в виде ишемии, венозной гиперемии
и стаза. Эти изменения вызваны в основном повышением вязкости крови
в связи с полицитемией.
• Нарушения микроциркуляции, главным образом интраваскулярные (замед­
ление кровотока в сосудах микроциркуляторного русла, стаз, турбулент­
ный ток крови), вызванные существенным повышением вязкости крови
и микротромбами в артериях, венах, микрососудах.
• Высокая частота тромбоза сосудов. Обусловлена полицитемией, повыш е­
нием вязкости крови, снижением скорости кровотока в сосудах, тромбоцитозом и тромбоцитопатиями, способствующими адгезии, агрегации
и агглютинации форменных элементов крови, а также высвобождению
из них прокоагулянтов.
• Частые геморрагии. Они вызваны нарушением структуры и функции эри­
троцитов и тромбоцитов (как следствие их опухолевого атипизма) и потре­
блением факторов гемокоагуляции.
П ом им о болезни В акеза, к первичны м эритроцитозам относят ряд
семейны х наследуемых н ем иелопролиф еративны х (т.е. не обусловлен­
ных опухолевой трансф орм ацией клеток эритроидного ряда) заболеваний
с малоизученны ми на сегодняш ний день этиологией и патогенезом. Все
эти заболевания характеризую тся увеличением количества эритроцитов
в единице объема крови, гиперволем ией и другими признакам и истинной
полицитемии.
Вторичные эритроцитозы
Вторичные эритроцитозы — состояния, являющ иеся симптомами других
болезней или патологических процессов. Устранение причин этих болезней
или процессов позволяет ликвидировать вторичные эритроцитозы, не прово­
дя специального лечения.
Вторичные эритроцитозы подразделяются на абсолютные и относитель­
ные.
Вторичные абсолютные эритроцитозы
Вторичные абсолютные эритроцитозы — состояния, характеризующиеся
увеличением числа эритроцитов в единице объема крови в связи с активацией
эритропоэза и выходом избытка эритроцитов из костного мозга в сосудистое
русло.
Причина вторичных абсолютных эритроцитозов
Непосредственная причина вторичного абсолютного эритроцитоза — уси­
ленное образование эритропоэтина и/или повышение чувствительности к нему
эритроидных клеток. В свою очередь, причиной этих нарушений наиболее
часто бывают:
— общая, как правило, хроническая гипоксия любого генеза. Гипоксия —
важнейший фактор, стимулирующий продукцию эритропоэтина;
—ишемия почки или обеих почек, реже печени, селезенки (при кистах в них,
отеке, стенозе артерий, воспалении), что стимулирует выработку эритропоэтинов;
— опухолевый рост, сопровождающийся избыточной продукцией эритропоэ­
тина, например новообразования почки (гипернефрома), печени, селе­
зенки, матки.
Проявления вторичных абсолютных эритроцитозов
Проявления вторичного абсолютного эритроцитоза приведены на рис. 22.5.
Рис. 22.5. Основные гематологические проявления вторичных абсолютных эритроцитозов
Они включают:
• костный мозг:
—увеличение количества пролиферирующих клеток эритроидного ростка
костного мозга (под влиянием эритропоэтина и /и ли в связи с повыше­
нием чувствительности к нему клеток-мишеней),
— возрастание численности эритроидных клеток разной степени зрелости
(от эритробластов до ретикулоцитов и эритроцитов);
• периферическую кровь:
— эритроцитоз и ретикулоцитоз;
— полицитемическая гиперволемия, Ш выше нормы, повышение вязкости
крови. При длительном значительном эритроцитозе происходит гипер­
трофия миокарда.
В отличие от истинной полицитемии, эритроцитозы, как правило, не сопро­
вождаются тромбоцитозом и лейкоцитозом.
Вторичные относительные эритроцитозы
Вторичные относительные эритроцитозы характеризуются увеличением числа
эритроцитов в единице объема крови без активации их продукции в костном
мозге и без повышения их абсолютного числа в крови.
Причины вторичных относительных эритроцитозов:
— снижение объема плазмы крови (гемоконцентрация), сопровождающее
потерю организмом жидкости (диарея, рвота, плазморрагия при ожого­
вой болезни, лимфоррагия). Этим обусловлено развитие полицитемиче­
ской гиповолемии;
— выброс в циркулирующую кровь эритроцитов из органов и тканей, депони­
рующих их (при стресс-реакции, острой гипоксии, гиперкатехоламинемии) с развитием полицитемической гиперволемии.
Проявления вторичных относительных эритроцитозов:
— повышение Ш (как результат гемоконцентрации);
—нормо- или гиповолемическая полицитемия (в основном за счет эритроци­
тоза);
—повышение вязкости крови.
АНЕМИИ
Анемия — уменьшение общего количества НЬ в организме, которое харак­
теризуется снижением уровня НЬ в единице объема крови (за исключением
острой кровопотери).
В больш инстве случаев анем ии сопровождаю тся и эритропенией.
Исключение составляют некоторые железодефицитные состояния и талас семии. При них количество эритроцитов может быть нормальным или даже
увеличенным.
Термин «анемия» отражает только изменения в крови, выявленные лабо­
раторными методами. Таким образом, анемия может характеризовать либо
конкретное заболевание (например, железодефицитная анемия, ЖДА), либо
быть одним из симптомов других патологических состояний.
Общие лабораторные признаки анемии
К наиболее общим признакам анемии относят:
— содержание НЬ периферической крови ниже 100 г/л;
— количество эритроцитов менее 4хЮ12/л;
—уровень железа в сыворотке крови менее 14,3 мкмоль/л.
Во врачебной практике возможно наличие одного признака анемии
или их сочетание (например, для талассемий не характерно уменьшение
количества эритроцитов).
С практической точки зрения основной и обязательной характеристикой ане­
мии является сниженное содержание НЬ в единице объема крови.
Сущность анемии и ее значение для организма определяются прежде всего
уменьшением кислородной емкости крови, что приводит к гипоксии гемического
типа. Именно с гипоксией связаны основные клинические симптомы и рас­
стройства жизнедеятельности у больных анемией.
От анемий следует отличать гидремии — состояния, обусловленные увели­
чением жидкой части крови (гемодилюция) при нормальном общем содержании
в организме НЬ и эритроцитов. Концентрация НЬ в единице объема крови
при этом снижена, что дает формальную картину анемии. В данном случае
говорят о ложной анемии, так как общее количество НЬ в крови не уменьша­
ется. Ложная анемия может наблюдаться после инфузии большого количества
жидкости, плазмы или сыворотки крови в сосудистое русло.
Необходимо помнить также о вероятности так назы ваемой скрытой
анемии. Так, при обезвоживании организма у пациентов с анемией (рвота,
понос, интенсивное и /и ли длительное потение без восполнения утраченного
объема жидкости) может отмечаться «сгущение» крови (гемоконцентрация),
при котором в единице ее объема количество НЬ может быть нормальным
или даже повы ш енны м, несмотря на снижение его общего содержания
в организме.
ВИДЫ АНЕМИЙ
Анемия, как правило, — симптом другого заболевания, поэтому строго
классифицировать анемии сложно. Тем не менее предложены классифика­
ционные критерии, позволяющие дифференцировать анемии по ряду каче­
ственных и количественных параметров (например, по причине, патогенезу,
типу кроветворения и др.).
Классификационные признаки анемии
• Причина:
— первичные (наследственные, врожденные);
—вторичные (приобретенные).
• Патогенез:
— постгеморрагические;
—гемолитические;
—дизэритропоэтические.
• Тип кроветворения при анемии:
— нормобластные (нормоцитарные);
— мегалобластные (мегалоцитарные).
• Регенераторная способность эритроидного ростка:
— регенераторные;
— гиперрегенераторные;
— гипорегенераторные;
—арегенераторные;
—апластические.
• Размер эритроцитов:
—нормоцитарные;
—микроцитарные;
—макроцитарные;
—мегалоцитарные.
• Острота развития анемии:
— острая (развивается в течение нескольких суток);
—хроническая (наблюдается в течение нескольких недель—лет).
Некоторые дополнительные наименования анемий приведены в статье
«Анемии» (см. приложение «Справочник терминов»).
Большое практическое значение для характеристики анемии имеют сле­
дующие критерии.
• Размер эритроцитов:
—микроцитарные анемии: средний диаметр эритроцита менее 6,7 мкм;
—нормоцитарные анемии: средний диаметр эритроцита 7 -8 мкм [средний
эритроцитарный объем 80—94 фемтолитра (фл)];
—макроцитарные анемии: средний диаметр эритроцита более 9,5 мкм.
• Степень насыщения эритроцитов гемоглобином или содержание сывороточ­
ного железа (среднее содержание НЬ в эритроците в норме составляет 27—33
пикограмма). В повседневной практике наиболее доступный метод опреде­
ления содержания НЬ в эритроцитах — расчет цветового показателя:
—нормохромия эритроцитов: значение цветового показателя колеблется
в диапазоне 0,8—1,0;
— гипохромия эритроцитов: значение цветового показателя менее 0,8;
—гиперхромия эритроцитов: при цветовом показателе более 1,0.
• Степень регенерации эритроцитов в костном мозге. Ее определяют по коли­
честву ретикулоцитов в периферической крови (по этому значению оцени­
вают и эффективность эритропоэза). В норме количество ретикулоцитов
в циркулирующей крови равно 0,5—1,5% (5—15%с);
—регенераторная анемия (иногда гиперрегенераторная, если количество
ретикулоцитов значительно превышает верхнюю границу нормы): уве­
личение количества ретикулоцитов более 1,5% свидетельствует об акти­
вации эритропоэза, например, при гемолитических анемиях или ЖДА;
—гипорегенераторная анемия: число ретикулоцитов в периферической крови
ниже нормы говорит о снижении эффективности эритропоэза (например,
апластические анемии, витамин В12-дефицитные анемии, лейкозы).
• Концентрация гемоглобина:
—легкая степень (НЬ 80-100 г/л);
— средняя степень (НЬ 60—80 г/л);
—тяжелая степень (НЬ ниже 60 г/л).
Возможно сочетание двух критериев, например гипохромные микроци­
тарные анемии, нормохромные нормоцитарные анемии (элемент «-хромный»
указывает на содержание НЬ в эритроцитах).
Механизмы развития анемий
В настоящей главе механизмы развития анемий рассматриваются в соот­
ветствии с их патогенетической классификацией. В связи с этим все анемии
подразделены на:
—постгеморрагические;
—гемолитические;
—дизэритропоэтические (обусловленные нарушениями эритропоэза).
Постгеморрагические анемии
Постгеморрагические анемии развиваются в результате острого или хро­
нического кровотечения.
Острая постгеморрагическая анемия
Острая постгеморрагическая анемия — нормохромная нормоцитарная
гиперрегенераторная анемия, возникающая вследствие острой кровопотери
в течение короткого периода времени.
Минимальная потеря крови, представляющая опасность для здоровья взрос­
лого человека, равна примерно 500 мл. Тяжесть клинической картины определя­
ется количеством потерянной крови, скоростью и источником кровотечения.
Причина острой постгеморрагической анемии — массированное кровотечение
из поврежденных крупных сосудов или полостей сердца (травмы и хирургические
вмешательства, внематочная беременность, нарушения гемостаза, различные
заболевания внутренних органов, сопровождающиеся острым кровотечением).
Проявления острой постгеморрагической анемии
Общие признаки острой постгеморрагической анемии:
—тахикардия;
— одышка;
— падение АД и венозного давления;
— бледность кожных покровов и слизистых оболочек.
Выраженность этих изменений может не соответствовать тяжести анемии,
так как нередко они появляются в ответ на причину кровотечения (например,
на боль или травму).
Изменения в периферической крови носят стадийный характер и зависят
от времени, прошедшего после кровотечения.
В первые часы и сутки после кровопотери выявляются:
— нормоцитемическая гиповолемия (эквивалентное уменьшение общего
содержания форменных элементов и плазмы крови);
— снижение объема циркулирующих эритроцитов;
—нормальные показатели Ш , числа эритроцитов, уровня НЬ в единице
объема крови.
На 2—3-и сутки после кровопотери наблюдаются:
—эритропения;
— содержание НЬ ниже нормы;
—уменьшение Ш;
—нормальный уровень цветового показателя [в связи с тем, что в крови цир­
кулируют зрелые эритроциты, находившиеся в сосудистом русле (в том
числе в депо) до кровопотери];
—тромбоцитопения (в результате потребления кровяных пластинок в про­
цессе тромбообразования, гемодилюции, а также утраты их при крово­
потере);
—лейкопения (вследствие потери лейкоцитов во время кровотечения
и последующей гемодилюции).
На 4—5-е сутки после кровопотери:
— содержание НЬ пониженное;
— эритропения;
— № снижен;
—гипохромия эритроцитов; цветовой показатель ниже 0,85 (результат
отставания скорости синтеза НЬ от темпа пролиферации эритроидных
клеток);
—ретикулоцитоз, иногда наличие полихроматофильных и оксифильных
эритробластов (как результат высокой регенераторной способности
костного мозга);
—тромбоцитопения;
—лейкопения.
Хронические постгем оррагические анемии
Причина хронических постгеморрагических анемий — длительные, повто­
ряющиеся кровотечения в результате:
—нарушения целостности стенок сосудов (например, при инфильтрации в них
опухолевых клеток, экстрамедуллярном кроветворении, выраженной веноз­
ной гиперемии, язвенных процессах в ЖКТ, коже, слизистых оболочках);
— эндокринопатий (например, при дисгормональной аменорее);
— расстройств в системе гемостаза (например, при нарушении сосудистого,
тромбоцитарного или коагуляционного механизма у пациентов с гемор­
рагическими диатезами).
Патогенез и проявления хронических постгеморрагических анемий обуслов­
лены в основном нарастающим дефицитом железа в организме. Они явля­
ются частным вариантом ЖДА. В связи с этим механизм и проявления
хронических постгеморрагических анемий рассматриваются далее в разделе
«Дизэритропоэтические анемии».
Гемолитические анемии
Гемолитические анемии — большая группа заболеваний, характеризую­
щихся снижением средней продолжительности жизни эршдшнишв (в норме
120 сут) и преобладанием гемолиза эритроцитов над их образованием.
Гемолиз (разрушение эритроцита) может быть:
—внесосудистым (в селезенке, печени или костном мозге);
—внутрисосудистым.
Виды гемолитических анемий
• По степени замещения разрушенных клеток новыми эритроцитами:
— компенсированные;
— некомпенсированные.
• По этиологическому фактору анемии:
— идиопатические (причина не известна);
— симптоматические или вторичные (например, вызванные приемом
ЛС).
• По течению:
— острые;
— подострые;
— хронические.
• По типу дефекта (табл. 22.2).
Таблица 22.2. Причины гемолитических анемий, обусловленные характером дефекта
эритроцита или патогенного воздействия
Наследственные
Мембранопатии эритроци­
тов первичные(например,
наследственные сфероцитоз
и эллиптоцитоз)
Ферментопатии эритроцитов
(например, недостаточность
Г-6-ФД* и/или пируваткиназы)
Гемоглобинопатии (напри­
мер, талассемия; серповидно­
клеточная анемия)
Приобретенные
Иммуная аутоагрессия (например, лекарственный
гемолиз, изо-, ауто-, аллоиммунный гемолиз)
Механическая травма эритроцитов (например,
в результате турбулентности тока крови при арте­
риальной гипертензии, стенозе аорты, искус­
ственных клапанах)
Внутрисосудистые коагулопатии (например, ДВС,
тромбоцитопеническая пурпура)
Инфекции (например, малярия или эндотоксины
при инфекции)
Мембранопатии вторичные (например, при парок­
сизмальной ночной гемоглобинурии)
* Г-6-ФД — глюкозо-6-фосфатдегидрогеназа.
Этиология гемолитических анемий
Гемолитические анемии возникают либо при дефектах самих эритроцитов
(внутриклеточные факторы), либо под воздействием внешних по отношению
к эритроцитам причин (внеклеточные факторы). Обычно внутриклеточные
факторы — наследуемые или врожденные, а внеклеточные — приобретенные.
Внеклеточные причины гемолитических анемий
М икроокружение эритроцитов представлено плазмой крови и эндотели­
ем сосудов. Присутствие в плазме крови ауто- или изоантител, токсичных
веществ или инфекционных агентов вызывает изменения стенки эритроцита,
что приводит к его разрушению. Это наблюдается при:
—изоиммунной гемолитической анемии при эритробластозе плода;
—дефектах эндотелия сосудов — микроангиопатиях (например, при гемо­
литической микроангиопатической анемии). У детей она может проте­
кать остро в виде гемолитико-уремического синдрома;
— гемоглобинурии пароксизмальной Холодовой;
— гемолизе эритроцитов энзимопатического происхождения;
—назначении некоторых ЛС (например, сульфаниламидов, противомаля­
рийных препаратов).
Внутриклеточные причины гемолитических анемий
Внутриклеточные дефекты, которые, как правило, наследуются (исключая
пароксизмальную ночную гемоглобинурию), включают:
—мембранопатии (например, наследуемый сфероцитоз и эллиптоцитоз,
гемоглобинурия пароксизмальная ночная);
—гемоглобинопатии (например, серповидно-клеточная анемия); известно
более 300 заболеваний, обусловленных точечными мутациями генов
глобинов. Дефект молекулы глобина нарушает его полимеризацию.
Изменяется мембрана, форма эритроцита, увеличивается подвержен­
ность гемолизу;
—энзимопатии, например, повыш енная активность аденозиндезаминазы
(генА Ш , 102700, 20я 13.11); недостаточность аденилаткиназы, Г-6-ФД,
гексокиназы, глутатионпероксидазы, глутатионредуктазы, глутатионсинтетазы, глю козо-6-ф осф атизомеразы , дифосфоглицератмутазы,
пируваткиназы, фосфофруктокиназы и др.
Патогенез гемолитических анемий
Общий механизм лизиса эритроцитов (рис. 22.6) заключается в дезорганиза­
ции фосфолипидно-белковой структуры их мембраны.
Рис. 22.6. Изменения в эритроцитах, ведущие к их гемолизу
Масштаб повреждений мембраны эритроцита может колебаться в широком
диапазоне — от микроразрывов до декомпозиции макромолекул и образования
пор. В двух последних случаях развивается каскад перечисленных ниже реакций.
• Повышение проницаемости мембран в клетках эритроидного ряда (от проэритробласта до зрелого эритроцита) для ионов и органических веществ.
• Утрата микро- и макромолекулярных веществ (К +, фосфатов, ферментов
и др.) клетками эритроидного ряда.
• Избыточное поступление в эритроциты Ш +, Са2+, органических соединений
и воды.
• Высвобождение в цитозоль ионов, микро- и макромолекулярных соединений,
ранее находившихся в митохондриях, эндоплазматической сети и других
органоидах.
• Увеличение осмоляльности внутриклеточной жидкости (за счет ионов, мета­
болитов, липидов, углеводов, белков и их соединений).
• Ток избытка жидкости в клетки по градиенту осмотического и онкотического давления.
• Гипергидратация эритроидных клеток, их набухание, утрата дискоидной
формы, округление их (сфероцитоз).
• Разрушение эритроидных клеток. При этом высвобождающийся из эритро­
цитов НЬ трансформируется в билирубин. Он циркулирует в крови, прони­
кает в ткани, а также выводится с экскрементами и мочой. Это провоцирует
развитие гемолитической желтухи со свойственными ей расстройствами
функций физиологических систем организма:
—внутрисосудистый гемолиз — наиболее гидратированные клетки гемолизируются в просвете сосудов;
—внутриклеточный гемолиз — менее гидратированные, но с пониженной
способностью к деформации клетки разрушаются в капиллярах тканей,
синусах селезенки, печени, поглощаются и лизируются макрофагами.
Гемолиз эритроцитов при первичных (наследуемых и врожденных) гемоли­
тических анемиях обусловлен генетическим парциальным или сочетанным
дефектом структуры их мембран (мембранопатии), ферментов (ферментопатии), гемоглобина (гемоглобинопатии).
Мембранопатии характеризуются нарушением белково-липидной структу­
ры и физико-химического состояния мембран в эритроидных клетках.
Причинами мембранопатии являются генетические дефекты мембранных
или околомембранных полипептидов клеток эритроидного ряда.
Патогенез мембранопатии. Для гемолитических анемий, обусловленных
мембранными дефектами, характерен синтез аномальных белков (белковоза­
висимые мембранопатии) либо аномальных липидов (липидзависимые мем­
бранопатии, например, при акантоцитозе).
Примеры гемолитических анемий, развивающихся в результате белковоза­
висимой мембранопатии:
—наследственны е формы сф ероцитоза (болезнь М инковского—
Ш оффара);
— наследуемые эллиптоцитоз, стоматоцитоз, пиропойкилоцитоз, синдром
«Юг-ноль».
Ферментопатии. Гемолитические анемии, обусловленные ферментопатиями, характеризуются нарушением белково-липидной структуры и физико­
химического состояния мембран эритроидных клеток и развиваются при генных
мутациях ряда ферментов.
Гемоглобинопатии. Описано много гемоглобинопатий, сопровождающихся
проявлениями гемолитической анемии (см. далее «Талассемии», а также ста­
тью «Гемоглобинопатии» в приложении «Справочник терминов»).
Проявления гемолитических анемий
Проявления гемолитических анемий разнообразны и в значительной сте­
пени определяются конкретным заболеванием. Наиболее общие проявления
представлены на рис. 22.7.
Рис. 22.7. Основные гематологические проявления гемолитических анемий
Дизэритропоэтические анемии
Дизэритропоэтические анемии характеризуются нарушением пролиферации и/или
дифференцировки эритроидных клеток, а также синтеза гемоглобина в них.
Эти анем ии дифференцирую т в зависимости от их происхождения
(рис. 22.8).
Рис. 22.8. Виды дизэритропоэтических анемий
Гипопластическая и апластическая анемии
Гйпо- и апластические анемии являются следствием повреждения стволовых
клеток, сочетающегося с подавлением функций костного мозга.
По происхождению эти анемии подразделяются на:
—первичные (наследуемые и врожденные, например анемия Фанкони);
—вторичные (приобретенные).
Причины и патогенез гипо- и апластических анемий
Причиной гипо- и апластических анемий являются факторы различной при­
роды:
—физической (например, ионизирующего облучения);
—химической, чаще всего ЛС, например хлорамфеникола (левомицетин*),
фенилбутазона (бутадион*), иммунодепрессантов, мепробамата, хлорпромазина (аминазин*), цитостатиков и др.;
— биологической (главным образом, вирусов, например, вызывающих гепа­
тит, инфекционный мононуклеоз и др., а также антиэритроцитарных АТ
и Т-цитотоксических лимфоцитов).
Так, воздействие физических факторов (например, высоких доз ионизиру­
ющей радиации) обусловливает гипоплазию костного мозга. Выраженность ее
зависит от дозы облучения. В основе гипоплазии кроветворной ткани лежит
необратимое повреждение и гибель стволовых клеток, вплоть до их полного
исчезновения, наблюдающегося при аплазии.
Химические и биологические факторы (например, вирусы и ЛС) тормозят
синтез нуклеиновых кислот и белка в стволовых клетках, нарушают их клеточное
и/или физико-химическое микроокружение; это ведет к расстройствам проли­
ферации, повреждению и гибели стволовых клеток.
Любой из указанных механизмов (или их комбинация) обусловливает нару­
ш ение пролиферации и/или гибель стволовых гемопоэтических клеток, вклю ­
чая эритропоэтические. Это и ведет к гипо- или апластическим анемиям.
Проявления гипо- и апластических анемий
Для гипо- и апластических анемий характерен комплекс изменений в кост­
ном мозге и периферической крови, показанный на рис. 22.9.
Анемия вследствие нарушения синтеза глобиновых
ДНК
Анемии, развивающиеся в результате нарушения синтеза глобиновых ДНК,
как правило, гиперхромные макроцитарные с мегалобластным типом кроветво­
рения.
Мегалобластный эритропоэз возникает вследствие нарушения синтеза Д Н К
в условиях дефицита витамина В ,2 (цианкобаламин) или фолиевой кислоты,
а также при недостаточности метионинсинтетазы и дигидрофолатредуктазы.
При макроцитарной мегалобластной анемии эритроидный росток костного
мозга представлен аномальными эритроидными клетками-мегалобластами.
В эту группу анемий входят пернициозная анемия и другие В12-дефицитные
анемии, а также фолиеводефицитная анемия. Указанные анемии протекают
Рис. 22.9. Основные гематологические проявления гипо и апластических дизэритропоэтических анемий
тяжело и с трудом поддаются лечению. Поэтому мегалобластные анемии ранее
называли пернициозными (витамин В]2-дефицитные анемии, в том числе
анемия Аддисона—Бирмера).
Патогенез мегалобластных анемий см. в приложении «Справочник тер­
минов» в статьях «Анемии витамин В ]2-дефицитные», «Анемия фолиево­
дефицитная», «Витамин В12», «Гиповитаминоз В12», «Кислота фолиевая»,
«Недостаточность фолиевой кислоты».
Гематологические проявления мегалобластных анемий приведены на рис. 22.10.
Рис. 22.10. Основные гематологические проявления мегалобластных анемий
Анемии, развивающиеся при нарушениях
обмена железа
К анемиям, развивающимся при нарушениях обмена железа, относят:
- ЖДА (сидеропенические);
—железорефрактерные (сидероахрестические).
Обмен ж ел еза в организме
Железо участвует в функционировании всех биологических систем орга­
низма. Суточная потребность в железе составляет для мужчин 10 мг, для ж ен­
щин — 18 мг (в период беременности и лактации — 38 и 33 мг соответственно).
Общее количество железа в организме составляет 4—4,5 г. Различают клеточ­
ное, внеклеточное железо и железо запасов (рис. 22.11).
Клеточное железо составляет значительную часть от общего количества желе­
за в организме. Оно участвует во внутреннем обмене железа и входит в состав
гемсодержащих соединений (гемоглобин, миоглобин, другие ферменты, напри­
мер цитохромы, каталазы, пероксидаза), негемовых ферментов (например,
НАДН-дегидрогеназа), металлопротеидов (в частности, аконитаза).
Железо, поступившее
с пищей в течение
суток (1-2 мг)
Потеря железа
с калом
(>0,7 мг/сут)
\
/ Общая
(
потеря
\ (1 мг/сут
\
)
)у
/
Кишечник
Другие потери
Плазма (общее содержание железа >20 мг)
Свободно
Железо, связанное
циркулирующее с трансферрином
железо (>7 мг)
(>4 мг)
Селезенка,
печень, мышцы
Железо
запасов
Железо
других
клеток
Другие
соединения
железа (>9 мг)
Другие органы
Железо
запасов
Клеточное железо
(ферменты,
цитохромы и т.д.)
Рис. 22.11. Схема обмена железа (Ре) в организме здорового мужчины с массой тела 70 кг
Внеклеточное железо. К нему относят свободное железо плазмы и железо­
связывающие сывороточные белки (трансферрин, лактоферрин), участвую­
щие в транспорте железа.
Железо запасов. Оно находится в организме в вице двух белковых соедине­
ний — ферритина и гемосидерина с преимущественным отложением их в пече­
ни, селезенке и мышцах и включается в обмен при недостаточности клеточного
железа.
Ж елезодеф ицитны е анемии
Дефицит железа в организме развивается в тех случаях, когда потери его
превышают 2 мг/сут.
Причинами этого могут быть:
— меньшее поступление железа в организм вследствие общего голодания,
значительного уменьшения в рационе продуктов питания, содержащих
железо, нарушения всасывания железа в Ж К Т (всасывается главным
образом двухвалентное железо, входящее в состав гема. Этот процесс
нарушается при хронических гастритах, энтеритах, резекциях желудка
и особенно тонкой кишки);
—увеличение потерь железа организмом при хронических, повторных кровопотерях (желудочных, кишечных, маточных и др.), а также массиро­
ванных кровоизлияниях;
— возрастающий расход железа организмом при беременности и последу­
ющем вскармливании ребенка (за этот период теряется в общей слож­
ности более 800 мг железа), особенно на фоне еще не проявляющегося
клинического дефицита железа).
Общая и клиническая характеристика ЖДА дана в статье «Анемия желе­
зодефицитная» (см. приложение «Справочник терминов»).
Патогенез железодефицитных анемий
Д ефицит железа в плазме крови и клетках организма обусловливает мень­
шее его содержание в митохондриях эритроидных клеток костного мозга. Это
тормозит синтез гема, соединение его с глобином и, следовательно, образование
НЬ (рис. 22.12).
Рис. 22.12. Основные звенья патогенеза железодефицитных анемий
Одновременно с этим нарушается синтез и других железосодержащих сое­
динений как в эритроидных клетках (каталаза, глутатионпероксидаза), так
и в клетках паренхиматозных органов (цитохромы, миоглобин, пероксидаза,
каталаза и др.).
Недостаток указанных ферментов в эритроцитах влечет за собой снижение
резистентности к повреждающему действию перекисных соединений, повыш ен­
ный их гемолиз и сокращение времени циркуляции в крови.
Проявления железодефицитных анемий
В костном мозге (рис. 22.13):
— сохраняется нормобластический тип кроветворения;
— часто (но не всегда) умеренная гиперплазия клеток красного ростка гемопоэза; увеличено число базофильных и полихроматофильных эритробластов при уменьшении количества оксифильных (признак торможения
эритропоэза);
—уменьшено содержание депонированного в костном мозге железа и числа
сидеробластов (нормобласты с гранулами железа).
В периферической крови:
—уменьшено количество эритроцитов, содержание НЬ (до 30—40 г/л, что обу­
словливает развитие гемической гипоксии) и цветовой показатель (до 0,6
и менее);
—количество ретикулоцитов различно — от нормального до пониженного
(при хроническом течении анемии) или повышенного (на начальных эта­
пах анемии);
—пойкилоцитоз, анизоцитоз (много микроцитов), наличие «теней» эритро­
цитов (в связи со сниженным содержанием в них НЬ);
— понижен уровень железа (Ре2+) в плазме крови (сидеропения) до 1,8—
7,2 мкмоль/л (при норме 12-30 мкмоль/л);
—лейкопения (за счет нейтрофилов);
—количество тромбоцитов обычно в пределах нормы.
В тканях и органах:
—признаки различных видов дистрофий. Они вызваны дефектами струк­
туры, активности железосодержащих ферментов и других соединений
(например, глутатионпероксидаз, каталазы, пероксидаз, цитохромов,
миоглобина). В связи с этим, а также вследствие развития тканевой
гипоксии выявляют мышечную слабость (миастения), шелушение,
трещины кожи и слизистых оболочек, повышенную ломкость ногтей,
выпадение волос, изменения стенки Ж К Т (сопровождающиеся гипотрофическим глосситом, гастритом, энтеритом).
Ж елезореф рактерны е анемии
Железорефрактерные (норфиринодефицитные, сидеробластные, сидероахрестические) анемии развиваются как результат нарушения процессов
включения железа в гем.
Общая и клиническая характеристика железорефрактерных анемий дана
в статье «Анемия сидеробластная» (см. приложение «Справочник терминов»).
По происхождению железорефрактерные анемии подразделяют на:
—первичные (наследственные и идиопатические);
—вторичные (приобретенные).
Первичные железорефрактерные анемии
Проявления первичных железорефрактерных анемий показаны на рис. 22.14.
Приобретенные (вторичные) железорефрактерные анемии
Причины:
—дефицит пиридоксина (витамин В6);
—хронические интоксикации (наиболее часто соединениями свинца, алко­
голем, антимикобактериальным средством изониазид).
Патогенез приобретенных железорефрактерных анемий
При дефиците витамина В6 нарушаются включение железа в молекулу гема
и синтез НЬ. В связи с этим увеличивается содержание железа в плазме крови
и клетках различны х органов.
Глава 22. Типовые формы патологии системы крови
Рис. 22.13. Основные гематоло! ические проявления железодефицитных анемий
Рис. 22.14. Основные гематологические проявления первичных железорефрактерных (порфиринодефицитных) анемий
-Ь
СО
При отравлении свинцом блокируются сульфгидрильные группы ферментов
синтеза протопорфиринов (в частности, сингетазы 8-аминолевулиновой кислоты,
декарбоксилазы уропорфириногена) и как следствие — тема. Нарушение син­
теза тема нередко (особенно при отравлении соединениями свинца) сочетается
со сниженной скоростью образования глобина (преимущественно (3-цепи).
Проявления приобретенных железодефицитных анемий
При свинцовом отравлении, как правило, наблюдаются изменения в систе­
ме крови, а также признаки поражения нервной системы и ЖКТ.
В костном мозге:
—увеличено количество эритробластов и сидеробластов.
В периферической крови:
— эритропения (эритроциты гипохромные, мишеневидные, с базофильной
пункгацией цитоплазмы);
— ретикулоцитоз;
— повышение уровня железа (до 60—80 мкмоль/л).
В клетках разных органов и тканей также обнаруживается железо, т.е. раз­
вивается гемосидероз.
В моче значительно увеличено содержание аминолевулиновой кислоты. Это
один из наиболее характерных признаков свинцового отравления (как след­
ствие блокады свинцом синтетазы 5-аминолевулиновой кислоты).
Поражение нервной системы характеризуется развитием:
— энцефалопатии (проявляющейся головной болью, снижением памяти,
судорогами);
— полиневритов (с расстройством движений и чувствительности);
—парезов.
Повреждение Ж КТ характеризуется:
— снижением аппетита;
— «свинцовой коликой» (схваткообразные сильные боли в животе);
— запором.
Для витамин В6-дефицитной анемии характерны:
— незначительная эритропения;
— выраженная гипохромия эритроцитов;
— анизоцитоз (макроцитоз);
— пойкилоцитоз;
—мишеневидные эритроциты;
—увеличенное содержание железа в сыворотке крови.
Анемии, развивающиеся вследствие нарушения
синтеза глобинов
К анемиям, развивающимся вследствие нарушений синтеза глобинов
(гемоглобинопатии), относится множество заболеваний, в том числе талассемии, болезнь нестабильного НЬ и серповидно-клеточная анемия.
Талассемии
Общая характеристика талассемий и варианты талассемий даны в статье
«Талассемии» (см. приложение «Справочник терминов»).
Патогенез талассемий
Поскольку одна из цепей глобина синтезируется в меньшем количестве
либо совсем отсутствует, нарушается закономерная для нормы количественная
сбалансированность двух цепей глобина (рис. 22.15).
Рис. 22.15. Основные звенья патогенеза талассемий
«Несбалансированная» (т.е. не имеющая пары) цепь агрегирует и выпадает
в осадок в цитозоле эритроидных клеток, в том числе в ретикулоцитах и эри­
троцитах периферической крови (тельца Хайнца).
Ядросодержащие эритроидные клетки, включающие несбалансированные
агрегированные цепи, разрушаются в костном мозге, а ретикулоциты и эри­
троциты, циркулирующие в крови, — в селезенке. В связи с повышенным
разрушением эритроидных клеток в костном мозге и селезенке часто развива­
ется эритропения. Вместе с тем при некоторых формах талассемии (например,
при малой (3-талассемии) выявляется эритроцитоз.
Проявления талассемий представлены на рис. 22.16.
Рис. 22.16. Основные гематологические проявления талассемий
Серповидно-клеточная анемия
Характеристика серповидно-клеточной анемии приведена в статье «Анемия
серповидно-клеточная» в приложении «Справочник терминов».
Лечение дизэритропоэтических анемий
Лечение дизэритропоэтических анемий направлено на:
—устранение или прекращение действия причинных факторов, вызывающих
нарушение пролиферации и дифференцировки эритроидных клеток
(этиотропная терапия);
—разрыв патогенетических звеньев анемических состояний: гипоксии, гемо­
сидероза, нарушений КОС (патогенетическое лечение);
—устранение последствий и неприятных симптомов анемий (симптоматиче­
ская терапия).
Патофизиология системы
лейкоцитов
В крови взрослого здорового человека в условиях покоя до приема пищи
содержится 4—9х109/л лейкоцитов. Большое количество лейкоцитов находит­
ся также за пределами сосудистого русла, в тканях, где они участвуют в реак­
циях иммунного надзора.
Характеристики разных лейкоцитов и лейкоцитопоэзов приведены в ста­
тьях «Нейтрофил», «Эозинофил», «Базофилы», «Лимфоциты», «Моноциты»
и «Гемопоэз» (см. приложение «Справочник терминов»).
Типовые изменения в системе лейкоцитов характеризуются сочетанными
или парциальными отклонениями по ряду параметров (рис. 22.17).
Рис. 22.17. Типовые изменения в системе лейкоцитов
ТИПОВЫЕ ИЗМЕНЕНИЯ КОЛИЧЕСТВА ЛЕЙКОЦИТОВ
В ЕДИНИЦЕ ОБЪЕМА КРОВИ: ЛЕЙКОПЕНИИ
И ЛЕЙКОЦИТОЗЫ
И лейкопении, и лейкоцитозы, как правило, не являются самостоятель­
ными заболеваниями, а относятся к реакциям, развивающимся при раз­
личных болезнях, патологических процессах и состояниях.
Излечение болезни, ликвидация патологического процесса или болезнен­
ного состояния позволяют более или менее быстро нормализовать обшее коли­
чество или численность отдельных форм лейкоцитов, соотношение их зрелых
клеток, устраняют признаки дегенерации. Таким образом, обычно лейкопении
и лейкоцитозы не требуют специального лечения.
Отклонения в численности лейкоцитов периферической крови, а также
качественные изменения в них в определенной степени позволяют судить
о наличии патологического процесса и динамике его течения, т.е. имеют диа­
гностическое значение (в отличие, например, от лейкозов — самостоятельных
заболеваний, имеющих определенные причины, механизмы развития, про­
явления и требующих специального лечения).
Лейкопении
Лейкопении — состояния, характеризующиеся уменьшением количества
лейкоцитов в единице объема крови ниже нормы (обычно менее 4х1в9/л).
Различают лейкопении:
—первичные (врожденные или наследственные);
— вторичные (приобретенные).
Первичные лейкопении
К первичным лейкопениям (в подавляющем большинстве случаев речь идет
о нейтропениях) относятся болезнь Костманна, синдромы «ленивых» лейкоци­
тов, Чедиака—Хигаси, врожденная алейкия, семейные нейтропении, периоди­
ческая наследственная нейтропения, хроническая гранулематозная болезнь.
Вторичные лейкопении
Наиболее частые причины вторичных (приобретенных) лейкопений:
—ионизирующая радиация (вызывающая подавление пролиферации гемопоэтических клеток и/или разрушение лейкоцитов);
—химические вещества (бензол, горчичный газ, инсектициды, обладающие
цитостатическим эффектом);
— некоторые лекарственные средства (ЛС) (к нейтропении или агранулоцитозу могут привести НПВС, цитостатики, противоопухолевые антибио­
тики, антиметаболиты, сульфаниламиды, барбитураты, алкилирующие
вещества и др.);
—аутоагрессивные иммуноглобулины (1§), лимфоциты, фагоциты (образу­
ющиеся при болезнях иммунной аутоагрессии, например при ревмати­
ческих болезнях);
— генерализованные инфекции (брюшной тиф, паратиф, грипп, корь, риккетсиозы, гепатиты).
Механизмы развития лейкопений
• Нарушение и/или угнетение процесса формирования лейкоцитов. Причины:
—генетический дефект лейкопоэтических клеток (например, аномалии
генов, контролирующих созревание лейкоцитов);
— расстройство механизмов регуляции лейкопоэза (например, при гипотиреоидных состояниях, гипокортицизме, снижении уровня лейкотриенов
или чувствительности к ним клеток лейкоцитарного ростка гемопоэза);
— недостаток компонентов, необходимых для лейкопоэза (например,
при значительном дефиците белков, фосфолипидов, аминокислот,
фолиевой кислоты, цианокобаламина).
Расстройство лейкопоэза может касаться всех ростков (например, под вли­
янием высокой дозы радиации) либо преимущественно одного или несколь­
ких из них: например, при длительном приеме мепробамата, хлорохина
(хингамин*), колхицина развивается агранулоцитоз, который характеризуется
значительным уменьшением количества гранулоцитов — нейтрофилов, эозинофилов и базофилов, а также моноцитов.
• Чрезмерное разрушение лейкоцитов в сосудистом русле или органах гемопоэ­
за. Причинами этого могут быть:
— проникающая радиация;
— противолейкоцитарные 1§. Они могут образовываться вследствие мута­
ций в геноме В-лимфоцитов, продуцирующих антитела (АТ) в ответ
на переливание донорской крови или в результате применения ЛС,
которые выступают в качестве гаптенов, например сульфаниламидов,
барбитуратов. Гаптены обусловливают образование АТ, вызывающих
агглютинацию и разрушение лейкоцитов.
• Перераспределение лейкоцитов в сосудистом русле (носит временный харак­
тер). Перераспределительная лейкопения наблюдается при:
— шоковых состояниях;
—тяжелой и длительной мышечной работе (при ней наблюдается ско­
пление лейкоцитов в капиллярах мышц, кишечника, печени, легких
и одновременное уменьшение их количества в других регионах сосуди­
стого русла);
— развитии феномена «краевого стояния» лейкоцитов, характеризующего­
ся адгезией большого количества их к эндотелию стенок микрососудов.
Такая картина нередко наблюдается на раннем этапе воспаления, охваты­
вающего значительную площадь (например, при роже или флегмоне);
— выходе множества лейкоцитов из сосудистого русла в ткани при их мас­
сивном повреждении (например, при перитоните, плеврите, пневмонии,
обширном механическом повреждении мягких тканей).
• Повышенная потеря лейкоцитов организмом. Причины:
— острая или хроническая кровопотеря;
— плазмо- и /и ли лимфоррагии (например, при обширных ожогах, хрони­
ческих гнойных процессах — остеомиелите, эндометрите, перитоните).
• Гемодилюционная лейкопения (встречается сравнительно редко). П ричина—
гиперволемия, развивающаяся в результате:
— трансфузии большого объема плазмы крови или плазмозаменителей
в сосудистое русло;
— реабсорбции жидкости из тканей в микрососуды по градиенту осмотиче­
ского или онкотического давления (например, при гиперальдостеронизме, гипергликемии, гиперальбуминемии).
Проявления лейкопений
Для лейкопений характерно:
—снижение содержания в единице объема крови лейкоцитов всех направ­
лений дифференцировки (лейкопения) или одного из них: лимфоцитов,
моноцитов, нейтрофилов, базофилов или эозинофилов (лимфоцито-,
моноцито-, эозино-, нейтропения соответственно);
—уменьшение количества молодых форм нейтрофилов (палочкоядерных,
метамиелоцитов) на начальных этапах развития лейкопенической реак­
ции, что свидетельствует об угнетении регенерации лейкоцитов в кро­
ветворной ткани. При прекращении действия причинного фактора
наблюдается постепенное увеличение числа молодых форм нейтрофи­
лов (сдвиг лейкоцитарной формулы влево), что является признаком
активации лейкопоэза;
— признаки дегенерации лейкоцитов. Дегенеративные изменения проявляют­
ся различными изменениями контура лейкоцитов (пойкилоцитоз), в част­
ности шиловидными выростами цитолеммы, наличием клеток разного
размера (анизоцитоз), сморщиванием или набуханием клеток, появлением
вакуолей, токсогенной зернистости и включений в цитоплазме, гиперсег­
ментацией или пикнозом ядер и их разрушением (кариорексис).
М ногие дегенеративные формы лейкоцитов при лейкопении сочетаются
иногда с уменьшением числа сегментоядерных лейкоцитов и умеренным уве­
личением содержания палочкоядерных и даже метамиелоцитов (эту картину
крови называют дегенеративным ядерным сдвигом влево). Если увеличивает­
ся количество сегментоядерных лейкоцитов с признаками дегенеративных
изменений в них без увеличения числа палочкоядерных клеток, то говорят
о дегенеративном ядерном сдвиге вправо.
Значение лейкопении
При выраженной лейкопении снижается резистентность организма, главным
образом противоинфекционная, а также противоопухолевая. Это обусловлено
тем, что лейкоциты участвуют в реализации гуморального и клеточного зве­
ньев иммунитета, а также фагоцитарной реакции. При лейкопениях часто
наблюдается инфицирование организма (с развитием ринитов, бронхитов,
плевритов, воспаления легких, конъюнктивитов и других форм инф екцион­
ных процессов), возможно также развитие новообразований.
Лейкоцитозы
Лейкоцитозы — состояния, характеризующиеся увеличением числа лейко­
цитов в единице объема крови выше нормы (более 9 х109/л).
Причины лейкоцитозов
По происхождению лейкоцитозы подразделяются на:
— эндогенные;
— экзогенные.
И те и другие могут быть инфекционными и неинфекционными.
Причинами лейкоцитозов наиболее часто бывают:
—физические факторы (например, периодическое воздействие на орга­
низм ионизирующей радиации в малых дозах);
—химические агенты (например, воздействие умеренных доз алкоголя,
гипоксии; прием ЛС, стимулирующих пролиферацию лейкопоэтиче­
ских клеток);
— биологические факторы; их большинство (например, продукты ж изне­
деятельности живых и компоненты погибших вирусов, бактерий, риккетсий; иммунные комплексы Аг—АТ; повышенный уровень отдельных
БАВ: лейкопоэтинов, гистамина, продуктов клеточного распада).
Механизмы развития лейкоцитозов
Развитие лейкоцитозов является следствием:
— активации нормального лейкопоэза и выхода лейкоцитов из гемопоэтической ткани в периферическую кровь;
—перераспределения лейкоцитов в сосудистом русле;
—опухолевой активации лейкопоэза при лейкозах и гематосаркомах;
—гемоконцентрации.
Активация нормального лейкопоэза. Причины:
—повышение уровня и/или активности гуморальных стимуляторов лейкопоэ­
за (например, лейкоцитарных факторов роста, колониестимулирующих
факторов);
—снижение содержания и/или активности ингибиторов пролиферации лей­
копоэтических клеток и индукторов их созревания. В результате увеличи­
вается количество пролиферирующих клеток лейкопоэтической ткани,
что сочетается, как правило, с дифференцировкой их в зрелые лейкоци­
ты. И менно это наблюдается, например, при воспалительных реакциях
и развитии аллергических процессов. Лейкоцитозы с таким механизмом
развития обозначают как регенераторные (истинные, абсолютные).
Перераспределение лейкоцитов в сосудистом русле. Этот феномен характе­
ризуется:
—скоплением избытка зрелых (!) лейкоцитов в каком-либо сосудистом
регионе организма;
—отсутствием признаков гиперплазии лейкопоэтической ткани;
—сохранением общего числа лейкоцитов в крови в пределах нормы.
Причинами феномена перераспределения лейкоцитов могут быть:
—значительная физическая нагрузка («миогенный лейкоцитоз»);
—шоковые состояния (при них увеличивается количество лейкоцитов в крови
микрососудов, снабжающих легкие, печень, стенки кишечника).
Перераспределительный лейкоцитоз носит временный характер и не сопро­
вождается увеличением числа молодых форм лейкоцитов. И менно поэтому
лейкоцитозы с таким механизмом развития называются ложными или отно­
сительными.
Гиперпродукция лейкоцитов при опухолевом поражении гемопоэтической
ткани (гемобластозы). Причины:
—увеличение общего числа лейкоцитов за счет активации пролиферации лейкозных (опухолевых) клеток;
—стимуляция деления и созревания нормальных лейкоцитов (как след­
ствие появления в организме чужеродных опухолевых антигенов).
Образующиеся в ответ на это в избытке нормальные лейкоциты осу­
ществляют иммунные реакции организма.
Гемоконцентрация. Причина — гиповолемия различного происхождения
(например, в результате повторной рвоты, диареи, полиурии). При общем
нормальном количестве лейкоцитов содержание их в единице объема крови
увеличено. Важно, что одновременно повышается в крови количество и дру­
гих форменных элементов крови (рис. 22.18).
Рис. 22.18. Основные механизмы развития лейкоцитозов
Проявления лейкоцитозов
Увеличение количества всех форм лейкоцитов или отдельных их видов
(лимфоциты, моноциты, разные гранулоциты) в значительной мере опреде­
ляется характером причинного фактора:
—при аллергических реакциях, как правило, происходит преимущественное
увеличение в крови числа эозинофилов (аллерген обусловливает высво­
бождение из лимфоцитов стимуляторов эозинофильного лейкопоэза:
ИЛ-5, ИЛ-17, фактора хемотаксиса эозинофилов ЕСР, эотаксина);
—при инфекциях, вызванных бактериями (в частности, стрептококками,
стафилококками), стимулируется миелопоэз и выброс в кровь гранулоцитов, в основном нейтрофилов;
—при внедрении в организм многих вирусов (например, возбудителей
коклюш а, гепатита) и некоторых микроорганизмов (например, воз­
будителей туберкулеза, сифилиса, бруцеллеза) происходит преиму­
щ ественная стимуляция лим ф оп оэза и увеличивается количество
лимфоцитов в периферической крови. Некоторые вирусы, бактерии
и простейш ие, вызывающие инфекции (например, инф екционны й
мононуклеоз, краснуху, бруцеллез, малярию), активизирую т также
моноцитопоэз и мобилизацию моноцитов из костного мозга в кровь
с развитием моноцитоза.
Изменения лейкоцитарной формулы нейтрофилов
при лейкоцитозах
Истинные лейкоцитозы (регенераторные, абсолютные), развивающ иеся
за счет усиления пролиферации клеток миелоцитарного ряда, сопровождаются
изменениями лейкоцитарной формулы. Эти изменения обусловлены увеличени­
ем или уменьшением в периферической крови количества молодых форм мие-
лоцитарных клеток (в основном нейтрофилов) и появлением таких их форм,
которые в норме в периферической крови отсутствуют. В таком случае говорят
об изменении соотношения зрелых и незрелых форм лейкоцитов — о ядерном
сдвиге гранулоцитов влево или вправо.
Применение указанных выше терминов («влево», «вправо») связано с рас­
положением названий молодых форм нейтрофилов (палочкоядерные, мета­
миелоциты, миелоциты, промиелоциты) в левой части лабораторного бланка,
а зрелых — в их правой части.
Ядерные сдвиги лейкоцитарной формулы нейтрофилов
Поскольку при микроскопии мазка крови основным критерием для иден­
тификации разных форм зрелости зернистых лейкоцитов является характер
ядра (форма, размер, интенсивность окраски), сдвиги лейкоцитарной форму­
лы обозначают как «ядерные» (рис. 22.19).
Рис. 22.19. Виды ядерных сдвигов нейтрофилов в лейкоцитарной формуле
Сдвиг влево характеризуется увеличением количества молодых и незре­
лых форм нейтрофилов.
Сдвиг вправо проявляется повышением числа сегментоядерных форм
нейтрофилов.
Сдвиг лейкоцитарной формулы нейтрофилов вправо
Такой сдвиг нередко сочетается с более частыми признаками дегенерации
лейкоцитов и меньшим содержанием палочкоядерных нейтрофилов.
Сдвиг лейкоцитарной формулы нейтрофилов влево
Сдвиги лейкоцитарной формулы нейтрофилов влево определяются появ­
лением незрелых форм нейтрофилов. Различают гипорегенераторный, реге­
нераторный, гиперрегенераторный и регенераторно-дегенераторный типы
сдвига влево.
• Гипорегенераторный ядерный сдвиг нейтрофилов влево. О нем говорят
при увеличении содержания палочкоядерных нейтрофилов выше нормы
(более 6%) и умеренном лейкоцитозе (обычно до 10—11хЮ9/л).
• Регенераторный ядерный сдвиг. Он характеризуется превышением нормы
процентного содержания палочкоядерных нейтрофилов, появлением
в периферической крови метамиелоцитов, лейкоцитозом до 13—18х 109/л.
• Гиперрегенераторный сдвиг лейкоцитарной формулы нейтрофилов. П рояв­
ляется значительным увеличением содержания палочкоядерных нейтро-
филов, наличием в периферической крови большого количества метамие­
лоцитов и появлением миелоцитов, увеличением общего числа лейкоцитов
до 20—25 х 109/ л . Вместе с тем общее количество лейкоцитов может быть
нормальным или даже сниженным. В отдельных случаях последнее наблю­
дается после длительного периода значительного лейкоцитоза и обуслов­
лено истощением миелоидного ростка гемопоэтической ткани.
• Регенераторно-дегенераторный ядерный сдвиг. Наблюдается при некоторых
инфекционных заболеваниях, хронических гнойных процессах, протека­
ющих со значительной интоксикацией. Характеризуется более или менее
выраженным увеличением содержания палочкоядерных нейтрофилов,
метамиелоцитов и миелоцитов, уменьшением количества сегментоядер­
ных нейтрофилов (как правило), признаками дегенеративных изменений
цитолеммы, цитоплазмы и ядра, увеличением общего числа лейкоцитов.
Индекс ядерного сдвига нейтрофилов
Указанные выше изменения в соотношении зрелых и незрелых форм ней­
трофилов можно оценить количественно, рассчитав индекс ядерного сдвига
нейтрофилов. Он отражает отношение процентного содержания суммы всех
молодых форм нейтрофилов (палочкоядерные, метамиелоциты, миелоциты,
промиелоциты) к их зрелым формам.
У здоровых взрослых людей индекс ядерного сдвига нейтрофилов колеб­
лется в диапазоне от 0,05 до 0,10. Увеличение его свидетельствует о ядерном
сдвиге нейтрофилов влево, уменьшение — о сдвиге вправо.
Перераспределительные и гемоконцентрационные (ложные) лейкоцитозы
не сопровождаются изменением лейкоцитарной формулы нейтрофилов.
При значительных лейкоцитозах в костном мозге и лимфатических узлах
отмечаются признаки гиперплазии лимфопоэтической ткани в виде увеличения
размеров лимфоидных фолликулов и их зародышевых центров.
Виды и значение лейкоцитозов
Дифференцируют физиологические и патологические виды лейкоцитозов
(рис. 22.20).
Ф изиологические лейкоцитозы
К ним относят большинство лейкоцитозов. Они характеризуются адап­
тивным характером и адекватностью ответа тем факторам, которые их вызыва­
ют. К физиологическим лейкоцитозам относят функциональные и защ итно­
приспособительные .
Функциональный лейкоцитоз развивается при выполнении организмом
определенной функции (например, лейкоцитоз наблюдается при беремен­
ности; в крови сосудов киш ечника после приема пищи; в крови, оттекающей
от мышц после длительной физической работы).
Защитно-приспособительный лейкоцитоз выявляется при воспалитель­
ных процессах, повреждении клеток и тканей (например, после инфарктов
или инсультов, травмы мягких тканей), стресс-реакции.
В названных и других подобных случаях лейкоцитоз сопровождается
активацией функций лейкоцитов, в том числе фагоцитарной — одной из важ­
нейших среди них. Благодаря этому повыш ается резистентность организма
к инфекционным и неинфекционным патогенным воздействиям.
Патологические лейкоцитозы
Наблюдаются при гемобластозах, особенно при лейкозах. Такая разновид­
ность лейкоцитоза, развивающаяся за счет увеличенного количества лейко­
цитов опухолевой природы, не имеет адаптивного значения для организма.
Гемобластозные (т.е. опухолевые) лейкоциты характеризуются нарушением
функциональной активности: снижена способность синтезировать и высво­
бождать цитокины, низка фагоцитарная активность. В связи с этим у паци­
ентов с лейкозами реакции иммунитета менее эффективны, нередко развива­
ются аллергические реакции и болезни иммунной аутоагрессии.
Рис. 22.20. Виды лейкоцитозов по их биологическому значению
Типовые изменения лейкоцитарной формулы
Лейкоцитарная формула — численное описание соотношения различных видов
циркулирующих в периферической крови лейкоцитов.
И зменения лейкоцитарной формулы являются следствием увеличения
или уменьшения содержания отдельных видов лейкоцитов и в связи с этим —
изменивш егося соотношения между ними.
Превышение нормы числа отдельных видов лейкоцитов обозначают терми­
нами «нейтрофилия» («нейтрофилез»), «базофилия», «эозинофилия» («эозинофилез»), «лимфоцитоз», «моноцитоз».
Уменьшившееся по сравнению с нормой число отдельных разновидностей
лейкоцитов называют нейтропенией, эозинопенией, лимфопенией (лимфоцитопения), моноцитопенией.
Агранулоцитоз — отсутствие или значительное снижение абсолютного числа
всех видов зернистых лейкоцитов (нейтрофилов, эозинофилов и базофилов). Это
состояние сочетается, как правило, с лейкопенией.
Термин «базопения» не употребляют, так как базофилы и в норме могут
отсутствовать в периферической крови.
Относительные и абсолютные изменения в лейкоцитарной формуле
При изменениях относительного (процентного) содержания того или иного
вида лейкоцитов в лейкоцитарной формуле говорят либо об относительной
нейтропении, эозинопении, лимфопении, моноцитопении (если уменьшается
процентное содержание лейкоцитов соответствующего вида), либо об относи­
тельной нейтпофилии эозинофилии, относительном моноцитозе, лимфоцитозе (если увеличивается их относительное содержание).
Изменения абсолютного содержания лейкоцитов в единице объема крови
называют абсолютной нейтропенией, эозинопенией, лимфопенией, моноцитопенией (если уменьшается их абсолютное число в единице объема крови)
или абсолютной нейтрофилией, эозинофилией, абсолютным моноцитозом
или лимфоцитозом (если возрастает число соответствующих разновидностей
лейкоцитов). Это обусловлено тем, что именно абсолютные величины отра­
жают истинное содержание тех или иных видов лейкоцитов в крови, а отно­
сительные — характеризуют только соотношение различных клеток между
собой в единице объема крови.
Характеризуя изменения в составе лейкоцитов, необходимо оценивать
как относительное, так и (обязательно!) абсолютное их содержание.
Во многих случаях направленность изменений совпадает. Часто встречает­
ся, например, относительная и абсолютная нейтрофилия или нейтропения.
Отклонение относительного (процентного) содержания клеток в едини­
це объема крови не всегда отражает изменение их истинного, абсолютного
количества. Так, относительная нейтрофилия может сочетаться с абсолютной
нейтропенией (подобная ситуация возникает, если относительная нейтро­
филия наблюдается в условиях значительной лейкопении: например, содер­
жание нейтрофилов равно 80%, а общее число лейкоцитов составляет лиш ь
1,0хЮ9/л).
Чтобы определить абсолютное число того или иного вида лейкоцитов
в крови, необходимо рассчитать эту величину исходя из известного общего
количества лейкоцитов и процентного содержания соответствующих клеток
(в приведенном примере 80% от 1,0хЮ9/л составит 0,8хЮ9/л . Это более
чем в два раза меньше 2,Ох 109/л — нижней границы нормального абсолютного
содержания нейтрофилов).
Сдвиги лейкоцитарной формулы нейтрофилов
Оценивая сдвиги лейкоцитарной формулы, исходят из того, что в пери­
ферической крови могут появляться нейтрофильные лейкоциты разной
степени зрелости (метамиелоциты, миелоциты, промиелоциты, миелобласты). При этом определяют (см. выше «Индекс ядерного сдвига») наличие
и степень изменения в соотношении зрелых и молодых форм указанных
гранулоцитов. Изменения обозначают как сдвиг лейкоцитарной формулы
нейтрофилов вправо или влево.
Значение сдвигов лейкоцитарной формулы нейтрофилов
Анализ лейкоцитарной формулы (выявление изменений абсолютного
содержания нейтрофилов, эозинофилов и других лейкоцитов, оценка направ­
ленности и выраженности нейтрофильного сдвига) позволяет определить
наличие и вид лейкоцитоза или лейкопении по клеточному составу, степень
сдвигов в содержании и соотношении отдельных форм лейкоцитов, возмож­
ный механизм их возникновения. Так, увеличение общего числа лейкоцитов
в сочетании с абсолютной нейтрофилией свидетельствует о регенераторном
(истинном) нейтрофильном лейкоцитозе. Если повышение общего числа лей­
коцитов сопровождается абсолютной нейтро- и эозинофилией, имеет место
регенераторный смешанный (нейтрофильно-эозинофильный) лейкоцитоз.
Уменьшение общего содержания лейкоцитов в сочетании с абсолютной лимфопенией — признак истинной лимфоцитарной лейкопении и т.д.
Наличие выраженного ядерного сдвига нейтрофилов влево при нейтрофильном лейкоцитозе обычно свидетельствует об истинной (регенераторной)
природе этого лейкоцитоза, а отсутствие такого сдвига чаще наблюдается
при перераспределительном механизме развития нейтрофильного лейкоцито­
за или при нейтрофильной лейкопении.
Патофизиология тромбоцитов
Подробная характеристика тромбоцитов и тромбоцитопоэза приведена
в статьях «Тромбоциты» и «Гемопоэз» (см. приложение «Справочник тер­
минов»).
Изменения в системе тромбоцитов, как правило, сопровождаются расстрой­
ством жизнедеятельности организма в целом и заключаются в том, что их коли­
чество в единице объема крови превышает норму (тромбоцитозы), либо
их количество в единице объема крови падает ниже нормального уровня (тромбоцитопении), либо изменяются функциональные свойства пластинок (тромбоцитопатии), либо, наконец, наблюдается сочетание указанных отклонений.
ТРОМБОЦИТОЗЫ
Тромбоцитозы — состояния, характеризующиеся увеличением количества
тромбоцитов в единице объема крови выше нормы (320—340х109/л).
Виды тромбоцитозов
По механизму развития различают тромбоцитозы:
— абсолютные;
— относительные:
о перераспределительные;
о гемоконцентрационные.
Абсолютные тромбоцитозы
Абсолютные (истинные, пролиферативные) тромбоцитозы характеризуются
возрастанием количества тромбоцитов в крови в результате их повышенного
образования.
Причины:
— генные дефекты. Классический пример — миелопролиферативный иди-
опатический тромбоцитоз;
— увеличение концентрации и/или активности стимуляторов тромбоцитопоэ­
за — тромбоспондина, тромбопоэтина, РАТ, ИЛ-3, ИЛ-6, ИЛ-11;
— опухолевая трансформация мегакариобластов под влиянием канцероге­
нов с последующей интенсификацией тромбоцитопоэза при гемобла-
стозах. Это наблюдается, например, при мегакариобластных лейкозах.
При этом возможно значительное (в 10—15 раз превышающее нормаль­
ный уровень) и длительное увеличение числа тромбоцитов в перифери­
ческой крови.
Относительные тромбоцитозы
Относительные (ложные, непролиферативные) тромбоцитозы не сопровожда­
ются увеличением общего количества тромбоцитов в крови.
Причины:
- перераспределение тромбоцитов в различных регионах сосудистого русла.
Так, количество тромбоцитов увеличивается в участках микрососудов
с поврежденными стенками (например, при васкулитах), в первые часы
после острой кровопотери, длительного стресса, ожогов, травмы (за счет
выброса крови из депо и выхода из костного мозга);
— гемоконцентрация: увеличение относительной массы тромбоцитов при неиз­
менном или сниженном объеме плазмы крови. Это может наблюдаться
в результате плазморрагии (например, при обширных ожогах) или при зна­
чительной потере жидкости (например, у пациентов с длительной диаре­
ей, рвотой, при продолжительном интенсивном потоотделении).
Значение тромбоцитозов
Адаптивное значение тромбоцитозов заключается в ускоренном образовании
тромбоцитарного сгустка, в дальнейшем тромба (например, при нарушении
целостности стенки сосуда) и в поддержании оптимального метаболизма
в клетках эндотелия и их целостности за счет выделения при контакте с ними
ангиогенных факторов.
Патогенное значение тромбоцитозов характеризуется избыточной актива­
цией коагуляции белков крови и процесса тромбообразования с нарушением
микроциркуляции в тканях (например, при тромбоцитозе у пациентов с мегакариобластным лейкозом).
ТРОМБОЦИТОПЕНИИ
Тромбоцитопении — состояния, характеризующиеся уменьшением количе­
ства тромбоцитов в единице объема крови ниже нормы, как правило, менее
180—150х1О9/л . К тромбоцитопениям относятся также самостоятельные
заболевания и некоторые синдромы, сопутствующие другим болезням.
Причины тромбоцитопений
Тромбоцитопении могут вызываться различными факторами физической,
химической и биологической природы (см. раздел «Этиология и патогенез»
в статье «Тромбоцитопении» приложения «Справочник терминов»).
Механизм развития тромбоцитопений
Механизм развития тромбоцитопений заключается в реализации одного
или нескольких из следующих процессов (рис. 22.21).
• Подавление тромбоцитопоэза. Этим обусловлено развитие абсолютной
гипорегенераторной тромбоцитопении. Такая картина может наблюдаться
при гемобластозах; метастазах новообразований в костный мозг, лучевой
болезни, применении некоторых ЛС (например, тиазидных диурети­
ков или химиотерапевтических препаратов), избирательно подавляющих
созревание мегакариоцитов; при дефиците витамина В12 или фолиевой
кислоты, врожденном дефиците мегакариоцитарных колониеобразующих
единиц в костном мозге (вследствие этого развивается амегакариоцитарная тромбоцитопения).
• Повышенное разрушение тромбоцитов (см. раздел «Этиология и патогенез»
в статье «Тромбоцитопении» приложения «Справочник терминов»).
• Массированное «потребление» тромбоцитов. Выявляется при генерализо­
ванном тромбообразовании (например, при ДВС-синдроме на этапе обра­
зования большого количества тромбов).
• Избыточное депонирование тромбоцитов в селезенке. Этот синдром обо­
значают термином «гиперспленизм». В норме в селезенке содержится
около 30% всего пула тромбоцитов. Увеличение размеров селезенки
(спленомегалия) обусловливает депонирование значительного количества
тромбоцитов с исключением их из системы гемостаза. При значительном
увеличении селезенки возможно депонирование 90% всего пула тромбо­
цитов. Оставшиеся в кровотоке тромбоциты имеют нормальную про­
должительность циркуляции. Гиперспленизм характеризуется умеренной
тромбоцитопенией, нормальным количеством мегакариоцитов в костном
мозге и значительным увеличением селезенки.
Рис. 22.21. Основные механизмы развития тромбоцитопений
Проявления тромбоцитопений
Костный мозг:
- гиперплазия костного мозга; характеризуется увеличением в нем количе­
ства мегакариобластов и мегакариоцитов. Наблюдается при повышен­
ном разрушении или генерализованном «потреблении» тромбоцитов;
— гипоплазия костного мозга; выявляется у пациентов с гемобластозами
(лейкозами), лучевой болезнью, метастазами опухолей (не относящихся
к гемобластозам) в костный мозг;
— снижение содержания гликогена и активности ряда ферментов (например,
лактатдегидрогеназы, Г-6-ФД) в мегакариобластах и мегакариоцитах,
что уменьшает продолжительность жизни тромбоцитов.
Периферическая кровь:
— уменьшение количества тромбоцитов и увеличение их размеров, обычно
при нормальном количестве эритроцитов, НЬ и лейкоцитов. В условиях
выраженного геморрагического синдрома возможно развитие анемии.
Система гемостаза
Проявления
на рис. 22.22.
тромбоцитопений
в системе
гемостаза
представлены
Рис. 22.22. Изменения в системе гемостаза при тромбоцитопениях
Лечение больных тромбоцитопениями
Этиотропный принцип терапии при тромбоцитопениях
Этот принцип лечения предусматривает прекращение (уменьшение степе­
ни) патогенного действия причины, вызывающей тромбоцитопению.
С этой целью:
—выполняют спленэктомию;
—удаляют гемангиомы;
—обеспечивают защиту от радиоактивного излучения;
—заменяют ЛС, вызывающие тромбоцитопению, на препараты, которые
не оказывают тромбоцитолитического действия;
—предупреждают попадание в организм веществ, обусловливающих тром­
боцитопению (этанол, соединения золота и др.);
—инактивируют и элиминируют противотромбоцитарные 1§ и др.
Патогенетический принцип лечения при тромбоцитопениях
Для уменьшения степени «потребления» и/или разрушения тромбоци­
тов, активации тромбоцитопоэза, нормализации содержания и активности
в крови про- и антиагрегантов, факторов свертывающей, противосверты­
вающей и фибринолитической систем проводят трансфузию тромбоцитов,
пересадку костного мозга, используют лимфо- и/или плазмаферез (удаление
из крови антитромбоцитарных АТ и лимфоцитов), а также иммунодепрессан­
ты; антикоагулянты, антиагреганты.
Симптоматическое лечение при тромбоцитопении
Для нормализации функций органов и их систем, нарушенных вследствие
тромбоцитопении, вливают кровь и/или тромбоцитарную массу, а также
устраняют постгеморрагическое состояние.
ТРОМБОЦИТОПАТИИ
Тромбоцитопатии — состояния, характеризующиеся нарушением одного
или нескольких свойств тромбоцитов (адгезивного, агрегационного, коа­
гуляционного) и, как правило, расстройствами гемостаза.
Тромбоцитопатиям (в отличие от тромбоцитопений) присущи стабильные,
длительно сохраняющиеся функциональные, биохимические и морфологи­
ческие изменения в тромбоцитах. Они наблюдаются даже при нормальном
количестве тромбоцитов и не исчезают при устранении тромбоцитопении
(если она имелась).
Виды тромбоцитопатий
Тромбоцитопатии подразделяют на:
—первичные (наследственные и врожденные);
—вторичные (приобретенные).
Первичные тромбоцитопатии
Наблюдаются при генных дефектах. Примеры — болезнь фон Виллебранда,
тромбастения Гланцманна, недостаточность тромбоксан-А-синтетазы.
Вторичные тромбоцитопатии
Развиваются при воздействии химических и биологических факторов.
• Химические факторы:
— избыток продуктов обмена веществ, в норме выводящихся почками; счи­
тают, что они (возможно, креатинин) деполимеризуют крупномолеку­
лярные полимеры фактора VIII;
—некоторые ЛС (подробнее см. в разделе «Этиология» статьи «Тром­
боцитопатии» в приложении «Справочник терминов»);
— гиповитаминозы (дефицит аскорбиновой кислоты, цианокобаламина).
• Биологические факторы:
—вещества, образующиеся в опухолевых клетках. Они нарушают деление
и созревание мегакариоцитов. Это наблюдается при различных формах
лейкозов или метастазах солидных опухолей в кроветворную ткань;
—продукты деградации фибриногена и фибрина (например, при ДВСсиндроме);
— повышенное содержание в плазме крови нормальных и аномальных белков,
например, при болезни Вальденстрема и миеломной болезни;
— повышенная концентрация в плазме крови факторов свертывающей систе­
мы, например, при переливании больших доз крови, плазмы, концен­
тратов прокоагулянтов.
Патогенез тромбоцитопатий
В основе развития как первичных, так и вторичных тромбоцитопатий
лежит расстройство следующих процессов (рис. 22.23).
• Нарушение контактной активности тромбоцитов. В большинстве случа­
ев этот механизм развития тромбоцитопатий включает одно, а иногда
несколько из указанных ниже звеньев:
— расстройства синтеза и/или накопления в гранулах тромбоцитов их содер­
жимого. Эти нарушения ведут к расстройствам гемостаза и состояния
эндотелия стенок сосудов;
— нарушение дегрануляции при взаимодействии тромбоцитов с агрегиру­
ющими факторами: АДФ, катехоламинами, тромбоксаном А2, коллаге­
ном и др. Эти расстройства, как и нарушения синтеза и/или накопления
в гранулах их компонентов, снижают контактную (адгезивную и агрегационную), а также прокоагулянтную активность тромбоцитов (способ­
ность инициировать процесс тромбообразования);
— аномалии физико-химических свойств и/или химического состава и струк­
туры мембран тромбоцитов. Чаще наблюдаются дефицит гликопротеинов,
нарушения структуры и соотношения различных фракций мембран­
ных фосфолипидов. Эти изменения также обусловливают нарушения
адгезивно-агрегационной активности тромбоцитов.
• Нарушения прокоагулянтной активности тромбоцитов. Этот механизм вклю­
чает:
— снижение синтеза, содержания и/или активности фосфолипидного фак­
тора 3 тромбоцитов (фактора коагуляции белков крови). Этот фактор
при совместном действии с другими прокоагулянтами обусловливает
переход протромбина в тромбин;
— нарушение высвобождения тромбоцитарного фактора 3 из тромбоцитов,
что обусловлено наследуемыми, врожденными или приобретенными
мембранопатиями, аномалиями канальцев и элементов цитоскелета
тромбоцитов. Это препятствует взаимодействию фактора 3 тромбоцитов
с плазменными факторами гемокоагуляции на поверхности тромбоци­
тов. Указанные аномалии приводят к нарушениям процесса свертыва­
ния белков крови и тромбообразования.
• Аномалии механизмов и контактной, и прокоагулянтной активности тромбо­
цитов одновременно. При синдроме Вискотта—Олдрича, например, нару­
шаются образование и хранение компонентов плотных гранул различного
типа тромбоцитов, а также освобождается их содержимое. Эти изменения
сопровождаются расстройством адгезивной, агрегационной и прокоагу­
лянтной активности тромбоцитов.
Рис. 22.23. Основные звенья патогенеза тромбоцитопатий
Парциальная или сочетанная реализация указанных выше механизмов обу­
словливает либо преимущественное нарушение контактной активности тром­
боцитов (их агрегация и/или адгезия), либо преимущественные расстройства
их прокоагулянтных свойств.
Проявления тромбоцитопатий
• Геморрагический синдром. Он проявляется внутренними и внешними кро­
вотечениями, а также кровоизлияниями в различные органы, ткани, кожу,
слизистые оболочки.
• Расстройства микрогемоциркуляции (изменения объема и скорости крово­
тока в сосудах микроциркуляторного русла, турбулентный его характер
и др.). Они нередко ведут к нарушениям обмена веществ в тканях (в связи
с развитием капилляротрофической недостаточности), различным дистро­
фиям, эрозиям и изъязвлениям.
• Значительные изменения функциональных свойств тромбоцитов (адгезивно­
го, агрегационного, прокоагуляционного).
• Дефекты гранул тромбоцитов (отсутствие или уменьшение их числа), напри­
мер, при синдроме «серых тромбоцитов», затрудненное высвобождение
их содержимого.
• Отклонения от нормы размера и формы мегакариоцитов и тромбоцитов.
Лечение при тромбоцитопатиях
Лечение больных с тромбоцитопатиями представляет сложную задачу
и у многих пациентов (особенно с наследственными и врожденными форма­
ми) проводится в течение всей жизни.
Этиотропный принцип лечения при тромбоцитопатиях
Направлен на прекращение действия (защита от воздействия) факторов
физического, химического, биологического характера, на лечение болезней,
патологических процессов и состояний, вызывающих тромбоцитопатию.
Патогенетический принцип терапии при тромбоцитопатиях
Чтобы предотвратить нарушения адгезивной, агрегационной и прокоа­
гулянтной активности тромбоцитов (уменьшить их степень), применяют
проагреганты, прокоагулянты и/или антифибринолитические препараты
(е-аминокапроновая кислота, парааминометилбензойная кислота); использу­
ют вещества, стимулирующие «реакцию высвобождения» (АТФ, магния суль­
фат, магния тиосульфат); переливают кровь, тромбоцитарную массу, белковые
препараты крови (фибриноген, тромбин и др.).
Симптоматический принцип лечения при тромбоцитопатиях
Для нормализации функций органов и тканей, нарушенных вследствие рас­
стройств микрогемоциркуляции, кровотечений и кровоизлияний при тромбо­
цитопатиях вводят растворы, нормализующие реологические свойства крови
(плазмозаменители, плазма), максимально быстро останавливают кровотече­
ние, устраняют постгеморрагические состояния.
НАРУШЕНИЯ СИСТЕМЫ ГЕМОСТАЗА
Система гемостаза — комплекс факторов и механизмов, обеспечивающих
оптимальное агрегатное состояние форменных элементов крови (рис. 22.24).
В узком (прикладном) смысле термин «гемостаз» (от греч. к а т а — кровь,
5(а т — остановка) применяют, чтобы обозначить собственно процесс оста­
новки кровотечения.
Система гемостаза включает факторы и механизмы трех категорий:
—обеспечивающие коагуляцию белков крови и тромбообразование —
свертывающая система;
—обусловливающие торможение или блокаду коагуляции белков плазмы
и процесс тромбообразования — противосвертывающая система;
— реализующие процессы лизиса фибрина — фибринолитическая система.
Биологическая роль системы гемостаза состоит в:
— обеспечении оптимальных реологических (агрегационных) свойств крови;
— реализации процесса коагуляции белков крови и тромбообразования;
— адгезии, агрегации и активации форменных элементов крови с образовани­
ем тромба при повреждении стенок сосудов или сердца. Это предотвращает
или уменьшает потерю крови организмом.
Типовые формы патологии системы гемостаза
Многочисленные нарушения системы гемостаза подразделяются на три
группы:
1) усиление свертываемости белков крови и тромбообразования — гипер­
коагуляция и развитие тромботического синдрома;
2) снижение свертываемости белков крови и тромбообразования — гипо­
коагуляция и развитие геморрагических синдромов;
3) фазное нарушение состояния системы гемостаза — фаза гиперкоагуляции,
сопровождаясь интенсивным потреблением прокоагулянтов, переходит
в фазу гипокоагуляции; развиваются коагулопатия потребления и тромбо­
геморрагические синдромы.
Фибрин
Рис. 22.24. Гемокоагуляционный каскад. Активация фактора X I I запускает внутрен­
ний механизм; высвобождение тканевого фактора и активация фактора V I I запу­
скают наружный механизм коагуляции. Оба пути приводят к активации фактора X
[Зубаиров Д.М ., 1995]
Тромботический синдром
Тромботический синдром, или тромбофилия (от гр. гкттЪош — ком,
сгусток, ркНео — люблю), — состояние, характеризующееся чрезмерной
(неадекватной) коагуляцией белков крови и тромбообразованием, вызы­
вающими ишемию тканей и органов.
Основные причины тромботического синдрома
• Повреждение стенок сосудов и сердца (например, при их механической
травме, атерогенезе, васкулитах, ангиопатиях у пациентов с СД);
• Патология форменных элементов крови (например, тромбоцитопатии,
гемолиз эритроцитов, чрезмерное повышение адгезии и агрегации тром­
боцитов и эритроцитов);
• Патология факторов системы гемостаза:
—абсолютное или относительное преобладание эффектов прокоагулянтных факторов;
—недостаточность антикоагулянтных и фибринолитических факторов,
например, при системном атеросклерозе, СД, гипертонической болезни
(ГБ), эндотоксинемиях, шоковых состояниях.
Механизмы развития тромботического синдрома
Механизмы гиперкоагуляции и тромботического синдрома представлены
на рис. 22.25. К этим механизмам относят:
• чрезмерную активацию прокоагулянтов и проагрегантов. Наиболее частые
причины:
—гиперлипопротеинемии. Известно, что липопротеины (ЛП) активируют
фактор Хагемана (фактор свертывания XII) и стимулируют активность
протромбиназы;
—повышенный уровень антифосфолипидных АТ (1§С, 1§М), например,
при антифосфолипидном синдроме. АТ к анионным фосфолипидам
стимулирует реакцию высвобождения из тромбоцитов, эндотелиоцитов,
кардиомиоцитов и некоторых других клеток прокоагулянтов и их после­
дующую активацию. Такой механизм тромбообразования выявляется,
например, при системной красной волчанке (СКВ) и ИБС;
—массированные травмы мягких тканей (например, при механическом
повреждении органов, тканей, конечностей, синдроме длительного раз­
давливания), ожоги большой площади, шоковые состояния, сепсис;
• увеличение концентрации в крови прокоагулянтов и проагрегантов (фибри­
ноген, протромбин, акцелерин, проконвертин, тромбин и др.). Наиболее
частые причины:
—гиперкатехоламинемия. Катехоламины активируют процесс синтеза
фибриногена (например, при патогенном стрессе или феохромоцитоме);
—гиперкортицизм (с гиперпродукцией глюкокортикоидов). Глюкокортикоиды стимулируют образование протромбина, проакцелерина,
фибриногена;
—атеросклероз стенок артерий (атеросклеротические изменения стенок
артерий потенцируют синтез фибриногена, протромбина, фактора
Хагемана, антигемофилических глобулинов и др.);
—септицемия (при ней стимулируется гиперпродукция тканевого тромбопластина).
Результатом чрезмерной активации и/или увеличения в крови прокоагу­
лянтов и проагрегантов может быть:
—гиперкоагуляция белков крови;
—адгезия, агрегация и активация форменных элементов крови;
—образование единичных тромбов;
—генерализованный тромбоз (наблюдается при синдроме ДВС);
• снижение содержания и/или угнетение активности прокоагулянтов и проагре­
гантов. Наиболее частые причины:
— наследственный дефицит антитромбина. Характеризуется снижением
синтеза антитромбина III, а также снижением его сродства к гепарину;
— печеночная, почечная или панкреатическая недостаточность. Эти состоя­
ния обусловливают снижение синтеза гепатоцитами антитромбина III;
—гиперлипопротеинемии. Они обусловливают снижение уровня гепарина
в крови (за счет его адсорбции на поверхности форменных элемен­
тов крови и иммунных комплексов, например, при СКВ или пурпуре
Шенлейна- Геноха);
— наследственная или приобретенная недостаточность протеинов С и 8.
Вторичный (приобретенный) дефицит этих белков наблюдается при
печеночной недостаточности, СД, лейкозах, массивных травмах, респи­
раторном дистресс-синдроме (РДС) взрослых;
• уменьшение уровня и/или подавление активности фибринолитических аген­
тов. Наиболее частые причины:
— подавление синтеза и выделение в кровь клетками активатора плазминогена. Наблюдается у пациентов с атеросклерозом, инфарктом миокарда,
ревматоидным артритом);
— наследственная или приобретенная гиперпродукция антиплазминов;
— уменьшение продукции фактора XII (например, при васкулитах или ДВС-
синдроме). Именно это послужило причиной смерти больного тромбофилией по фамилии Хагеман (его именем назван фактор XII).
Рис. 22.25. Основные механизмы гиперкоагуляции и тромботического синдрома
Последствия гиперкоагуляции и тромбоза
• Нарушения центральной, органотканевой гемоциркуляции и микроциркуля­
ции с исходом в инфаркт. При этом характер и тяжесть нарушений крово­
обращения определяются видом сосуда, пораженного тромбозом (артерии
или вены, микрососуды или магистральные стволы), количеством тромбированных сосудов, наличием коллатералей и условий для их развития, ско­
ростью и масштабом процесса тромбообразования, значимостью для орга­
низма, органа или ткани. Наиболее опасны тромбы в сосудах мозга, сердца,
легкого, поджелудочной железы, надпочечников, кишечника.
• Расстройства кровообращения, не завершающиеся инфарктом. Они обуслов­
ливают гипоксию тканей и органов (первично-циркулярного типа), раз­
витие дистрофических изменений и снижение их функций, гипотрофию
и гипоплазию тканевых и клеточных элементов, сдавление ткани дистальнее места пристеночного венозного тромба (расширенной веной и отеч­
ной тканью), образование тромбоэмболов (чаще в связи с разрушением
венозного тромба).
Геморрагические заболевания и синдромы
Геморрагические заболевания и синдромы представляют собой патологиче­
ские состояния, характеризующиеся повышенной кровоточивостью в резуль­
тате недостаточности одного или нескольких элементов гемостаза.
Этиология геморрагических синдромов и состояний
Выделяют наследственные и приобретенные геморрагические формы пато­
логии. Причинами их являются различные факторы.
Наследственные формы геморрагических синдромов и состояний вызываются
генетически детерминированными патологическими изменениями:
— сосудистой стенки;
— мегакариоцитов и тромбоцитов;
— адгезионных белков плазмы крови;
— плазменных факторов свертывающей системы крови.
Приобретенные геморрагические формы патологии в большинстве случаев
вызваны патогенными факторами иммунопатологических процессов, токсикоинфекций, дистрофий и другими:
— стенок кровеносных сосудов (с развитием генерализованных васкулитов);
— мегакариоцитов и тромбоцитов (с формированием тромбоцитопатий);
— адгезионных белков плазмы крови;
—факторов свертывающей системы крови (с развитием коагулопатий).
Виды геморрагических форм патологии
По происхождению различают следующие виды геморрагических заболева­
ний и синдромов:
— васкулиты;
— тромбоцитопении;
— тромбоцитопатии;
— коагулопатии, ДВС.
Васкулиты. Они обусловлены первичным поражением сосудистой стенки
с вторичным развитием коагулопатий и тромбоцитопатий. К этой группе
относят наследственную геморрагическую телеангиэктазию Рандю—Ослера,
синдром Элерса—Данло, гигантские гемангиомы при синдроме Казабаха—
Мерритта, геморрагический васкулит Шенлейна-Геноха, эритемы, геморра­
гические лихорадки и др.
Тромбоцитопении. Развиваются в результате первичного поражения мегакариоцитарно-тромбоцитарного ростка гемопоэза, перераспределения тромбо­
цитов и их депонирования в селезенке, повышенного разрушения (например,
при СКВ или идиопатической тромбоцитопенической пурпуре), избыточного
потребления тромбоцитов и образования тромбов (при ДВС, тромботической
тромбоцитопенической пурпуре), применения некоторых ЛС.
Тромбоцитопатии. Характеризуются наличием аномальных тромбоцитов
и нарушением их функций. Наиболее распространенные среди них — тромбастения Гланцманна и болезнь фон Виллебранда.
Коагулопатии. Обусловлены нарушениями свертываемости крови:
—наследственными (при гемофилии А, гемофилии В, болезни фон
Виллебранда, дефиците факторов свертываемости крови);
— приобретенными (при витамин К-зависимых коагулопатиях в условиях
недостаточности функции печени, нарушения всасывания витамина
К или его алиментарной недостаточности; ДВС; патологии печени,
приводящей к дефициту многих факторов свертывания; при нарушении
стабилизации фибрина, повышенном фибринолизе; при других приоб­
ретенных расстройствах свертывания, например при амилоидозе, при­
водящем к дефициту фактора X).
Диссеминированные тромбогеморрагические синдромы (ДВС и др.). Являются
следствием комплексных нарушений в различных звеньях системы гемостаза.
Тйпы кровоточивости
Выделяют несколько типов геморрагических синдромов и состояний.
Капиллярный, или микроциркуляторный (петехиально-синячковый). Он харак­
теризуется петехиальными высыпаниями, синяками и экхимозами на кожных
покровах и слизистых оболочках. Часто сочетается с повышенной кровоточи­
востью слизистых оболочек (носовые кровотечения, меноррагии). Возможно
развитие тяжелых кровоизлияний в головной мозг. Этот тип кровоточи­
вости характерен для тромбоцитопений и тромбоцитопатий, болезни фон
Виллебранда, недостаточности факторов протромбинового комплекса (VII,
X, V и II), некоторых вариантов гипо- и дисфибриногенемий, для умерен­
ной передозировки антикоагулянтов. При наследственных тромбоцитопатиях
обычно наблюдается синячковый тип кровоточивости (петехиальная сыпь
не характерна).
Гематомный. Проявляется болезненными, напряженными кровоизлияниями
в подкожную клетчатку, мышцы, крупные суставы, в брюшину и забрюшинное
пространство. Гематомы могут привести к сдавлению нервных стволов, разру­
шению хрящей и костной ткани, нарушению функций опорно-двигательного
аппарата. Иногда развиваются почечные и желудочно-кишечные кровотечения.
Характерны длительные кровотечения при порезах, ранениях, после удаления
зубов и хирургических вмешательств, часто влекущие за собой анемию. Этот
тип кровоточивости наблюдается и при некоторых наследственных нарушениях
свертываемости крови (гемофилия А и В, выраженная недостаточность фактора
VII), приобретенных коагулопатиях, сопровождающихся появлением в крови
ингибиторов факторов VIII, IX, УШ+У, и при передозировке антикоагулянтов,
а также при наследственной тромбоцитопатии с отсутствием пластиночного
фактора 3.
Смешанный капиллярно-гематомный. Характеризуется петехиально-синячковыми высыпаниями, сочетающимися с обширными плотными кровоиз­
лияниями и гематомами. Наблюдают при наследственных (выраженная недо­
статочность факторов VII и XIII, тяжелая форма болезни фон Виллебранда)
и приобретенных (острые синдромы ДВС, значительная передозировка пря­
мых и непрямых антикоагулянтов) нарушениях.
Васкулитно-пурпурный тип кровоточивости. Проявляется геморрагическими
или эритематозными (на воспалительной основе) высыпаниями; при этом
возможно развитие нефрита и кишечных кровотечений.
Ангиоматозный тип кровоточивости. Характеризуется повторными строго
локализованными кровотечениями, вызванными регионарной сосудистой
патологией. Наблюдают при телеангиэктазах, ангиомах, артериовенозных
шунтах.
Основные причины кровоточивости
Основные
на рис. 22.26.
причины
гипокоагуляции
и
кровоточивости
показаны
Рис. 22.26. Основные причины гипокоагуляции белков крови и геморрагического
синдрома
Механизмы гипокоагуляции
Главные механизмы формирования состояний и синдромов гипокоагуля­
ции и кровоточивости представлены на рис. 22.27.
Основные механизмы гипокоагуляции крови
и геморрагического синдрома
г
Уменьшение
содержания
в крови
прокоагулянтов
\ г
Недостаточное
образование
активированных
прокоагулянтов
ЛГ
'
Повышение
Увеличение содержания
содержания и/или
и/или чрезмерное
избыточная активация повышение активности
антикоагулянтов
фибринолитиков
Рис. 22.27. Основные механизмы гипокоагуляции белков крови и геморрагического
синдрома
Геморрагические состояния и синдромы вызываются патологией сосудов
(вазопатии), тромбоцитов (тромбоцитопатии), системы гемостаза (коагуло­
патии).
Геморрагические заболевания, обусловленные патологией сосудов
К типичным заболеваниям этой группы относят болезнь Рандю-Ослера,
пурпуру Шенлейна—Геноха, первичные геморрагические васкулиты.
Болезнь Рандю—О слера-Вебера (телеангиэктазия наследственная геморра­
гическая, ангиома наследственная геморрагическая, болезнь Ослера-Вебера,
болезнь Ослера): наследственная ОЛ) ангиопатия, сопровождается развитием
множественных телеангиэктазий, геморрагий, ЖДА, особенно после насту­
пления полового созревания.
Геморрагический васкулит (иммунокомплексный васкулит, болезнь Шенлейна—Геноха): кровоточивость, обусловленная поражением стенок сосудов малого
диаметра иммунными комплексами и компонентами системы комплемента (см.
статью «Пурпура» в приложении «Справочник терминов»).
Геморрагические синдромы, обусловленные патологией тромбоцитов
К геморрагическим формам патологии, обусловленным тромбоцитопениями и тромбоцитопатиями, относят идиопатическую тромбоцитопеническую
пурпуру, тромбастению Гланцманна, синдром Бернара-Сулье (см. раздел
«Патофизиология тромбоцитов» и соответствующие статьи в приложении
«Справочник терминов»).
Геморрагические формы патологии, обусловленные нарушениями свертыва­
ющей системы крови
Они развиваются в результате нарушений, возникающих в одном
или нескольких звеньях системы коагуляции белков крови (этапы гемокоагуляционного каскада, см. рис. 22.24).
Виды коагулопатий
Геморрагические синдромы и состояния могут быть наследственными
или приобретенными.
Примеры наследственных коагулопатий:
— дефицит компонентов фактора VIII (гемофилия А, болезнь фон Вилле­
бранда) или фактора IX (гемофилия В). Это наиболее распространен­
ные наследственные коагулопатии (более 95% случаев). См. статью
«Гемофилия» в приложении «Справочник терминов»;
— дефицит факторов VII, X, V и XI;
—дефицит других факторов (XII — при синдроме Хагемана, II — при гипо-
протромбинемии, I — гиподисфибриногенемии, XIII — при дефиците
фибринстабилизирующего фактора).
Примеры приобретенных коагулопатий:
— синдром ДВС;
— дефицит или угнетение активности факторов протромбинового комплекса
(II, VII, X, V), наблюдающиеся при заболеваниях печени, обтурационной желтухе, дисбактериозах кишечника, передозировке антагонистов
витамина К (кумарины, фенилин), геморрагическая болезнь новорож­
денных;
— коагулопатии, вызванные наличием в крови иммунных ингибиторов факто­
ров свертывания (чаще всего АТ к фактору VIII);
— кровоточивость, обусловленная гепаринизацией или введением фибринолитиков [стрептокиназа, урокиназа, алтеплаза (актилизе*)] и дефибрини-
рующих препаратов.
Диссеминированное внутрисосудистое свертывание белков крови
Тромбогеморрагические состояния характеризуются мозаичной (во вре­
мени и по месту преимущественной локализации в организме) сменой фазы
гиперкоагуляции и тромбоза фазой гипокоагуляции, фибринолиза и гемор­
рагического синдрома.
Клинически наиболее значимым проявлением тромбогеморрагических
состояний является диссеминированное внутрисосудистое свертывание (ДВС)
крови.
ДВС — патогенетически сложное состояние, возникающее при различных
заболеваниях и терминальных состояниях.
ДВС характеризуется рассеянным внутрисосудистым свертыванием белков
крови, агрегацией ее форменных элементов, активацией и истощением ком­
понентов свертывающей и фибринолитической системы, блокадой сосудов
микроциркуляции в органах с последующим микротромбообразованием.
В конечной фазе ДВС развиваются два противоположных явления (они наблю­
даются в разных участках сосудов и в разное время, сменяя друг друга):
- повышенное тромбообразование;
- тяжелый геморрагический синдром.
ДВС-синдром чреват смертью пациента (летальность колеблется в диа­
пазоне 30—60%) и требует проведения неотложных специализированных
врачебных мероприятий.
Причины ДВС-синдрома (рис. 22.28)
Рис. 22.28. Основные причины ДВС-синдрома
• Различные повреждения тканей и высвобождающиеся при этом факторы,
стимулирующие гемостаз (активирующие внешний механизм свертывания,
см. рис. 22.24), — наиболее частые причины ДВС-синдрома:
— акушерская патология (преждевременная отслойка, предлежание и раз­
рывы плаценты; эмболия околоплодными водами; атонические маточ­
ные кровотечения; антенатальная гибель плода; плодоразрушающие
операции; пузырный занос, аборт во II триместре беременности);
— повышенный гемолиз форменных элементов крови, в том числе и внутрисосудистый (трансфузии компонентов крови, кризы гемолитических
анемий, отравление гемолитическими ядами, гемолитико-уремический
синдром);
— онкологические заболевания (новообразования, гемобластозы);
— массивные повреждения тканей (ожоги, отморожения, электротравмы,
синдром длительного сдавливания, огнестрельные ранения, переломы
трубчатых костей, особенно осложненные жировой эмболией, опера­
тивные вмешательства);
— острые и подострые воспалительно-деструктивные процессы (панкреонекроз, перитониты, деструктивные пневмонии).
• Повреждение эндотелия сосудов (оно запускает внутренний механизм
свертывания): расслаивающаяся аневризма аорты, прогрессирующий ате-
росклероз сосудов, системные заболевания (СКВ, ревматоидный артрит),
гемолитико-уремический синдром, острый гломерулонефрит, различные
лихорадки и аллергические реакции.
• Инфекции. Бактериальные токсины повреждают эндотелий. Продукты
жизнедеятельности микроорганизмов, а также воспалительные изменения
органов, медиаторы воспаления активируют тканевые факторы гемокоа­
гуляции. Сепсис сам по себе представляет сочетание генерализованного
инфекционного заболевания и синдрома ДВС.
Патогенез
Массивное поступление в кровь тканевого тромбопластина активирует
свертывание белков крови и тромбоцитарный гемостаз. Это приводит к мно­
жественному тромбообразованию (гиперкоагуляционная стадия синдрома
ДВС), а затем к истощению факторов свертывания (гипокоагуляционная
стадия синдрома ДВС).
Внутрисосудистое свертывание крови сменяется гипокоагуляцией и тяже­
лым геморрагическим синдромом. Пусковой механизм синдрома ДВС — акти­
вация коагуляционного гемостаза (рис. 22.29).
Рис. 22.29. Общая схема патогенеза синдрома ДВС
ДВС-синдром характеризуется, как правило, «взрывным» началом и про­
грессирующим течением. В ходе его развития выделяют три фазы (стадии):
гиперкоагуляции, коагулопатии потребления, гипокоагуляции.
Стадия гиперкоагуляции синдрома ДВС
Стадия гиперкоагуляции и тромбообразования (гиперкоагуляционнотромботическая стадия) кратковременна. В этой стадии активируются оба
пути свертывания крови: внутренний, активируемый повреждением эндотелия,
и внешний, запускаемый тканевыми факторами (например, тромбопластино­
подобными веществами, продуктами протеолиза и др.).
Внутрисосудистое свертывание белков крови (включая фибринообразование), а также адгезия и агрегация тромбоцитов приводят к возникновению
микротромбов. Тромбообразование обусловливает нарушение микроциркуля­
ции в тканях, что сопровождается развитием гипоксии и нарушением их тро­
фики. Основные звенья патогенеза и проявления стадии гиперкоагуляции
представлены на рис. 22.30.
Стадия коагулопатии потребления
Стадия коагулопатии потребления характеризуется повышенным потреблени­
ем и истощением факторов свертываемости и тромбоцитов, развитием гипофибриногенемии и недостаточностью антикоагулянтов.
Основные звенья патогенеза и проявления стадии коагулопатии потребле­
ния показаны на рис. 22.31.
Стадия гипокоагуляции
Стадия гипокоагуляции (гипокоагуляционно-геморрагическая фаза) харак­
теризуется геморрагическим синдромом. В основе его развития — три основных
процесса:
— быстрое истощение компонентов свертывающей системы крови (про­
тромбин и фибриноген), физиологических антикоагулянтов (антитром­
бин III, протеины С, 8);
— уменьшение содержания тромбоцитов вследствие их потребления тромбами;
— усиленный фибринолиз (в ответ на повышенное образование фибрина).
При благоприятном течении ДВС-синдрома, своевременных и адекватных
лечебных мероприятиях возможно блокирование механизма синдрома и его
«обратное» развитие. Происходит восстановление кровообращения в поражен­
ных зонах, снижение продукции тромбина, повышение концентрации гемостатических факторов, нормализация содержания тромбоцитов. Основные звенья
патогенеза и проявления стадии гипокоагуляции показаны на рис. 22.32.
Проявления синдрома ДВС
Проявления ДВС-синдрома складываются из симптомов как основной пато­
логии, так и самого синдрома.
Первая (гиперкоагуляционная) стадия при остром его течении протекает
быстро и может в считанные минуты смениться гипокоагуляцией. О первой
стадии синдрома говорят тогда, когда на фоне основного заболевания (патоло­
гические процессы, перечисленные выше) появляются признаки полиорганной
недостаточности вследствие тромбозов, не характерных для фоновой патологии
(например, цианоз, одышка, кашель, застойные хрипы; олигурия, анурия;
желтуха, спутанность сознания).
В гипокоагуляционной стадии ДВС выявляются петехии и экхимозы (в местах
инъекций, наложения манжеты тонометра, трения одеждой), кровотечения
из операционных ран, метроррагии, носовые и желудочно-кишечные кровоте­
чения, кровоизлияния в кожу, слизистые оболочки, паренхиматозные органы.
Рис. 22.30. Основные шенья патогенеза и проявления стадии I ДВС-синдрома (стадии I ниеркоагулиции и тромбообразования)
Патофизиология, клиническая патофизиология
Проявпения:
-гилертромбопластинемия;
- гиперпротромбинемия;
-укорочение времени свертывания крови;
-бледность кожи и слизистых оболочек;
- тахипноэ
Проявления:
- гипофибриногенемия;
-уменьшение концентрации в крови антитромбина;
-нарастание уровня продуктов деградации фибрина в крови;
-значительная тромбоцитопения;
-кровотечение из поврежденных сосудов;
-кровоизлияния
Рис. 22.31. Основные звенья патогенеза и проявления стадии II ДВС-синдрома (ста­
дии коагулопатии потребления)
Проявления:
- значительная гипофибриногенемия;
- критическое падение уровня антитромбина III в крови;
- существенное повышение содержания продуктов деградации фибрина;
- критическая тромбоцитопения;
- нарастающее кровотечение;
-кровоизлияния в неповрежденные ткани;
- полиорганная недостаточность
Рис. 22.32. Основные звенья патогенеза и проявления стадии III ДВС-синдрома (ста­
дии гипокоагуляции)
В результате кровоизлияния в надпочечники может развиться острая надпо­
чечниковая недостаточность (ОПН, синдром Уотерхауса-Фридериксена).
При выраженной кровопотере часто наблюдается гиповолемический шок,
усугубляющий тканевую гипоксию и ацидоз.
Основной диагностический тест при синдроме ДВС (в том числе до появ­
ления клинических проявлений) — изменение показателей коагуляционного
гемостаза:
— стадия гиперкоагуляции: концентрации тромбопластина и протромбина
увеличены; время свертывания менее 4 мин; паракоагуляционные тесты
не изменены; спонтанная агрегация тромбоцитов повышена;
— стадия коагулопатии потребления: концентрация фибриногена менее 2 г/л;
паракоагуляционные тесты положительны; концентрация продуктов дегра­
дации фибрина увеличена; тромбиновое время более 30—35 с, протромбиновое время более 20 с; концентрация антитромбина III менее 75%;
— стадия гипокоагуляции: время кровотечения увеличено; концентрация
фибриногена менее 1,5 г/л; паракоагуляционные тесты часто отрицатель­
ные; концентрация продуктов деградации фибрина более 2х102 мг/л; тром­
биновое время более 35 с; протромбиновое время более 22 с; концентрация
антитромбина III составляет 30—60%; содержание тромбоцитов снижено.
Принципы лечения ДВС-синдрома
Успех лечения во многом зависит от ранней диагностики. Больного пере­
водят в реанимационное отделение, при необходимости проводят ИВЛ.
Этиотропная терапия
Поскольку ДВС-синдром — «вторая болезнь», лечение направлено на устра­
нение или снижение патогенного действия причинного фактора: «первой
болезни» (например, антибактериальная терапия при сепсисе, устранение аку­
шерской патологии, ликвидация последствий гемолиза эритроцитов и т.п.).
Патогенетическое лечение ДВС-синдрома
Коррекция содержания и/или активности факторов системы гемостаза: в фазе
гиперкоагуляции при отсутствии активного кровотечения применяют анти­
коагулянты (обычно гепарин натрия в/в), свежезамороженную плазму крови;
при геморрагическом синдроме, сочетающемся с тромбоцитопенией, вводят
тромбоцитарную массу.
Восстановление объема крови (вводят изотонический раствор натрия хлори­
да, компоненты крови; при этом следует избегать перегрузки сердца объемом
и развития отека легкого).
Коррекция газового состава крови и КОС (ингаляция кислорода, введение
растворов натрия гидрокарбоната).
Нормализация почечного кровотока (при артериальной гипотензии приме­
няют симпатомиметики; при развитии ОПН — гемодиализ).
Снижение концентрации в крови иммунных комплексов, продуктов фибринолиза и бактериальных токсинов (с помощью плазмафереза).
Симптоматическое лечение синдрома ДВС
Симптоматическая терапия синдрома имеет целью облегчение состояния
пациента. Для этого устраняют неприятные, тягостные ощущения (болевые,
психоэмоциональные и др.), а также проводят мероприятия с целью устранить
недостаточность функции органов и физиологических систем.
Профилактика тромбогеморрагических синдромов
Профилактика повторного развития синдрома заключается в том, чтобы
ликвидировать условия, провоцирующие развитие ДВС, или предупредить
их возникновение (лечение основного заболевания, введение гепарина
натрия при гиперкоагуляции, повторные трансфузии свежезамороженной
плазмы).
Прогноз ДВС-синдрома
Прогноз ДВС-синдрома во многом зависит от эффективности лечения
основного заболевания, своевременности диагностики, адекватности лечеб­
ных мероприятий. Летальность при ДВС составляет 40-60%.
Основные причины смерти при тромбогеморрагических синдромах: ОПН,
дыхательная недостаточность, кровоизлияние в мозг, надпочечники, острая
кровопотеря, приводящая к развитию шока и комы.
Гемобластозы
Гемобластозы — опухоли, возникающие из кроветворных клеток гемопоэтической ткани.
Гемобластозы занимают первое место как причина смерти среди всех
болезней системы крови.
Гемобластозы подразделяются на:
— лейкозы (опухоли, системно поражающие гемопоэтические клетки всех
ростков костного мозга);
— лимфомы, лимфосаркомы (внекостномозговые плотные, растущие в виде
узла или нескольких узлов, опухоли только из лимфопролиферативных
кроветворных клеток);
— миелопролиферативные новообразования.
В клинической практике, согласно МКБ-10, используют не родовое понятие
«гемобластоз» или «лейкоз», а названия конкретных их нозологических форм
(разных стадий и определенных иммуно-, фено- и генотипов), каждая из кото­
рых подразумевает конкретную программу лечения. Так, к злокачественным
новообразованиям «лимфоидной, кроветворной и родственных им тканей»
(коды МКБ: С81-С96) относят болезнь Ходжкина (лимфогранулематоз), неходжкинские лимфомы (лимфосаркомы), злокачественные иммунопролиферативные болезни (в том числе макроглобулинемию Вальденстрема), множественную
миелому, лимфолейкоз, миелолейкоз, истинную полицитемию и ряд других
опухолей. Для лимфоидных гемобластозов предложена классификация К.ЕАЬ
(Кеукес! Еигор1ап-Атепсап с1а$§Шса1юп оГ Ьутр1ю1с1 пеор1а$т$, см. статью
«Классификация КЕАЬ> в приложении «Справочник терминов»),
В клинической литературе лимфомы (растущие вне костного мозга очаговые
злокачественные опухоли из кроветворных клеток) обозначают как гематосарко­
мы (син. — лимфосаркомы). К гематосаркомам относят миело-, эритро-, мегакариосаркомы, а также морфологически недифференцируемые гематосаркомы.
При метастазировании гематосарком в костный мозг опухолевый процесс
приобретает генерализованный характер. Этот феномен обозначают термином
«лейкемизация гематосарком».
Характеристики отдельных лимфом приведены в статье «Лимфомы» (см.
приложение «Справочник терминов»). В этом же приложении рассмотрена
этиология, патогенез, проявления и принципы терапии других гемобластозов:
истинной полицитемии, лимфогранулематоза (болезнь Ходжкина), миеломной болезни, макроглобулинемии Вальденстрема.
ОБЩАЯ ХАРАКТЕРИСТИКА ЛЕЙКОЗОВ
Лейкоз — системное опухолевое поражение гемопоэтических клеток кост­
номозговой ткани.
Для обозначения лейкозов не рекомендуется применять предложенный
Р. Вирховым термин «лейкемия» (белокровие), так как к лейкозам относятся,
помимо опухолей из лимфо- и миелопоэтических клеток, и новообразования
из клеток эритро- и мегакариоцитарных ростков. Кроме того, лейкозы не
всегда характеризуются увеличением числа лейкоцитов в крови — оно может
быть также нормальным или даже сниженным.
Этиология лейкозов
Причины лейкозов
Причинами лейкозов являются те же группы факторов, которые вызы­
вают опухоли (см. главу 18 «Типовые нарушения тканевого роста. Ново­
образования», т. 1).
Факторы риска лейкозов
Наследственное предрасположение. Описано доминантное и рецессивное
наследование хронического лимфолейкоза (ХЛЛ), а также низкая заболева­
емость этим лейкозом в одних этнических группах и высокая — в других.
Чаще наследуется не сам лейкоз, а нестабильность генома — сниженная
резистентность хромосом к действию мутагенов, что предрасполагает родо­
начальные миелоидные или лимфоидные клетки к опухолевой (лейкозной)
трансформации.
Применение ЛС с цитостатическим действием. Вероятность возникновения
острых лейкозов у больных, лечившихся цитостатиками, повышается в сотни раз.
Воздействие на организм проникающей радиации и/или рентгеновского излу­
чения (в том числе с лечебной целью). Минимальный интервал времени
до возникновения лейкоза после облучения составляет 5—10 лет, а после
химиотерапии — 2 года (максимум в течение 6-10 лет).
Патогенез лейкозов
Трансформация нормальных гемопоэтических клеток в опухолевые —
результат изменений в генетической программе.
Основную роль при этом играют хромосомные нарушения. В большинстве
случаев они определяют прогноз болезни и тип специфического лечения.
Нестабильность генома клеток гемобластоза приводит к появлению в перво­
начальном опухолевом клоне новых субклонов, среди которых в процессе жиз­
недеятельности организма, а также под воздействием лечения «отбираются»
наиболее автономные (феномен получил название «опухолевая прогрессия»).
Этим феноменом объясняют прогредиентность течения лейкозов, их «уход»
из-под контроля цитостатиков и генерализацию процесса.
Этиология, патогенез, проявления и принципы лечения различных лей­
козов (лейкоз острый, ХМЛ, ХЛЛ) рассмотрены в разделе «Лейкозы» насто­
ящей главы. Характеристика отдельных видов лимфом приведена в статье
«Лимфомы» приложения «Справочник терминов». В этом же приложении
рассмотрена этиология, патогенез, проявления и принципы лечения дру­
гих гемобластозов: истинной полицитемии, лимфогранулематоза (болезнь
Ходжкина), миеломной болезни, макроглобулинемии Вальденстрема.
Опухолевый атипизм гемобластозов
Лтипизм гемобластозов — совокупность существенных качественных
и количественных признаков, отличающих биологические свойства гемобластозных клеток от нормальных и других патологически измененных
(но не трансформированных) клеток и тканей. В основе формирования
атипизма гемобластозов лежит процесс опухолевой прогрессии.
Опухолевая прогрессия гемобластозов
Опухолевая прогрессия по своему существу является механизмом, при кото­
ром нарастает степень злокачественности клеток гемобластозов в результате
изменений их генетической программы.
Повышенная и постоянная изменчивость различных свойств гемобла­
стозов, приводящая к их гетерогенности и автономности, создает условия,
позволяющие их клеткам все больше приспосабливаться к условиям среды,
включая и возможность «ускользания» от цитостатической терапии.
Признаки опухолевой прогрессии гемобластозов
Проявления опухолевой прогрессии гемобластозов приведены на рис. 22.33.
Рис. 22.33. Признаки опухолевой прогрессии гемобластозов
Опухолевая прогрессия ведет к формированию и/или нарастанию степени
атипизма роста, обмена, структуры и функции лейкозов.
Атипизм роста гемобластозов
Проявления атипизма роста лейкозов представлены на рис. 22.34.
Рис. 22.34. Основные проявления атипизма роста лейкозов
Костный мозг при лейкозах:
— наличие клеток, относящихся к двум качественно разным типам гемопоэза — нормальному и опухолевому;
— увеличение количества делящихся гемопоэтических клеток («омоложение»
состава гемопоэтических клеток); сопровождается нарастанием количе­
ства атипичных бластных и молодых нормальных клеток гемопоэтической ткани.
Периферическая кровь при лейкозах:
— лейкемия (белокровие, от греч. 1еикоз — белый, к а т а — кровь): уве­
личение в крови количества лейкозных клеток; наблюдается часто,
но не всегда и характеризуется увеличением количества лейкозных кле­
ток различной степени зрелости (бластных, созревающих).
В зависимости от общего количества лейкоцитов и наличия бластных кле­
ток в единице объема крови при лейкозах выделяют четыре формы лейкоза:
— лейкемическую: количество лейкоцитов превышает 30—50хЮ9/л, боль­
шое количество бластных форм лейкозных клеток;
— сублейкемическую: количество лейкоцитов выше нормы (но до
30—50x109/л), большое количество бластных клеток;
— лейкопеническую: количество лейкоцитов ниже нормы, сравнительно
небольшое количество бластных лейкозных клеток;
— алейкемическую: количество лейкоцитов в диапазоне нормы, бластные
клетки отсутствуют; атипичные лейкоциты, их бластные и молодые
формы находятся лишь в ткани костного мозга.
Проявления лейкозов:
— лейкемический «провал» (лейкемические «ворота» — Ыа1из к и к а е т к ш ,
лейкемическое «зияние»). Выявляется при остром миелобластном лей­
козе (ОМЛ) и характеризуется наличием в периферической крови бласт­
ных, молодых и зрелых форм лейкозных клеток и отсутствием одной
или нескольких переходных форм гемопоэза. С этим и связано название
признака — лейкемический «провал»;
—анемия — спутник большинства лейкозов, в особенности острых;
— тромбоцитопения и меньшая свертываемость крови;
—геморрагический синдром. Характеризуется частыми кровотечениями,
в том числе в полости тела и полые органы (желудок, кишечник, пище­
вод, мочевой пузырь и др.), а также кровоизлияниями.
Атипизм обмена веществ при гемобластозах
Атипизм обмена характеризуется отклонением от нормы биохимических
характеристик клеток и/или продуктов их метаболизма.
Помимо общих признаков опухолевого атипизма обмена углеводов, белков,
липидов, ионов, минералов, кислотно-щелочного равновесия (КЩР) (см.
главу 18 «Типовые нарушения тканевого роста. Новообразования», т. 1),
для лейкозов характерен ряд специфических признаков:
—прекращение синтеза лейкозными клетками отдельных ферментов (напри­
мер, кислой фосфатазы, миелопероксидазы) и, как следствие, катализи­
руемых ими процессов;
—развитие пара- и диспротеинемий (парапротеинемия наблюдается
при миеломной болезни, макроглобулинемии Вальденстрема, болезни
тяжелых цепей Франклина).
Атипизм структуры гемобластозов
Характеризуется развитием признаков клеточного (см. главу 18 «Типовые
нарушения тканевого роста. Новообразования», т. 1) и тканевого атипизма (про­
являющегося наличием двух типов клеток в гемопоэтической ткани и пери­
ферической крови — нормальных и опухолевых). (Подробнее характеристику
атипизма структуры лейкозов см. в учебниках по патологической анатомии.)
Атипизм функций гемобластозов
При лейкозах нарушаются функции как трансформированных (лейкоз­
ных), так и нормальных лейкоцитов. Это приводит к существенным наруше­
ниям фагоцитарной активности, механизмов реализации клеточного и гумо­
рального иммунитета.
В совокупности указанные нарушения обусловливают развитие иммуноде­
фицита, в том числе ослабление противоинфекционной устойчивости и антибластомной резистентности организма.
ЛЕЙКЕМОИДНЫЕ РЕАКЦИИ
Лейкемоидные реакции — состояния, характеризующиеся изменениями
в крови, органах гемопоэза и организме в целом, сходные с теми, которые
наблюдаются при гемобластозах, главным образом — при лейкозах.
Свое название «лейкемоидные» эти реакции получили в связи с тем,
что изменения в гемопоэтической ткани и периферической крови напоминают
изменения при лейкозах. Однако лейкемоидные реакции никогда не транс­
формируются в тот лейкоз, с которым они сходны гематологически. Различия
между лейкемоидными реакциями и лейкозами приведены в табл. 22.3.
Таблица 22.3. Отличия лейкемоидных реакций от лейкозов
Лейкозы
Лейкемоидные реакции
Причины
Канцерогены
Возбудители инфекций
БАВ, активирующие выход форменных эле­
ментов крови из органов гемопоэза
Состояния, ведущие к повышенному «потре­
блению» форменных элементов крови
Иммунопатологические состояния
Механизмы развития
Трансформация нормальной гемо­
Активация нормального гемопоэза и посту­
поэтической клетки в опухолевую
пление в сосудистое русло избытка формен­
ных элементов крови
Подавление нормального гемопоэза и тормо­
жение выхода в сосудистое русло форменных
элементов крови
Проявления
Костный мозг
Очаговая гиперплазия нормальных гемопоэти­ Генерализованная гиперплазия опухо­
левых гемопоэтических клеток
ческих клеток (при пролиферативных лейке­
Обычно (но не всегда, например,
моидных реакциях)
Гипоплазия гемопоэтической ткани (при цито- при алейкемической форме) много
бластных и незрелых лейкозных клеток
пенических формах лейкемоидных реакций)
Периферическая кровь
Цитопения сочетается с наличием
Наличие бластных и незрелых форм лейкоци­
тарного, тромбоцитарного или эритроцитарно- в крови бластных лейкозных клеток
го гемопоэза (при пролиферативных реакциях) Признаки дегенерации клеток
обычно отсутствуют (наблюдаются
Лейко-, эритро- и/или тромбоцитопения
(при цитопенических лейкемоидных реакциях) при В-лимфолейкозах)
Лейкемический «провал» при ОМЛ
Признаки дегенерации форменных элементов
крови
ЛЕЙКОЗЫ
Ниже рассмотрены острые и хронические формы лейкозов, их патогенез,
критерии дифференцировки стадий, проявления и принципы лечения гемо­
бластозов.
Острые лейкозы
Клеточным субстратом острых лейкозов являются бластные опухолевые клетки.
Острый лейкоз без лечения приводит к смертельному исходу в течение
нескольких недель или месяцев. При правильном и своевременном лечении
прогноз часто благоприятен.
Острые лейкозы подразделяются на миелоидные (ОМЛ), лимфоидные
(ОЛЛ), монобластные, миеломонобластные, эритромиелобластные и мегакариобластные.
Острые лейкозы — злокачественные заболевания кроветворной системы.
Их морфологический субстрат — бластные опухолевые клетки. Наиболее
часто выявляются ОЛЛ и ОМЛ.
Частота острых лейкозов
У мужчин острые лейкозы выявляются в среднем в 13,2 случаях на 100 ООО,
у женщин соответственно в 7,7 на 100 ООО.
ОЛЛ чаще развивается в детском возрасте и после 40 лет. Частота ОМЛ
близка во всех возрастных группах.
Проявления острых лейкозов
Развитие острых лейкозов характеризуется:
— пролиферацией клона опухолевых клеток с характерными цитогенетиче­
скими нарушениями;
— угнетением нормального кроветворения;
— выходом бластных клеток в кровь;
— метастазированием лейкозных клеток в другие кроветворные (селезенка,
печень, лимфатические узлы) и некроветворные (кожа, ЦНС, яички,
легкие) органы.
Виды острого лейкоза
Существенным фактором диагностики и последующего лечения любого
лейкоза является типирование заболевания у конкретного больного. Критерии
типирования лейкозов представлены на рис. 22.35.
В основу дифференцировки острых лейкозов положены внешний вид
и цитохимические особенности бластных клеток, их иммунофенотип и гене­
тические особенности. Так, франко-американо-британский (РАВ) подход
к типированию лейкозов основан на оценке морфологии лейкозных клеток
(строение ядра, соотношение размеров ядра и цитоплазмы).
Острый миелобластный лейкоз
Варианты ОМЛ приведены в табл. 22.4.
Рис. 22.35. Критерии типирования лейкозов
Таблица 22.4. Варианты острого миелобластного лейкоза, ВОЗ, 1999
_____________________________________Варианты_____________________________________
ОМЛ с 1(8; 21); (д22; д22)__________________________________________________________
ОМЛ с 1(15; 17); (д22; д!1—12)_____________________________________________________
Острый миеломонобластный лейкоз
ОМЛ с патологической костномозговой эозинофилией (т у (16); (р13д22) или I
(16; 16); (Р13;д11)_________________________________________________________________
ОМЛ с дефектами 11д23 (МЬЬ)___________________________________________________
Острый эритроидный лейкоз_____________________________________________________
Острый мегакариоцитарный (мегакариобластный) лейкоз________________________
Острый базофильный лейкоз_____________________________________________________
Острый панмиелоз с миелофиброзом_____________________________________________
Острые бифенотипические лейкозы______________________________________________
ОМЛ с мультилинейной дисплазией______________________________________________
Вторичный ОМЛ_________________________________________________________________
Об уровне дифференцировки опухолевых клеток свидетельствует экс­
прессия на поверхности их ядерной мембраны, в цитоплазме и на цито­
плазматической мембране специфических дифференцировочных Аг, «класте­
ров дифференцировки», обозначаемых аббревиатурой СО (от англ. с1ш1ег о/
сИ//егепИаИоп). Иммунофенотипическая характеристика миелобластных лейко­
зов приведена в табл. 22.5.
Таблица 22.5. Иммунофенотипическая характеристика миелобластных лейкозов
Иммуновариант
лейкоза
М0 (острый малодифференциро­
ванный лейкоз)
М, (ОМЛ
без созревания)
М2 (ОМЛ с созре­
ванием)
Реже коэкспрессируемые антигены
НЬА-ОК+, СБ15+/-, С 0 1 3 +/ , СБЗЗ+/- Коэкспрессия лимфоид­
ных, миелоидных или
эритроидных маркёров
Доминирующий клеточный фенотип
Н1А-ОК+/-, С 0 3 8 +/-, СОПа+/-.
К Р В -Г /-, СБ53+, СОПЬ+/-, С 015+/-,
С Б7+/ -
СБЮ
НЬА-ОЯ+/“, СБ72+ (ИПОЮ),
СЭ38+/- СБ53+, ЯРВ-1++/-,
СО!1а+/-, СЭ11Ь++/_, С 0 15 ++/“
СБ1, СЭ2, СБ7, СБ10
Окончание табл. 22.5
Иммуновариант
лейкоза
М3 (острый промиелобластный
лейкоз)
М4 (острый миеломонобластный
лейкоз)
М5 (острый монобластный лейкоз)
М6 (острый эритромиелоз)
М7 (острый мегакариобластный
лейкоз)
Доминирующий клеточный фенотип
СБ53+, КРВ-1+/- С011Ь+/- -С Б 1 5 +/ - НЬА-БК+/--Т Н У -1 -, С Б38-, С02~,
С Б З-, С Б 4-, С Б 8-, СБ19”, СЭ72Н1А-БК+, С Б15+, СБ38+, СБ11Ь+
Реже коэкспрессируемые антигены
Маркёры зрелых миелоидных клеток отсут­
ствуют
Аг эритробластов
(3,4%)
Н1А-БК+, СБ11Ь+, СБ15+, СБ38“
Аг эритробластов (15%)
Гликофорин А+, Аг эритробластов+,
Н 1Л -БК +/-, С Б38-
СБ7, СБ15
СВ38+,С 0 4 1 +,Н 1 А -Б К +/- , СТ>1+/~,
С Б 4-, С Б 8-, С Э П Ь -, С Б 15-, СБЗЗ-.
С Б 10-, С Б 34-, С Б71-
Примечание: + — выраженная экспрессия Аг; +/_ — вариабельная экспрессия Аг; - —
отсутствие экспрессии Аг.
Острый лимфобластный лейкоз
Развитие В- и Т-лимфоцитов и типы ОЛЛ приведены на рис. 22.36, имму­
нологические и цитогенетические варианты ОЛЛ — в табл. 22.6.
Рис. 22.36. Развитие В- и Т-лимфоцитов и типы острого лимфобластного лейкоза:
ОЛЛ — острый лимфобластный лейкоз, САЬЬА — общий антиген ОЛЛ; Тс1Т — тер­
минальная дезоксинуклеотидилтрансфераза; ТА — антиген тимуса; с1§ — цитоплаз­
матический 1§; з1§ — мембранный 1§
Таблица 22.6. Иммунологические и цитогенетические варианты острых лимфобласт­
ных лейкозов
Тип и частота
Пре-пре-В ОЛЛ
(ранний пре-пре-В:
5—10%, обычный:
40-45% )
Пре-В ОЛЛ (20%)
В-ОЛЛ (4-5%)
Т-ОЛЛ (20-31%)
Фенотип
Цитогенетические
нарушения
Н Ь А -О К , Тс1Т+, СБ34+, СБ10+/-.
СБ19+, с1§- &!§-, СП20-/+,
СБ24+/-
1(12; 21) в 20-25% ,
1 (9; 22), аномалии Щ 23
НЬА—БК , СЭ19, СБ20+/-, СБ24,
СБ9, СБ10, СБ34<->, с1§М,
Тс1Т'/-
1 (1; 19)
Н Ь А -Б К , СБ19, СБ20, СЭ22,
С 024, СБ10+/- СВ34<->,
Тс1Т(-\ $1§
Н Ь А -О К /+, С 01, С Б2, сСБЗ,
СБ5, СЭ7, С Б 4 /С 0 8 , СОЮ+/СБ34-/+, СБ45+/-, Тс1Т
1 (8; 14), 1 (2; 8), 1 (8; 22)
1 (1; 14) в 15-25%
Примечание: с!§ — цитоплазматический 1§; з1§ — мембранный 1§; Тс1Т — терминаль­
ная дезоксинуклеотидилтрансфераза; + — выраженная экспрессия Аг;
— вариа­
бельная экспрессия Аг; - — отсутствие экспрессии Аг.
Общие проявления острых лейкозов
Клиническая картина обычно сходна у всех типов острых лейкозов. Начало
их, как правило, внезапное, реже — постепенное.
Состояние пациента, как правило, тяжелое. Оно обусловлено интоксикаци­
ей, геморрагическим синдромом (результаттромбоцитопении), ДН (вследствие
сдавления дыхательных путей увеличенными внутригрудными лимфатически­
ми узлами). Для острых лейкозов характерны нижеследующие симптомы.
• Анемический (гипоксический) синдром: бледность, одышка, сердцебиение,
сонливость.
• Снижение резистентности к инфекциям (бактериальным, грибковым
и вирусным). У пациентов с лейкозами выявляют как легкие (локальные)
формы инфекций (например, кандидозные стоматиты, гингивиты, пора­
жения слизистых оболочек, вызванные вирусом простого герпеса), так
и тяжелые генерализованные процессы (пневмонии, сепсис).
• Геморрагический синдром. При осмотре пациентов обнаруживают петехии
и экхимозы на коже (самопроизвольные, в местах инъекций или механи­
ческого трения). Возможны тяжелые носовые и внутренние кровотече­
ния (метроррагии, желудочно-кишечные кровотечения, кровоизлияния
в мозг).
• ДВС-синдром (характерен для промиелоцитарного лейкоза).
• Интоксикация организма. Проявляется лихорадкой, снижением массы тела
и аппетита, слабостью, повышенной потливостью.
• Болезненность костей. В наибольшей мере выражена в трубчатых костях
и позвоночнике, в области суставов (артралгии). Обусловлена опухолевой
гиперплазией костномозговой гемопоэтической ткани.
• Лимфаденопатия. Возможно увеличение любой группы лимфатических
узлов в связи с пролиферацией в них лейкозных лимфоидных клеток.
При этом выявляются множественные, плотные, эластичные, округлые
пакеты лимфатических узлов, которые могут быть спаяны друг с другом,
размером от 1 до 8 см; при пальпации они безболезненны.
• Увеличение печени и селезенки (связано с метастазированием лейкозных
клеток в эти органы и образованием экстрамедуллярных очагов гемопоэза
в них).
• Поражение ЦНС (нейролейкемия). Часто выявляется при ОЛЛ и значительно
ухудшает прогноз. Нейролейкемия обусловлена метастазированием лейкоз­
ных клеток в оболочки головного и спинного мозга или в вещество мозга.
• Гипертрофия тимуса (обусловлена метастазами лейкоза в вилочковую желе­
зу; может вызвать сдавление органов средостения).
Лабораторные признаки острых лейкозов
Анализ периферической крови
При острых лейкозах в периферической крови обнаруживают:
— нормохромную нормоцитарную анемию;
— различное количество лейкоцитов (нормальное, пониженное или увели­
ченное);
— нейтропению (вне зависимости от общего количества лейкоцитов);
— абсолютный и относительный лимфоцитоз;
— тромбоцитопения (как правило).
Пунктат костного мозга
Пункция костного мозга — основной метод исследования при лейкозах.
Его применяют для подтверждения диагноза и идентификации (морфологи­
ческой, иммунофенотипической, цитогенетической) типа лейкоза.
Миелограмма (количественная характеристика клеточных форм костного
мозга) при острых лейкозах имеет характерные особенности:
— число бластных клеток более 5%, вплоть до тотального бластоза;
— морфология бластов специфична для разных типов лейкоза;
— количество промежуточных форм опухолевых клеток увеличено;
— лимфоцитоз;
— красный росток кроветворения угнетен (за исключением острого эритро-
миелоза);
— количество мегакариоцитов уменьшено (за исключением острого мегака-
риобластного лейкоза).
Цитохимическое исследование костного мозга — основной метод при диа­
гностике форм острых лейкозов. Его проводят с целью выявить специфические
для различных бластов ферменты. Так, при ОЛЛ определяется положительная
реакция на гликоген, отрицательная реакция на липиды, пероксидазу, хлорацетатэстеразу. При ОМЛ — положительная реакция на миелопероксидазу,
липиды, хлорацетатэстеразу.
Иммунофенотипирование бластов позволяет определить с помощью моно­
клональных АТ наличие или отсутствие кластеров дифференцировки бласт­
ных клеток (СЭ-маркёры). Оно необходимо для точной диагностики ОЛЛ
(см. табл. 22.6 и рис. 22.36), а также в случаях, когда нет возможности провести
дифференциальную диагностику острых морфологически недифференциру­
емых ОЛЛ и ОМЛ.
Цитогенетическое исследование лейкозных клеток позволяет определить
хромосомные аномалии, уточнить диагноз и прогноз лейкоза.
Лечение острых лейкозов
Специфическая химиотерапия. Она направлена на достижение и закрепле­
ние ремиссии заболевания, состоит из нескольких этапов, различна для ОЛЛ
и ОМЛ и проводится по стандартным схемам (в зависимости от результатов
типирования заболевания).
Сопутствующая (сопроводительная) терапия. Ее проводят для:
—профилактики или устранения инфекций, обусловленных агранулоцитозом;
—снижения степени интоксикации при лизисе опухолевых лейкоцитов;
— уменьшения побочных эффектов химиотерапии.
Заместительная терапия. Проводится при угрожающей тромбоцитопении,
тяжелой анемии, нарушениях свертывания крови.
Трансплантация стволовых кроветворных клеток или костного мозга.
Прогностически неблагоприятные признаки лейкозов:
—мужской пол;
—возраст старше 50 лет;
—мегакариобластный и эритробластный типы ОМЛ;
—РЬ-позитивный ОЛЛ;
—лейкемическая форма дебюта лейкоза (лейкоцитоз свыше 50х] 09/л);
—тромбоцитопения менее 50хЮ9/л;
—нейролейкемия;
—неадекватное предшествующее лечение («предлеченность»).
Исходы острых лейкозов
• Смерть пациента. Наиболее частые причины смерти при острых лейкозах:
—нейролейкемия;
—геморрагический синдром (ДВС);
—почечная недостаточность (при синдроме лизиса опухолевых клеток);
—сердечная недостаточность;
—инфекционные осложнения (сепсис);
—острая надпочечниковая недостаточность (вследствие гипотрофии коры
надпочечников при отмене длительно применяемых глюкокортикоидов).
• Ремиссия заболевания после курса лечения.
• Выздоровление. О полном выздоровлении говорят при отсутствии рециди­
ва в течение 5 лет после полного курса терапии.
• Рецидив заболевания. Может быть ранним и поздним: ранний — во время
лечения; поздний — после завершенного лечения.
Хронические лейкозы
Субстрат хронических лейкозов — относительно дифференцированные
клетки кроветворной ткани. Пациенты могут жить без лечения в течение
нескольких месяцев или лет.
Хронические лейкозы нередко трансформируются в острые формы.
Хронические лейкозы подразделяются на:
—миелоидные (хронический миелолейкоз — ХМЛ);
—лимфоидные (ХЛЛ и волосатоклеточный лейкоз);
—моноцитарные (миеломоноцитарные);
—эритроцитарные (истинная полицитемия);
—мегакариоцитарные.
Хронический лимфолейкоз
Большинство (85%) ХЛЛ возникает из предшественников В-лимфоцитов.
Хронический В-клеточный лимфолейкоз (В-ХЛЛ) — опухоль из СБ5+позитивных В-клеток, первично поражающая костный мозг.
Заболеваемость варьирует в различных географических регионах и этниче­
ских группах, но болеют в основном пожилые люди. В-ХЛЛ составляет около
25% всех лейкозов, встречающихся в пожилом возрасте.
Детская заболеваемость казуистична. У молодых заболевание протекает
с явными признаками злокачественности, молодой возраст считается одним
из признаков плохого прогноза.
Мужчины болеют вдвое чаще женщин.
Механизм развития хронических лимфолейкозов
На уровне предшественницы В-клетки выявляется хромосомная аберра­
ция, приводящая к трисомии хромосомы 12 либо к структурным нарушениям
хромосом 6, 11, 13 или 14.
Патологические опухолевые клетки частично дифференцируются — лишь
до уровня рециркулирующих В-клеток или В-клеток памяти. Последующие
деления генетически нестабильных лимфоцитов приводят к появлению новых
мутаций и, соответственно, новых субклонов лейкозных клеток. Лейкоз может
сопровождаться появлением моноклональных 1§М или 1§С.
Виды хронических лимфолейкозов
ХЛЛ подразделяют на В- и Т-формы.
Проявления хронических лимфолейкозов
• Лимфоцитоз.
• Лимфаденопатия (увеличение лимфатических узлов).
• Спленомегалия. При этом симптоме прогноз лейкоза становится неблаго­
приятным (пациенты обычно живут 4—7 лет).
• Гемолитическая анемия (преимущественно иммунная аутоагрессивная).
• Тромбоцитопения (иммуноаутоагрессивного генеза).
• Анемический и геморрагический синдромы (обусловлены поражением кост­
ного мозга и появлением АТ к эритроцитам и тромбоцитам).
• Инфекционные осложнения: бактериальные, грибковые, вирусные инфек­
ции (вызваны гипогаммаглобулинемией, нарушениями в клеточном звене
иммунитета).
• Аллергические реакции немедленного типа (например, после вакцинации
или укусов комаров).
Лабораторные признаки хронических лимфолейкозов
Периферическая кровь
• Лейкоцитоз, абсолютный лимфоцитоз.
• Наличие в мазке крови «теней Боткина—Гумпрехта» (полуразрушенные ядра
лейкозных лимфоцитов как проявление опухолевого атипизма) — харак­
терного признака лейкозных клеток при ХЛЛ.
• Нормоцитарная нормохромная анемия.
• Ретикулоцитоз.
• Тромбоцитопения.
При иммунофенотипировании:
— экспрессия Аг СБ5 и СБ23 опухолевыми клетками;
— уменьшение содержания всех классов 1§;
— наличие в моче белка Бенс-Джонса.
При серологическом исследовании: наличие антиэритроцитарных и/или антитромбоцитарных 1§.
Пунктат костного мозга:
• избыточное число лимфоцитов в кроветворной ткани (вплоть до тотальной
лимфатической метаплазии);
• трисомия 12, деления 1 ^ , 13д, 14д, 6^, 16р при цитогенетическом анализе
опухолевых клеток.
Лечение больных хроническими лимфолейкозами
Радикальных способов лечения ХЛЛ в настоящее время нет.
• Лучевая терапия — один из основных методов лечения ХЛЛ.
• Спленэктомия — метод купирования панцитопении при ХЛЛ. Показана
при развитии иммунопатологических осложнений.
• Введение глюкокортикоидов. Монотерапия глюкокортикоидами при ХЛЛ
применяется в случаях тяжелых иммуных аутоагрессивных осложнений,
так как они усугубляют имеющийся иммунодефицит и могут стать при­
чиной фатальных септических осложнений.
• Применение алкилирующих химиотерапевтических средств (например,
хлорамбуцила, циклофосфамида). Используют при прогрессирующем
течении ХЛЛ.
• Аналоги пурина (например, флударабин) — приводит к ремиссиям у боль­
ных с тяжелой прогрессирующей и опухолевой формами ХЛЛ.
• Полихимиотерапевтические схемы. При устойчивости к алкилирующим
средствам назначают комбинацию препаратов, включающую циклофосфамид, винкристин, преднизолон и др.
• Использование биологических агентов: моноклональных АТ к Аг опухо­
левых клеток, например ритуксимаба (мабтера*), алемтузумаба (кэмпас*)
и их комбинаций в полихимиотерапевтических схемах.
Хронический миелолейкоз
ХМЛ — опухоль, развивающаяся из полипотентной клетки-предшественницы.
Ее пролиферация и дифференцировка приводит к расширению ростков
кроветворения, представленных (в отличие от острых лейкозов) преимуще­
ственно зрелыми и промежуточными формами лейкозных клеток.
Распространенность
ХМЛ — наиболее распространенный из всех лейкозов. На него приходится
около 20% случаев гемобластозов у взрослых и примерно 5% у детей.
Этиология и патогенез
Канцерогенные агенты на уровне клетки-предшественницы вызывают
транслокацию I (9; 22).
Этим обусловлено появление так называемой филадельфийской хромосо­
мы и экспрессия мутантного гена Ьсг-аЫ, который кодирует белок р210, обла­
дающий свойствами тирозинкиназы.
Экспансия РЬ-позитивных клеток в костном мозге, периферической крови
и экстрамедуллярных зонах объясняется не столько их высокой пролиферативной активностью, сколько расширением пула гранулоцитарных предшествен­
ников, утерявших чувствительность к регуляторным стимулам и изменениям
микроокружения. Это приводит к их диссеминации, нарушению продукции
цитокинов и подавлению нормального гемопоэза.
Транслокация I (9; 22) является диагностической для ХМЛ. При ее отсут­
ствии заболевание, характеризующееся клиническими, морфологическими
и цитохимическими признаками ХМЛ, определяется как атипичный ХМЛ.
Стадии и диагностика
В клиническом течении ХМЛ выделяют развернутую и терминальную стадии.
Развернутая стадия хронического миелолейкоза
Диагноз развернутой стадии ХМЛ устанавливают исходя из наличия «немо­
тивированной» природы нейтрофильного лейкоцитоза со сдвигом формулы
до миелоцитов и промиелоцитов, существенно повышенного соотношения
лейкоциты/эритроциты в костном мозге, «филадельфийской» хромосомы
в гранулоцитах крови и клетках костного мозга.
В трепанате костного мозга уже в этот период, как правило, наблюдается
почти полное вытеснение жира опухолевыми клетками миелоидной ткани.
Терминальная стадия ХМЛ
Для этой стадии характерно появление и интенсивное нарастание признаков
подавления нормальных ростков кроветворения:
— анемия;
— тромбоцитопения с геморрагическим синдромом;
— гранулоцитопения, осложняющаяся инфекцией и некрозами слизистых
оболочек.
Основной гематологический признак терминальной стадии ХМЛ — власт­
ный криз: увеличение содержания бластных клеток в костном мозге и крови (сна­
чала миелобластов, затем — морфологически недифференцируемых бластов).
В терминальной стадии более чем в 80% случаев появляются анеуплоцдные клоны
кроветворных клеток, содержащих аномальное число хромосом. Продолжительность
жизни больных в этой стадии обычно не превышает 6—12 мес.
Лечение хронического миелобластного лейкоза
• Химиотерапия (позволяет снизить количество лейкоцитов крови до
20x109/л).
• Облучение селезенки (проводят при значительной спленомегалии; умень­
шает степень цитопении).
• Применение блокатора мутантной тирозинкиназы (р210) — иматиниба (гливек*, 8Т1-571). Препарат обеспечивает полную ремиссию заболевания
без эрадикации опухолевого клона.
• Трансплантация стволовых клеток крови или красного костного мозга
(более чем в 70% случаев способствует ремиссии).
Глава 23
ТИПОВЫЕ ФОРМЫ ПАТОЛОГИИ
СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ
Сердечно-сосудистая система (ССС) — компонент системы крово- и лим­
фообращения. В нее, помимо сердца и сосудов (т.е. собственно ССС), входят
кровь и лимфа.
Система крово- и лимфообращения — одна из интегрирующих систем
организма. В ней различают центральное и периферическое звено.
Центральное кровообращение осуществляется на уровне сердца, а также
крупных сосудов и обеспечивает поддержание системного давления крови,
направление движения крови из артериального русла в венозное и далее
к сердцу, демпфирование значительных (систолических и диастолических)
колебаний АД при выбросе крови из желудочков сердца.
Периферическое кровообращение (органотканевое, местное, регионарное)
осуществляется в сосудах отдельных органов и тканей и обеспечивает ток
крови в них в соответствии с их постоянно меняющейся функциональной
активностью и уровнем пластических процессов.
Периферическое кровообращение включает и кровообращение в сосудах
микроциркуляторного русла. Оно осуществляется в артериолах, прекапиллярах, капиллярах, посткапиллярах, венулах и артериоловенулярных шунтах
органов и тканей. В целом это звено системы кровообращения осуществляет
доставку крови к тканям, транскапиллярный обмен метаболитами, 0 2 и С 02,
а также транспорт крови от тканей.
В норме ССС оптимально обеспечивает текущие потребности органов
и тканей в кровоснабжении, а уровень системного кровообращения определя­
ется деятельностью сердца, тонусом сосудов и состоянием крови (величиной
ее общей и циркулирующей массы, а также реологическими свойствами).
Нарушения функции сердца, сосудистого тонуса или изменения в системе
крови могут привести к недостаточности кровообращения.
Не достаточ ность
кровообращения
Недостаточность кровообращения — состояние, при котором система крово­
обращения не обеспечивает потребности тканей и органов в кровоснабжении.
Причины недостаточности кровообращения
К основным причинам недостаточности кровообращения относят:
— расстройства сердечной деятельности;
— нарушения тонуса стенок кровеносных сосудов;
— изменения ОЦК и/или реологических свойств крови.
Виды недостаточности кровообращения
Все разновидности недостаточности кровообращения классифицируют
по критериям компенсированности расстройств, остроте их развития и тече­
ния, выраженности их признаков.
Компенсированность расстройств кровообращения
По компенсированности все расстройства кровообращения подразде­
ляют на:
—компенсированные (признаки расстройств кровообращения выявляются
при нагрузке);
—некомпенсированные (признаки нарушения кровообращения обнаружи­
ваются в покое).
Острота развития и течения недостаточности кровообращения
По остроте развития и течения выделяют:
— острую недостаточность кровообращения (развивается в течение часов
и суток); наиболее частые причины ее — инфаркт миокарда, острая сер­
дечная недостаточность (ОСН), некоторые аритмии (пароксизмальная
тахикардия, выраженная брадикардия, мерцательная аритмия и др.),
шок, острая кровопотеря;
— хроническую (развивается на протяжении нескольких месяцев
или лет); причинами ее являются перикардиты, длительно текущие
миокардиты, миокардиодистрофии, кардиосклероз, пороки сердца,
гипер- и гипотензивные состояния, анемии, гиперволемии различно­
го генеза.
Выраженность признаков недостаточности кровообращения
По выраженности признаков выделяют три стадии недостаточности кро­
вообращения.
• Стадия I (начальная), или недостаточность кровообращения I степени.
Признаки (выявляются при физической нагрузке и отсутствуют в покое):
—уменьшение скорости сокращения миокарда;
—снижение фракции выброса;
—одышка;
—сердцебиение;
—быстрая утомляемость.
• Стадия II, или недостаточность кровообращения II степени (умеренно
или значительно выраженная недостаточность кровообращения). Указанные
для начальной стадии признаки недостаточности кровообращения обнаружи­
ваются не только при физической нагрузке, но и в покое.
• Стадия III (конечная), или недостаточность кровообращения III степени.
Характеризуется значительными нарушениями сердечной деятельности
и гемодинамики в покое, а также развитием существенных дистрофических
и структурных изменений в органах и тканях.
Факторы риска
сердечно-сосудистой патологии
Сердечно-сосудистые заболевания занимают первое место среди причин
инвалидизации и гибели человека («убийца №1»).
Высокий уровень заболеваемости, инвалидизации и летальности от болез­
ней ССС в значительной мере определяется широкой распространенностью
различных форм патологии сердца, и прежде всего ИБС. В промышленно
развитых странах 15—20% взрослого населения страдает ИБС. Последняя,
в свою очередь, является причиной внезапной смерти 2/3 пациентов, умерших
от сердечно-сосудистых заболеваний. Около половины страдающих этими
болезнями становятся инвалидами в трудоспособном возрасте. Постоянно
увеличивается заболеваемость и смертность от них среди молодого населения
(до 35 лет), а также в сельских местностях.
К основным факторам, определяющим высокую заболеваемость сердечно­
сосудистой патологией, относятся:
— повторные и затяжные стрессорные эпизоды;
— хроническая гиподинамия;
— интоксикация алкоголем, курение, избыточное потребление чая, кофе
и других «бытовых допингов»;
— некачественное, несбалансированное питание и переедание, ожирение.
Всего известно не менее 50 факторов риска, существенно влияющих
на возникновение кардиоваскулярной патологии.
Типовые формы патологии
сердечно-сосудистой системы
Патологические процессы ССС объединяются в две группы типовых форм
патологии.
• Нарушения центрального кровообращения. Они обусловлены патологией
сердечной деятельности и магистральных сосудов.
• Расстройства периферического кровообращения, включая нарушения микро­
циркуляции.
Большинство расстройств сердечной деятельности относится к трем группам
типовых форм патологии:
1) коронарной недостаточности;
2) аритмиям;
3) сердечной недостаточности.
КОРОНАРНАЯ НЕДОСТАТОЧНОСТЬ
Иоронариая недостаточность — типовая оормэ патологии сердца. Характе­
ризуется тем, что потребность миокарда в кислороде и субстратах метаболиз­
ма превышает их приток по коронарным артериям, а также нарушается отток
от миокарда продуктов обмена веществ, БАВ, ионов и других агентов.
Главный патогенетический фактор коронарной недостаточности — ишемия
миокарда.
Клинически коронарная недостаточность проявляется как ишемическая
болезнь сердца (ИБС). При поражении венечных артерий может развиться
стенокардия, инфаркт миокарда или внезапная сердечная смерть.
Виды коронарной недостаточности
Все разновидности коронарной недостаточности подразделяются на обра­
тимые и необратимые (рис. 23.1).
Обратимые нарушения коронарного кровотока
Обратимые (транзиторные) нарушения коронарного кровотока клиниче­
ски проявляются:
—различными формами стенокардии;
—состояниями после реперфузии (реваскуляризации) миокарда.
Стенокардия
Стенокардия — типовая форма коронарной недостаточности в результате
ишемии миокарда; характеризуется ощущением боли, чаще в области грудины
слева.
Боль нередко иррадиирует в область левой лопатки и левого плеча.
Разновидности стенокардии
• Стенокардия стабильного (типичного) течения. Наиболее часто встреча­
ющаяся разновидность стенокардии. Обычно бывает следствием сниже­
ния коронарного кровотока до критического уровня. Эпизоды стенокар­
дии развиваются в результате увеличения работы сердца.
• Стенокардия нестабильного течения (нарастающая, нестабильная). Характе­
ризуется нарастающими по частоте, длительности и тяжести эпизодами
стенокардии, нередко даже в покое. Эти эпизоды являются обычно резуль­
татом разрушения атеросклеротической бляшки и развития тромба на месте
дефекта, эмболии коронарной артерии, спазма ветви венечной артерии
сердца. Нередко эти эпизоды пролонгированы во времени и завершаются
инфарктом миокарда. В связи с этим такие эпизоды называют предын­
фарктными состояниями.
• Вариантная стенокардия (стенокардия Принцметала). Результат длительного
спазма коронарных артерий. Существенно, что повторные (даже кратко­
временные — до 3—8 мин) эпизоды стенокардии могут привести к форми­
рованию небольших участков некроза миокарда с последующим развити­
ем мелкоочагового кардиосклероза.
Клинические разновидности стенокардии приведены в статье «Болезнь.
Ишемическая болезнь сердца».
Состояния после реперфузии (реваскуляризации) миокарда
Они развиваются у пациентов с ИБС в результате хирургического воз­
обновления или значительного усиления коронарного кровотока (напри-
102
Патофизиология, клиническая патофизиология
Рис. 23.1. Виды, клинические формы и исходы коронарной недостаточности
мер, после аортокоронарного шунтирования, чрескожной внутрисосудистой
ангиопластики, стентирования коронарных артерий) или медикаментозного
восстановления тока крови в коронарных артериях (например, в результате
тромболизиса, дезагрегации форменных элементов крови с помощью тромбои фибринолитиков или дезагрегантов).
Необратимые нарушения коронарного кровотока
Необратимое прекращение или длительное значительное уменьшение при­
тока крови по коронарной артерии в каком-либо регионе сердца завершается,
как правило, инфарктом миокарда.
Инфаркт миокарда — типовая форма коронарной недостаточности.
Представляет собой очаговый некроз миокарда в результате острого и зна­
чительного дисбаланса между потребностью его в кислороде, субстратах мета­
болизма и их абсолютно или относительно недостаточной доставкой.
Наиболее частая причина инфаркта миокарда — тромбоз венечной артерии,
развившийся на фоне атеросклеротических изменений (до 90% всех случаев).
Один из клинических диагностических критериев инфаркта миокарда —
сильный болевой синдром («кинжальная боль» за грудиной) продолжитель­
ностью более 15—20 мин, не купирующийся нитроглицерином.
При развитии инфаркта миокарда возникают изменения сегмента 5Т
и зубца Т — депрессия или подъем сегмента 8Т и инверсия зубца Т. Подъем
сегмента 8 Т — характерный признак инфаркта миокарда, свидетельствующий
о повреждении миокарда. Через 8-12 ч после начала боли на ЭКГ возникает
важный признак инфаркта миокарда — патологический зубец 0 (характеризу­
ющий наличие некроза миокарда).
При инфаркте миокарда нередко развиваются опасные для жизни ослож­
нения:
-О С Н ;
— кардиогенный шок;
— отек легких;
— постинфарктный синдром;
— разрыв стенки желудочка или межжелудочковой перегородки, аневризма
стенок сердца;
— недостаточность митрального клапана;
—аритмии (синусовая брадикардия, АВ-блокада, желудочковые наруше­
ния ритма сердца, наджелудочковые нарушения ритма сердца, в том
числе фибрилляция предсердий);
—тромбоэмболия. Клинически тромбоэмболия артерий может проявляться
гемипарезами (эмболия артерий мозга), стойкой артериальной гипер­
тензией и гематурией (почечные артерии), болями в животе (брыжееч­
ные артерии), болями в ногах (бедренные артерии) и тромбоэмболией
легочной артерии.
Если инфаркт не приводит к смерти пациента, то погибший участок сердца
замещается соединительной тканью — развивается крупноочаговый кардио­
склероз.
Причины коронарной недостаточности
Многочисленные состояния и факторы, способные вызвать коронарную
недостаточность, объединены в три основные, как правило, взаимосвязанные
группы. Они представлены на рис. 23.2.
П ричины коронарной н е д о статочн ости
4
г
Уменьшающие и/или
прекращающие приток
крови к миокарду по
коронарным артериям
V
Повышающие расход
миокардом кислорода
и/или субстратов
обмена веществ
Снижающие содержание
кислорода и/или субстратов
обмена веществ
в крови и миокарде
/V
Коронарогенные
У
Некоронарогенные
Рис. 23.2. Группы причин коронарной недостаточности
Уменьшение и/или прекращение притока крови к миокарду как причина коро­
нарной недостаточности
Ниже перечислены факторы, приводящие к абсолютному снижению при­
тока крови к миокарду (рис. 23.3) по коронарным артериям (в практике врача
они встречаются наиболее часто).
Факторы, уменьшающие или прекращающие
приток крови к миокарду по коронарным артериям
Атеросклероз
коронарных
артерий
Агрегаты форменных
элементов крови и тромбы
в коронарных артериях
Спазм
коронарных
артерий
Уменьшение притока
к сердцу крови и сни­
жение перфузионного
давления в коронарных
артериях
Коронарная недостаточность |*~
Рис. 23.3. Факторы, уменьшающие или прекращающие приток крови к миокарду
по коронарным артериям
• Атеросклеротическое поражение коронарных артерий. У 92% пациентов
со стенокардией на коронароангиограммах выявляются значительные
локальные сужения просвета как минимум одной из венечных артерий
сердца.
• Агрегация форменных элементов крови (главным образом эритроцитов и тром­
боцитов) и образование тромбов в венечных артериях сердца. Этим процес­
сам в значительной мере способствуют:
—атеросклеротические изменения в стенках сосудов;
—турбулентный характер кровотока в венечных сосудах;
—повышение содержания и активности факторов свертывающей системы
крови, высвобождающихся из поврежденных клеток крови и сосудистой
стенки.
Указанные факторы дополнительно стимулируют агрегацию и адгезию тром­
боцитов, эритроцитов и лейкоцитов, высвобождение из них БАВ, потен­
цирующих, в свою очередь, клеточную агрегацию и тромбообразование
как в просвете, так и на внутренней поверхности коронарных артерий.
• Спазм коронарных артерий. Развитие коронарной недостаточности в резуль­
тате сосудистого спазма доказано ангиографически. Решающее значение
в развитии коронароспазма имеют катехоламины. Значительное увели­
чение их содержания в крови или повышение адренореактивных свойств
сосудов миокарда, как правило, сопровождается всеми клиническими,
электрокардиографическими и биохимическими изменениями, свой­
ственными стенокардии.
• Уменьшение притока крови к сердцу и снижение в связи с этим перфузионного
давления в коронарных артериях. К этому приводит значительная бради-
или тахикардия, трепетание и мерцание предсердий и/или желудочков
сердца, недостаточность аортальных клапанов, острая артериальная гипо­
тензия, сдавление коронарных артерий сердца (опухолью, рубцом, ино­
родным телом).
Как правило, коронарная недостаточность является результатом действия
комплекса взаимосвязанных факторов:
— сужения просвета коронарных артерий в результате утолщения их стенок
(вследствие атеросклеротических изменений, гипертрофии мышечной
оболочки, фиброзных изменений, отека и др.);
— сокращения гладкомышечных клеток (ГМК) коронарных артерий и умень­
шения их просвета под влиянием катехоламинов, тромбоксана А2, ПГР2
и других вазоконстрикторов;
— сужения и закрытия просвета сосуда агрегатами форменных элементов
крови и тромбами.
Эти представления суммированы в концепции о динамическом стенозе коро­
нарных артерий (рис. 23.4).
Рис. 23.4. Основные звенья в механизме динамического стеноза венечных артерий сердца
Повышение расхода миокардом кислорода и/или субстратов обмена веществ
как причина коронарной недостаточности
К основным факторам, вызывающим значительно большее потребление
миокардом кислорода и субстратов обмена веществ (рис. 23.5) и тоже приво­
дящим к коронарной недостаточности, относят следующие.
Рис. 23.5. Факторы, увеличивающие потребление миокардом кислорода и субстратов
обмена веществ
• Существенное повышение в сердце уровня катехоламинов (например,
при стрессе или феохромоцитоме). К механизмам кардиотоксического
действия избытка катехоламинов в миокарде (рис. 23.6) относят:
—чрезмерное потребление 0 2 и субстратов метаболизма миокардом.
Последнее связано с положительным хроно- и инотропным эффектом
катехоламинов и значительным возрастанием в связи с этим функции
сердца;
—снижение эффективности ресинтеза АТФ и в связи с этим расхода кис­
лорода и субстратов окисления. Последнее вызвано:
о повреждением мембранного аппарата кардиомиоцитов (прежде всего,
плазматической мембраны, митохондрий);
о инактивацией ферментов тканевого дыхания, гликолиза, пентозофосфатного шунта (при этом мембраны и ферменты альтерируются сво­
бодными радикалами, продуктами нарушенного метаболизма и перекисного окисления липидов; образование этих продуктов стимулиру­
ют катехоламины, а также активированные гидролазы лизосом);
о разобщением окислительного фосфорилирования, в основном вслед­
ствие избыточного накопления в кардиомиоцитах (под влиянием кате­
холаминов) ВЖК и ионов кальция;
—уменьшение (в сравнении с потребным) величины коронарного кро­
вообращения. Это является результатом укорочения (в условиях катехоламиновой тахикардии) диастолы, в течение которой приток крови
к миокарду максимален, большего напряжения миокарда и сдавления
в связи с этим коронарных сосудов, усиления агрегации клеток крови
в просвете микрососудов.
В целом, развитие коронарной недостаточности при чрезмерной акти­
вации симпатико-адреналовой системы характеризуется повышенным рас­
ходом 0 2 и метаболитов гиперфункционирующим миокардом, а также
ограничением их притока к миокарду по коронарным артериям.
• Значительное возрастание работы сердца. Наиболее часто является след­
ствием чрезмерной физической нагрузки, длительной тахикардии, острой
артериальной гипертензии, выраженной гемоконцентрации, значитель­
ной гиперволемии.
Важно, что чрезмерное увеличение работы сердца, а также причины, вызвавшие
ее, как правило, одновременно приводят и к активации симпатико-адреналовой
системы. Последнее сопровождается высвобождением избытка катехоламинов
и их кардиотоксическим действием.
Уменьшение содержания кислорода и/или субстратов
метаболизма в крови и клетках миокарда как причина коронарной
недостаточности
Состояния, при которых значительно уменьшается содержание кислорода
и/или субстратов обмена веществ в крови и клетках миокарда, приведены
на рис. 23.7.
Избыток катехоламинов
>
1
Чрезмерное расходование
Менее эффек­
миокардом кислорода
тивное действие
механизмов
и субстратов обмена
ресинтеза АТФ
веществ за счет
положительных хронои инотропного эффектов
Г
Уменьшение
объема
коронарного
кровотока
У
Vу
----------------------------------------- ►] 1 ювреждение миокарда
Образование
избытка активных
форм кислорода
и перекисей липидов
---------------- —
Рис. 23.6. Механизмы кардиотоксического действия избытка катехоламинов в миокарде
Рис. 23.7. Состояния, при которых в крови и/или кардиомиоцитах уменьшается
содержание кислорода и субстратов метаболизма
Механизмы повреждения сердца
при коронарной недостаточности
Недостаток кислорода и субстратов обмена веществ в миокарде, а также
нарушение оттока продуктов нарушенного метаболизма, ионов, БАВ в усло­
виях коронарной недостаточности активизируют ряд типовых механизмов
повреждения миокарда (рис. 23.8).
Рис. 23.8. Механизмы повреждения сердца при коронарной недостаточности
Общие механизмы повреждения клеток подробно рассматриваются в раз­
деле «Общие механизмы повреждения» (см. главу 5 «Повреждение клетки»,
т. 1). Указанные механизмы (расстройство энергетического обеспечения кардиомиоцитов, повреждение их мембран и ферментов, дисбаланс ионов и жид­
кости, изменение механизмов регуляции сердечной деятельности) действуют
как в зоне ишемии, так и за ее пределами, хотя в последней в существенно
меньшей мере.
Расстройство энергообеспечения кардиомиоцитов
при коронарной недостаточности
Механизмы расстройств энергообеспечения кардиомиоцитов при коро­
нарной недостаточности представлены на рис. 23.9.
В аэробных условиях основными субстратами для синтеза АТФ служат
жирные кислоты (65—70%), глюкоза (15—20%) и МК (10-15%). Роль амино­
кислот, кетоновых тел (КТ) и пирувата в энергообеспечении миокарда срав­
нительно невелика.
В условиях нарастающей ишемии в миокарде истощается связанный с миоглобином «резерв» кислорода и снижается интенсивность окислительного
фосфорилирования. В результате указанных изменений в кардиомиоцитах
уменьшается содержание АТФ и креатинфосфата (рис. 23.10).
Нарушение аэробного синтеза АТФ приводит к активации гликолиза
и накоплению в миокарде лактата. Одновременно быстро уменьшаются запа­
сы гликогена.
Активация гликолитического метаболизма углеводов приводит к метабо­
лическому ацидозу
Развитие внутри- и внеклеточного ацидоза существенно изменяет прони­
цаемость мембран для метаболитов и ионов, подавляет активность ферментов
энергообеспечения, синтеза клеточных структур, транспорта субстратов мета­
болизма и катионов.
Ишемия миокарда, мин
I
Несколько секунд
♦
Начало снижения
уровня АТФ
1 -2
V
Прекращение сократительной
функции
I
2 0 -4 0
«10
«20
V
+
Снижение уровня АТФ Снижение уровня АТФ Необратимое повреждение
кардиомиоцитов
(«50% от нормального) (=90% от нормального)
Рис. 23.10. Изменения в клетках миокарда в зависимости от продолжительности ишемии
Глава 23. Типовые формы патологии сердечно-сосудистой системы
Рис. 23.9. Механизмы нарушения энергетического обеспечения миокарда при коронарном недостаточности (период ишемии
и начальный этап рсперфузии): АНТ — адениннуклеотидтрансфераза
109
В отдаленных от зоны ишемии участках сердца процессы энергетического
обеспечения миокарда страдают в значительно меньшей мере.
Главные последствия, вызванные расстройствами процессов энергообеспече­
ния кардиомиоцитов:
— снижение сократительной функции миокарда;
— нарушения кровообращения в органах и тканях;
— развитие сердечных аритмий. Нарушения ритма сердца, в свою очередь,
нередко становятся причиной внезапной смерти пациентов с коронарной
недостаточностью.
П овреждение мембран и ферментов кардиомиоцитов
при коронарной недостаточности
Основные свойства миокарда (автоматизм, возбудимость, проводимость,
сократимость), а также их регуляция в значительной мере зависят от состояния
мембран и ферментов кардиомиоцитов. В условиях ишемии их повреждение
является следствием действия нескольких общих факторов. Основные меха­
низмы повреждения клеточных мембран и ферментов рассмотрены в разделе
«Общие механизмы повреждения» главы 5 «Повреждение клетки» (подраздел
«Повреждение мембран и ферментов») в томе 1 и приведены на рис. 23.11.
Дисбаланс ионов и жидкости в миокарде
при коронарной недостаточности
Ионный дисбаланс (рис. 23.12, см. также раздел «Дисбаланс ионов и воды
в клетке» в главе 5 «Повреждение клетки», т. 1) развивается вслед за рас­
стройствами энергообеспечения кардиомиоцитов и повреждением их мембран
и ферментов или одновременно с ними.
Дисбаланс ионов и жидкости, развивающийся при ишемии миокарда,
относится как к общему содержанию ионов и жидкости в клетках и ткани
миокарда в целом, так и к внутри- и внеклеточному соотношению отдельных
ионов (прежде всего Ыа+, К+, С1~, Са2+), а также к внутриклеточному соот­
ношению и распределению (компартментализация) отдельных ионов.
Общее содержание ионов в клетках ишемизированного миокарда суще­
ственно возрастает.
Внутри- и внеклеточное соотношение и распределение отдельных ионов,
а также жидкости в кардиомиоцитах значительно изменяется (см. рис. 5.9 в
томе 1; рис. 23.13).
Наиболее важны следующие изменения содержания ионов и жидкости:
— увеличение [К+] вне кардиомиоцитов (вследствие снижения активности
№ +-/К +-АТФазы, дефицита АТФ, повышения проницаемости плазма­
тической мембраны);
— повышение содержания ионов № + в кардиомиоцитах;
— увеличение [Са2+] в клетках миокарда;
— расстройство регуляции объема клеток миокарда.
Потеря К+ кардиомиоцитами сопровождается повышением его содержа­
ния в интерстициальной жидкости и крови. В связи с этим гиперкалиемия
является одним из характерных признаков коронарной недостаточности, осо­
бенно при инфаркте миокарда.
Ишемия миокарда, начальный этап его реперфузии
Изменение
общего содержания
ионов и жидкости
в клетках и ткани
миокарда
внутри- и внеклеточного
соотношения отдельных
ионов
внутриклеточного
соотношения и
распределения
ионов
Глава 23. Типовые формы патологии сердечно-сосудистой системы
Рис. 23.11. Механизмы повреждения мембран и ферментов клеток миокарда при коронарной недостаточности (период ишемии
и начальный этап реперфузии)
Рис. 23.12. Дисбаланс ионов и жидкости в миокарде при коронарной недостаточности (период ишемии и начальный этап реперфузии)
111
Рис. 23.13. Нарушение внутри- и внеклеточного соотношения ионов и жидкости в клет­
ках миокарда при его ишемии
Указанные факторы влекут за собой накопление избытка жидкости в клет­
ках миокарда и существенное увеличение их объема.
Последствия дисбаланса ионов и жидкости в миокарде при коронарной
недостаточности представлены на рис. 23.14.
Рис. 23.14. Последствия дисбаланса ионов и жидкости в миокарде при коронарной
недостаточности (период ишемии и начальный этап реперфузии)
Дисбаланс ионов и жидкости вызывает нарушение ряда фундаментальных про­
цессов (прежде всего электрогенеза и сократительных характеристик), протекаю­
щих в клетках миокарда (см. рис. 5.11 в томе 1). При ишемии миокарда страдают
все процессы мембранного электрогенеза: возбудимость клеток миокарда, авто­
матизм ритмогенеза и проведение импульсов возбуждения. В связи с существен­
ными отклонениями мембранного электрогенеза развиваются аритмии сердца.
Расстройства механизмов регуляции сердечной деятельности
при коронарной недостаточности
Функции сердца в целом, а также характер и степень повреждения его кле­
ток при коронарной недостаточности изменяются в результате не только пря­
мой их альтерации патогенными факторами ишемии. В значительной мере эти
изменения обусловлены и расстройствами механизмов регуляции сердечной
деятельности. Эти расстройства развиваются на одном (реже) или нескольких
(чаще) уровнях регуляции:
—взаимодействия БАВ (гормоны, нейромедиаторы) с рецепторами;
—образования клеточных (вторых) посредников регуляторных влияний;
—метаболических клеточных реакций, регулируемых внутриклеточными
посредниками.
Подробнее эти вопросы рассмотрены в разделе «Расстройства регуляции
внутриклеточных процессов» (см. главу 5 «Повреждение клетки», т. 1).
Коронарная недостаточность характеризуется стадийно изменяющейся актив­
ностью симпатического и парасимпатического механизма регуляции.
На начальном этапе ишемии миокарда, как правило, наблюдается значи­
тельная активация симпатико-адреналовой системы. Это сопровождается уве­
личением содержания в миокарде норадреналина и особенно адреналина.
Вследствие этого развивается тахикардия, увеличивается величина сердечного
выброса, как правило, снижающегося сразу после того, как начался приступ
коронарной недостаточности.
Параллельно с этим могут усиливаться и парасимпатические влияния
(о чем свидетельствует возросшее содержание ацетилхолина в миокарде),
но степень их усиления меньше, чем симпатических.
Значительно позже (через несколько десятков минут, иногда часов после
начала коронарной недостаточности) нередко регистрируется меньшее содержа­
ние в миокарде норадреналина и сохраняется повышенный уровень ацетилхоли­
на. Одновременно отмечается брадикардия, снижается величина сердечного
выброса, скорость сокращения и расслабления миокарда.
В условиях коронарной недостаточности (особенно при длительной ишемии
и на начальном этапе реперфузии миокарда) развивается феномен гормональнонейромедиаторной диссоциации катехоламинов (соотношение нейромедиатора
норадреналина и гормона адреналина). Для этого феномена характерно:
- значительное увеличение в ишемизированном миокарде концентрации адре­
налина и проявление его кардиотоксического действия. Механизм проокси-
дантного действия избытка адреналина в сердце приведен на рис. 23.15;
- одновременное уменьшение содержания в миокарде норадреналина.
Избыток алоеналина в миокаоле---------------------’
Аутоокисление Увеличение транспорта
адреналина
Са2' в кардиомиоциты
в адренохром
Трансформация
ксантиндегидрогеназы
1 г
•Г
г
Подавление
активности
ферментов
антиоксидантной
защиты сердца
Усиление
гидролиза АТФ
Накопление
избытка
адениннуклеозидов
’Г
ГО
с "
I!
К Ц
X О
1 °
<
Ксантин
мочевая кислота
*■ Генерация избытка активных форм кислорода
_______
I______________
Накопление избытка субстратов СПОЛ
--------------------------------
_________ ^_________
| Интенсификация СПОЛ |
Рис. 23.15. Механизмы прооксидантного эффекта избытка адреналина в миокарде
Феномен гормонально-нейромедиаторной диссоциации катехоламинов сопро­
вождается потенцированием ишемического и реперфузионного повреждения мио­
карда (см. «Эффекты постокклюзионной реперфузии миокарда»).
Коронарная недостаточность сопровождается и другими изменениями
нейрогуморальной регуляции функции сердца, но они весьма индивидуальны
(в зависимости от длительности эпизода коронарной недостаточности, числа
их в анамнезе, возраста пациента, выраженности сердечной недостаточности
и т.д.) и рассматриваются в клинических руководствах.
Эффекты постокклюзионной реперфузии миокарда
Наиболее распространенная форма коронарной недостаточности — стено­
кардия — характеризуется спонтанным или вызванным медикамеитозпо
чередованием более или менее длительного периода ишемии миокарда
и периода возобновляющегося коронарного кровотока — реперфузии.
Состояния, обусловленные постокклюзионной (постстенотической) репер­
фузией миокарда, в последние десятилетия значительно участились в связи
с внедрением в клиническую практику различных ангиопластических (см.
статью «Ангиопластика» в приложении «Справочник терминов») и/или меди­
каментозных (фибринолизис, дезагрегация клеток крови, вазодилатация и др.)
методов, позволяющих устранить стеноз или окклюзию магистральных ветвей
коронарных артерий.
Постишемическая реперфузия миокарда
Возобновление тока крови (реперфузия) — самый эффективный способ
прекратить действие патогенных факторов ишемии миокарда и устранить
последствия их влияния на сердце.
Доказано, что постишемическая реперфузия:
- препятствует развитию инфаркта миокарда;
—предотвращает формирование аневризмы в ранее ишемизированной
зоне сердца;
- способствует образованию соединительной ткани в стенке аневризмы,
если она развилась;
- потенцирует восстановление сократительной функции сердца.
Вместе с тем, начальный этап постокклюзионной реперфузии коронарных сосу­
дов нередко сопровождается существенными расстройствами функции сердца:
- развитием аритмий, включая фибрилляцию желудочков, что чревато
смертью пациента;
— преходящей дестабилизацией центрального и органотканевого кровообра­
щения;
—дисбалансом биохимических параметров миокарда;
— нарушением электрофизиологических свойств сердца.
Таким образом, на раннем этапе реперфузии возможно пролонгирование
и даже потенцирование повреждения реперфузируемого участка сердца.
В связи с этим сформулировано положение о том, что коронарная недоста­
точность является совокупностью двух синдромов — ишемического и реперфузионного, а не только одного — ишемического, как считалось ранее.
Реперфузионное повреждение миокарда
Постокклюзионная реперфузия коронарных артерий оказывает наряду
с основным репаративным, восстановительным эффектом также и патогенное
действие на миокард. Последнее является суммарным следствием:
—пролонгирования его ишемического повреждения;
—дополнительной альтерации миокарда факторами реперфузии и реоксигенации.
К основным механизмам дополнительного реперфузионного повреждения кле­
ток миокарда относят:
— усугубляющееся нарушение энергетического обеспечения клеток реперфузируемого миокарда на этапах ресинтеза, транспорта, утилизации энер­
гии АТФ (см. рис. 23.9). Основные причины этого:
о гипергидратация, набухание и разрушение митохондрий в реперфузируемом миокарде (является результатом осмотического отека органелл,
перерастяжения и разрыва их мембран в связи с избыточным накопле­
нием в них Са2+ и жидкости);
о разобщающий эффект избытка Са2+;
о выход АДФ, АМФ и других пуриновых соединений из митохондрий
кардиомиоцитов в межклеточную жидкость;
— нарастание степени повреждения мембран и ферментов клеток миокарда
(см. рис. 23.11); причины:
о реперфузионная (кислородзависимая) интенсификации липопероксидного процесса;
о кальциевая активация протеаз, липаз, фосфолипаз и других гидролаз;
о осмотическое набухание и разрыв мембран клеток миокарда и их орга­
нелл;
—возрастание дисбаланса ионов и жидкости в кардиомиоцитах (см.
рис. 23.12); причины:
—реперфузионные расстройства процессов энергообеспечения и повреж­
дение мембран и ферментов, что обусловливает накопление избытка
№ + и Са2+ в клетках миокарда и, как следствие, жидкости в них;
— снижение эффективности регуляторных (нервных, гуморальных) воздей­
ствий на сердце (в норме способствующих интеграции и нормализации
внутриклеточных процессов);
— нарастание степени гормонально-нейромедиаторной диссоциации.
Рациональная терапия постишемических реперфузионных состояний позволяет:
—предотвратить развитие инфаркта миокарда или значительно уменьшить
объем пораженного участка миокарда;
—стимулировать процессы репарации в сердечной мышце;
— нормализовать сократительную функцию сердца;
—восстановить оптимальные параметры кровообращения в организме.
Изменение основных показателей функции сердца
при коронарной недостаточности
Коронарная недостаточность сопровождается характерными изменениями
ЭКГ и сократительной функции сердца.
• Изменения ЭКГ при коронарной недостаточности:
— в покое примерно у половины пациентов, которые не переносили ранее
инфаркт миокарда, каких-либо характерных отклонений не выявляется;
— в момент болевого приступа, как правило, регистрируется снижение
(депрессия) сегмента 5Т(он становится горизонтальным либо дугообраз­
ным) и инверсия зубца Т (часто, но не всегда); при вариантной стенокар­
дии выявляется преходящий подъем сегмента 5Т .
• Изменения показателей сократительной функции сердца:
— ударный и сердечный выброс, как правило, снижается; степень снижения
обычно коррелирует со степенью ишемии миокарда, размером и топо­
графией поврежденной зоны сердца; причиной этого является снижение
величины ударного выброса, связанное в основном с «выключением»
ишемизированного региона миокарда из сократительного процесса.
Одним из механизмов компенсации снижения ударного выброса сердца
является тахикардия, обусловленная активацией симпатико-адреналовой
системы (в ответ на падение величины сердечного выброса), а также
повышением давления крови в полых венах и предсердиях;
— конечное диастолическое давление в полостях сердца обычно возрастает.
Основные причины этого:
о снижение сократительной функции поврежденного миокарда;
о уменьшение степени диастолического расслабления миокарда, что
вызвано субконтрактурным состоянием его в связи с избытком Са2+
в цитозоле и миофибриллах кардиомиоцитов (на начальном этапе коро­
нарной недостаточности содержание Са2+ в саркоплазме увеличивается
в основном в связи с его выходом из митохондрий и саркоплазматической сети; на более поздних — в связи с поступлением ионов кальция
из внеклеточной жидкости через поврежденную плазмолемму);
— скорость систолического сокращения и диастолического расслабления мио­
карда существенно снижается. Основные причины:
о дефицит энергии АТФ;
о повреждение мембран миофибрилл, саркоплазматической сети и сар­
коплазмы;
о снижение активности Са2+-зависимых АТФаз.
При отсутствии противопоказаний изменения на ЭКГ исследуются на фоне
функциональных (физических или функциональных) нагрузок.
АРИТМИИ СЕРДЦА
Аритмии — типовая форма патологии сердца.
Аритмии сердца характеризуются нарушением частоты и периодичности
генерации возбуждения и/или последовательности возбуждения предсер­
дий и желудочков.
Дополнительный материал по патофизиологии аритмий изложен в прило­
жении «Справочник терминов» (статьи «Аритмия сердца», «Блокада сердца»,
«Брадикардия», «Гиперкалиемия», «Кардиоверсия», «Тахикардия», «Трепетание»,
«Фибрилляция», «Экстрасистола», «Экстрасистолии», «Электрокардиография»,
«Элекгрокардиостимуляция», а также статьи и подстатьи по упомянутым в этом
разделе нозологическим единицам).
Виды аритмий, их этиология и патогенез
Аритмии являются следствием нарушения основных свойств сердца:
—автоматизма;
—проводимости;
—возбудимости;
—их комбинированных нарушений.
Расстройства сократимости лежат в основе развития сердечной (миокар­
диальной) недостаточности и не являются непосредственной причиной нару­
шений ритма сердца.
Аритмии в результате нарушения автоматизма
Автоматизм — свойство ткани сердца спонтанно генерировать П Д. Автоматизм
определяется своеобразием формирования МП в клетках-водителях ритма.
Оно заключается в спонтанном медленном уменьшении величины диасто­
лической поляризации. Эта разность потенциалов, возникающая вследствие
снижения выхода из клетки ионов калия и медленного поступления в нее
ионов натрия во время диастолы, в конце концов резко увеличивает про­
ницаемость мембраны для № ', что вызывает формирование электрического
импульса (ПД).
Изменение нормального автоматизма обусловлено нарушением функ­
ций синусно-предсердного узла, водителей ритма второго и третьего поряд­
ка. Возникновение патологического автоматизма (эктопическая активность)
может наблюдаться в ткани предсердий, желудочков, пучке Гиса, волокнах
Пуркинье при частичной деполяризации кардиомиоцитов.
Триггерная активность (ранняя и поздняя постдеполяризация) обуслов­
ливает возникновение эктопических импульсов: ранняя постдеполяризация
появляется во время 3-й фазы ПД, поздняя — после ее окончания.
При ранней постдеполяризации эктопические импульсы формируются в фазе
ранней реполяризации на фоне медленного ритма. Механизм постдеполяризации
запускается в результате увеличившейся продолжительности ПД, например,
при удлинении интервала <2—Т, низком внутриклеточном содержании ионов
калия. Именно так развивается желудочковая тахикардия типа «пируэт» {Iокайе
с!е ро1п1е.ч, произносится как «торсад де пуант») в условиях доминирования пара­
симпатических влияний на сердце (при неврозах или гипотиреозе).
При поздней постдеполяризации эктопические импульсы возникают на фоне
ускоренного ритма. Основной причиной их считают перегрузку кардиомиоци­
тов ионами кальция в результате избыточных адренергических влияний на них
при гипертрофии миокарда и сердечной недостаточности, интоксикации сер­
дечными гликозидами, реперфузии (восстановление нарушенного кровотока
в сосудах сердца с помощью тромболитиков). Примерами возникновения
аритмий в результате поздней постдеполяризации являются желудочковые
экстрасистолы и тахикардии при гликозидной интоксикации, желудочковые
экстрасистолы при сердечной недостаточности, катехоламинзависимые пред­
сердные и желудочковые тахикардии.
Виды номотопных и гетеротопных аритмий
В зависимости от места (топографии) генерации аномального импульса воз­
буждения выделяют номотопные и гетеротопные аритмии (рис. 23.16, см. также
«Система проводящая сердца» в приложении «Справочник терминов»).
Рис. 23.16. Виды аритмий в результате нарушения автоматизма сердца
Номотопные аритмии. Возникают в синусно-предсердном узле. К ним
относятся синусовые тахикардия, брадикардия, аритмия и синдром слабости
синусно-предсердного узла.
Гетеротопные аритмии. Это эктопические ритмы. Они возникают вне
синусно-предсердного узла и обусловлены преобладанием автоматизма ниже­
лежащих центров ритмогенеза.
Проявляются гетеротопные аритмии:
—миграцией наджелудочкового водителя ритма;
—предсердным медленным ритмом;
—АВ-ритмом (узловой ритм);
—идиовентрикулярным редким (желудочковый) ритмом (гетеротопный
ритм сердца, при котором водитель ритма расположен в миокарде желу­
дочков);
—идиовентрикулярным ускоренным ритмом сердца, когда ЧСС равна
60—120 в минуту (возникает при патологической циркуляции возбужде­
ния по миокарду желудочков);
—АВ-диссоциацией: полным прекращением проведения возбуждения
от предсердий к желудочкам, когда предсердия и желудочки сокращают­
ся независимо друг от друга (полный поперечный блок);
—«выскакивающими» импульсами.
Патогенез и проявления номотопных аритмий
Синусовая тахикардия
Синусовая тахикардия характеризуется учащающейся в покое генерацией
в синусно-предсердном узле импульсов возбуждения (как правило, более 100
в минуту) с одинаковыми интервалами между ними.
Электрофизиологический механизм развития номотопных аритмий — уско­
рение спонтанной диастолической деполяризации плазмолеммы в клетках
синусно-предсердного узла.
Причины номотопных аритмий:
- активация влияния на сердце симпатико-адреналовой системы. При этом
усиливается выброс нейромедиатора норадреналина из терминалей
симпатической нервной системы и гормона адреналина из мозгового
вещества надпочечников. Такая ситуация наиболее часто наблюдается
при эмоциональном стрессе, физических нагрузках, неврозах, острой
артериальной гипотензии (сопровождающейся активацией афферент­
ной импульсации с барорецепторов), сердечной недостаточности (вслед­
ствие повышения притока крови к правому предсердию и включения
рефлекса Бейнбриджа), гипертермии, лихорадке;
- меньшее влияние на сердце парасимпатической нервной системы. Это
может быть следствием повреждения центральных нервных образова­
ний (подкорковых ядер, ретикулярной формации, ядер продолговатого
мозга), проводящих путей, парасимпатических ганглиев и нервных ство­
лов, холинорецепторов миокарда, что обусловливает снижение холинреактивных свойств сердца;
— прямое действие повреждающих факторов различной природы (физиче­
ские, химические, биологические) на клетки синусно-предсердного узла.
Последнее часто наблюдается при миокардитах, инфаркте миокарда,
перикардитах, механической травме, кардиосклерозе.
Синусовая брадикардия
Синусовая брадикардия проявляется замедленной в покое генерацией
синусно-предсердным узлом импульсов возбуждения по сравнению с нормой
(как правило, 40-60 в минуту) с одинаковыми интервалами между ними.
Основной электрофизиологический механизм развития синусовой брадикардии — замедление процесса спонтанной диастолической деполяризации мем­
бран в клетках синусно-предсердного узла.
Причины синусовой брадикардии:
- активация воздействия парасимпатической нервной системы на сердце.
Наблюдается при раздражении ядер блуждающего нерва (например,
вследствие повышения внутричерепного давления при менингитах,
энцефалитах и т.п.) или его окончаний, повышении внутрижелудочкового давления и тонуса миокарда, надавливании на глазные яблоки
(рефлекс Ашнера—Даньини), а также в зоне проекции бифуркации
сонной артерии (рефлекс Херинга) и в области солнечного сплете­
ния;
— ослабление симпатико-адреналового воздействия на сердце. Синусовая
брадикардия может развиваться при неврозах, повреждении мозговых
структур (например, гипоталамуса), проводящих путей, внутрисердечных ганглиев и окончаний симпатических нервных волокон в миокарде,
при снижении адренореактивных свойств сердца;
— непосредственное воздействие повреждающих факторов на клетки синусно­
предсердного узла. Такими факторами могут быть механическая травма,
кровоизлияние или инфаркт в зоне синусно-предсердного узла, токси­
ны и ЛС (хинин, препараты наперстянки, опиаты, холиномиметики),
метаболиты (непрямой билирубин, желчные кислоты).
Указанные выше факторы могут обусловить не только развитие синусовой
брадикардии, но и (при их значительной силе или длительности действия)
значительно более редкие импульсы (менее 50 в минуту) или прекращение
генерации импульсов синусно-предсердным узлом. Такие состояния называют
соответственно синдромом слабости синусно-предсердного узла и остановкой
синусно-предсердного узла (синоатриальная блокада III стадии).
Синусовая аритмия
Синусовая аритмия — нарушение сердечного ритма, характеризующее­
ся неравномерными интервалами между отдельными ПД, исходящими
из синусно-предсердного узла.
Электрофизиологический механизм возникновения синусовой аритмии —
колебания скорости (увеличение, снижение) медленной спонтанной диасто­
лической деполяризации пейсмекерных клеток.
Наиболее частые причины синусовой аритмии:
—флюктуация (усиление/ослабление) парасимпатических влияний
на сердце;
—нарушение соотношения симпатико-адреналовых и парасимпатических
воздействий на миокард;
—колеблющееся содержание в крови газов ( 0 2 и С 02) и/или метаболитов
(лактат, пируват, желчные кислоты);
—применение отдельных ЛС (наперстянка, опиаты, холино- и симпатолитики, холино- и симпатомиметики);
—действие физических факторов непосредственно на клетки синусно­
предсердного узла (травма, кровоизлияние, новообразование и т.п.).
Синусовая аритмия проявляется:
—чередованием периодов нормального ритма и периодов тахи- и бради­
кардии или медленным восстановлением синусового ритма после эпи­
зода тахикардии (последнее служит проявлением синдрома слабости
синусно-предсердного узла).
Синусовая аритмия наблюдается при различных формах невроза, энцефа­
литов, стенокардии, отравлений и т.п.
Синдром слабости синусно-предсердного узла
Синдром слабости синусно-предсердного узла (синдром брадикардиитахикардии) — неспособность синусно-предсердного узла обеспечивать ритм
сердца, адекватный уровню жизнедеятельности организма.
Основной электрофизиологический механизм возникновения аритмии — нару­
шение (нередко временное прекращение) автоматизма синусно-предсердного
узла, особенно фаз реполяризации и спонтанной диастолической деполяри­
зации и возникновение на этом фоне гетеротопных (эктопических) очагов
ритмической активности.
Причины синдрома слабости синусно-предсердного узла:
— преобладание парасимпатических влияний на сердце; развиваются у пациентов
с невротическими состояниями (психастения, истерия, невроз навязчивых
состояний), неправильной дозировкой ЛС (например, р-адреноблокаторов,
антагонистов кальция, некоторых противоаритмических средств);
— нарушение адрено- и холинореактивных свойств клеток синусно-предсерд­
ного узла; чаще выявляется уменьшение их адрено- и/или повышение
холинореактивности;
—прямое повреждение области синусно-предсердного узла сердца (ишемия,
кровоизлияния, опухоли, травмы, воспалительные процессы).
Главные ЭКГ-проявления слабости синусно-предсердного узла:
—периодическая или постоянная синусовая брадикардия, сменяющаяся
синусовой тахикардией, трепетанием или мерцанием предсердий;
—медленное восстановление синусового ритма после прекращения сину­
совой тахикардии, эпизоды остановки синусно-предсердного узла.
Характерные изменения гемодинамики при слабости синусно-предсердного узла:
—при синусовой тахикардии и в аналогичном периоде синусовой аритмии
наблюдается усиление сердечного выброса (за счет повышения ЧСС)
и незначительное возрастание систолического АД;
—при синусовой брадикардии и в аналогичном периоде синусовой арит­
мии возможно снижение величины сердечного выброса; при этом ударный
выброс несколько увеличивается в связи с удлинением диастолы и воз­
растанием кровенаполнения камер сердца;
—снижение АД и потеря сознания в связи с ишемией мозга при ЧСС 35
и ниже (например, при синдроме слабости синусно-предсердного узла).
Прекращение генерации импульсов синусно-предсердного узлом (син­
дром остановки синусно-предсердного узла) более чем на 10—20 с вызы­
вает потерю сознания и развитие судорог. Это состояние известно
как синдром Морганьи—Адамса—Стокса. Патогенетическая основа син­
дрома — ишемия мозга.
Изменения коронарного кровотока при синусовых нарушениях ритма сердца:
—значительное уменьшение сердечного выброса при выраженной бради­
кардии может обусловить падение перфузионного давления в венечных
артериях и развитие коронарной недостаточности;
—длительная и выраженная синусовая тахикардия, особенно при наличии
коронаросклероза, также может привести к коронарной недостаточно­
сти в результате возросших метаболических потребностей миокарда.
Патогенез и проявления эктопических аритмий сердца
Снижение активности или прекращение деятельности синусно-пред­
сердного узла в результате его функционального или органического повреж­
дения создает условия для включения автоматических центров второго и тре­
тьего порядка.
Эктопический (по отношению к синусно-предсердному узлу) очаг с его
более редким ритмом принимает на себя функцию пейсмекера. В связи
с этим нарушения ритма такого типа носят название гетеротопных, пассивных
или замещающих (синусовый ритм) аритмий.
• Предсердный медленный ритм. Эктопический водитель ритма при нем
находится, как правило, в левом предсердии. На ЭКГ выявляются редкие
(менее 70—80 в минуту) импульсы возбуждения. Предсердный медленный
ритм может наблюдаться при неврозах, приобретенных (ревматических)
или врожденных пороках сердца и кардиомиопатиях.
• Атриовентрикулярный (АВ) ритм (узловой ритм) наблюдается в тех случаях,
когда импульсы в синусно-предсердном узле или вообще не возникают,
или генерируются с меньшей частотой, чем в клетках АВ-узла. Источником
ПД может быть верхняя, средняя или нижняя часть АВ-узла. Чем выше
локализация пейсмекера, тем более выражено его влияние и тем больше
частота генерируемых им импульсов.
• Миграция наджелудочкового водителя ритма. Характеризуется перемещением
пейсмекера из синусно-предсердного узла в нижележащие отделы (преиму­
щественно в АВ-узел) и обратно. Это, как правило, происходит при подав­
лении автоматизма синусно-предсердного узла в результате преходящего
повышения влияний блуждающего нерва. Ритм сердца при этом зависит
от нового источника импульсов и потому становится неправильным.
• Идиовентрикулярный желудочковый ритм. Он развивается как замещающий
при подавлении активности центров первого и второго порядка. Импульсы
генерируются обычно в пучке Гиса верхней части межжелудочковой пере­
городки, в одной из его ножек и их разветвлениях (ритм ножек пучка Гиса)
и реже — в волокнах сети Пуркинье. В связи с этим выделяют клинически
значимые разновидности идиовентрикулярного ритма:
- идиовентрикулярный редкий (желудочковый) ритм — гетеротопный ритм
сердца, при котором водитель ритма расположен в миокарде желудоч­
ков;
— идиовентрикулярный ускоренный ритм сердца (ЧСС 60—120 в минуту).
Возникает при патологической циркуляции возбуждения по миокарду
желудочков. Идиовентрикулярный ритм расценивают как ускоренный
при наличии трех желудочковых комплексов и более с частотой 50—100
в минуту. Обычно он возникает при инфаркте миокарда, протекает бес­
симптомно и не требует вмешательства.
• АВ-диссоциация — полное прекращение проведения возбуждения от пред­
сердий к желудочкам; при этом предсердия и желудочки сокращаются
независимо друг от друга (полный поперечный блок).
• Диссоциация с интерференцией. Заключается в одновременной, но несогла­
сованной работе двух генераторов сердечного ритма: как правило, номотопного (синусового) и гетеротопного (чаще АВ, реже желудочкового).
• «Выскакивающие» сокращения — регистрация отдельных (замещающих)
сокращений сердца под влиянием импульсов, генерируемых центрами
автоматизма второго или третьего порядка на фоне временного снижения
автоматической функции синусно-предсердного узла. Типичный при­
мер — наджелудочковые экстрасистолы.
Аритмии в результате нарушений проводимости
Проводимость — способность кардиомиоцитов проводить импульсы воз­
буждения. Нарушения при проведении возбуждения проявляются различными
сердечными блокадами или аритмиями, возникающими в результате механиз­
ма гееп1гу. Характеристики этих видов блокад и аритмий приведены в ста­
тьях «Блокада сердца», «Аритмии сердца», «Синдром Вольфа—П аркинсонаУайта», «Синдром Клерка—Леви—Кристеско» в приложении «Справочник
терминов».
Повторный вход импульса. Петля мащ уо-ге-еп1гу возникает в дополни­
тельных проводящих путях (при синдроме преждевременного возбуждения
желудочков) или в АВ-соединении. Для возникновения геепТгу необходима
однонаправленная блокада проведения импульса и анатомический или функ­
циональный барьер для формирования петли ге-еп й у. Кроме того, развитие
ге-еп(гу характеризуется замедленным проведением импульса и коротким
рефрактерным периодом.
Виды нарушений проводимости
Разновидности расстройств проводимости представлены на рис. 23.17.
Виды нарушений проводимости импульса в сердце
По изменению скорости
проведения импульса
1
Замедление,
блокада
1
'
Ускорение
По продолжительности
нарушения проводимости
^
Временное || Постоянное |
По локализации
нарушения
Синоатриальное)
Интраатриальное
ИнтравентрикулярнЪеЦ Атриовентрикулярное
Рис. 23.17. Разновидности нарушений проводимости в сердце
Нарушения проводимости по изменению скорости проведения импульсов
возбуждения подразделяются на две группы:
—сопровождающиеся замедлением и/или блокадой проведения импуль­
сов;
—сопровождающиеся ускорением проведения импульсов возбуждения.
Замедление и/или блокада проведения импульсов
Замедление, вплоть до блокады, проведения возбуждения — следствие функ­
циональных или органических изменений в проводящей системе сердца.
Причиной замедления и блокады проведения импульсов возбуждения обычно
являются:
— более сильное парасимпатическое влияние на сердце и/или его более выра­
женные холинореактивные свойства. Ввиду возросшего тонического воз­
действия блуждающего нерва на миокард существенно замедляется ско­
рость волны возбуждения по проводящей системе, особенно на уровне
АВ-узла (отрицательный дромотропный эффект ацетилхолина);
— непосредственное повреждение клеток проводящей системы сердца различны­
ми факторами физического, химического и биологического происхожде­
ния. Наиболее часто это наблюдается при инфаркте миокарда, миокардитах,
кровоизлияниях, операционных (кардиохирургических) травмах миокарда,
опухолях, рубцах, интоксикации алкоголем, никотином, медикаментами
(препараты наперстянки, хинидина, антагонистов кальция, блокаторы
Р-адренорецепторов и др.), действии бактериальных ядов (при дифтерии,
скарлатине, брюшном тифе), вирусной инфекции, нарушении трансмемб­
ранного распределения ионов (чаще всего гиперкалиемии).
Характеристика отдельных видов аритмий при замедлении или блокаде про­
ведения импульсов возбуждения
Нарушение синоатриального (синусно-предсердного) проведения импуль­
са. Торможение или блокада передачи импульса возбуждения от синусно­
предсердного узла к предсердиям обусловливает выпадение отдельных сердеч­
ных сокращений. В результате замедляется частота и нарушается регулярность
сердечных сокращений.
Расстройства внутрипредсердного проведения. В связи с несимметричным
расположением синусно-предсердного узла по отношению к предсердиям
возбуждение их в норме происходит неодномоментно (вначале — правого
и с некоторым запозданием — левого). Возрастание гетерохронии возбужде­
ния предсердий в условиях патологии может вызвать в различной степени внутрипредсердное торможение или блокаду проведения синусовых импульсов.
Нарушения АВ-проведения. Характеризуются замедлением или бло­
кадой проведения возбуждения из предсердий в желудочки. Развиваются
в результате удлинения рефракторного периода после ПД клеток предсердия
и АВ-системы. Различают несколько разновидностей АВ-блокады.
Внутрижелудочковые (интравентрикулярные) нарушения проведения. Чаще
наблюдается нарушение проведения в одной из ножек пучка Гиса. Затем оно
распространяется по неповрежденной системе ножек пучка и только после
этого к другому желудочку (главным образом, через межжелудочковую пере­
городку). Такой маршрут возбуждения обусловливает неодновременное, дискордантное сокращение желудочков.
Расстройства гемодинамики при замедлении или блокаде проведения импуль­
сов возбуждения
Нарушения гемодинамики зависят от длительности эпизода аритмии,
характера основного заболевания и уровня повреждения проводящей систе­
мы сердца. Так, замедление или кратковременная блокада синусно-предсердного
проведения обусловливает уменьшение минутного выброса сердца, снижение
АД, развитие ишемии органов и тканей. Если блокада длится несколько
минут и не сопровождается развитием замещающего гетеротопного (узлового
или желудочкового) ритма, то это ведет к асистолии и гибели пациента.
Нарушение внутрипредсердного и внутрижелудочкового проведения импульса
возбуждения само по себе существенно не изменяет частоты и ритма сердеч­
ных сокращений. В связи с этим системные гемодинамические расстройства
определяются основным заболеванием сердца (миокардиты, пороки клапанов,
инфаркт миокарда и т.п.).
При полной АВ-блокаде нарушения гемодинамики обусловлены главным
образом степенью желудочковой брадикардии и основной сердечной патоло­
гией. В результате значительной брадикардии часто отмечается застой веноз­
ной крови и снижение сердечного выброса.
Блокада проведения импульса на любом уровне проводящей системы серд­
ца (чаще при полной АВ-блокаде) может осложниться развитием синдрома
Морганьи-Адамса—Стокса. Патогенетическая основа синдрома — значительное
снижение, вплоть до прекращения, эффективной работы сердца. Клинически
синдром проявляется внезапной потерей сознания, отсутствием пульса и сер­
дечных тонов. Возможны эпилептиформные судороги. Длится приступ обыч­
но 5—20 с, редко 1—2 мин.
Снижение коронарного кровотока при замедлении или блокаде проведения
импульсов возбуждения выявляют в тех случаях, когда имеется существенное
падение системного АД. Последнее обусловливает уменьшение перфузион­
ного давления в венечных артериях сердца и может привести к коронарной
недостаточности в результате замедленной доставки кислорода и субстратов
метаболизма к миокарду.
Ускоренное проведение возбуждения между предсердиями и желудочками
или отдельными участками сердца
Причины ускоренного проведения возбуждения (рис. 23.18)
Синдромы ускоренного предсердно-желудочкового проведения
(В ол ьф а-П аркин со н а-У ай та; К л ер ка -Л ев и -К р и стеско )
Рис. 23.18. Основные причины ускоренного проведения возбуждения в сердце
•Дополнительные (минуя АВ-узел) пути проведения возбуждения (рассмо­
трены в статье «Пучок»). В этом случае синусовые импульсы поступают
в желудочки по дополнительным пучкам проводящей ткани и по основ­
ному АВ-узлу. По дополнительным пучкам возбуждение распространя­
ется быстрее и достигает желудочков раньше того же импульса, прохо­
дящего через АВ-узел. Таким образом, импульс, распространяющийся
по дополнительному пучку, преждевременно активирует часть желудочков.
Остальная часть их возбуждается позднее импульсом, проходящим через
АВ-узел. Это и обусловливает развитие тахикардии.
• Повышенная возбудимость гетеротопных очагов. Согласно теории повышен­
ной возбудимости, в миокарде желудочков имеются участки, способные
к преждевременному (опережающему) возбуждению. Эти участки могут
стать очагами гетеротопной ритмической активности под воздействием
ряда факторов: импульса возбуждения, распространяющегося по прово­
дящей системе сердца; электрического или механического (например,
при растяжении миокарда избытком притекающей крови) раздражения,
активации симпатико-адреналовых влияний на сердце.
Клинические проявления ускоренного проведения импульсов возбуждения:
—синдром Вольфа—Паркинсона—Уайта; он характеризуется развитием
пароксизмальной тахикардии (примерно в 50-80% случаев), мерцания
или трепетания (20—30% случаев) предсердий и/или желудочков;
—синдром Клерка—Леви—Кристеско; характеризуется преждевременным
возбуждением желудочков, ускорением интервала Р —К / 0 и увеличением
ЧСС.
Расстройства гемодинамики при ускорении проведения импульсов возбуждения:
—снижение ударного и сердечного выброса; обусловлено пониженным
наполнением камер сердца кровью в условиях тахикардии, мерцатель­
ной аритмии, трепетания предсердий;
—пониженное АД; вызвано снижением сердечного выброса и ОПСС.
Коронарный кровоток, как правило, снижен в большей или меньшей степе­
ни. Это чревато развитием коронарной недостаточности.
Нарушения ритма в результате комбинированных расстройств свойств сердца:
возбудимости, проводимости и автоматизма
Возбудимость — свойство клеток воспринимать сигнал и реагировать на него
возбуждением. Возбудимость сердечной мышцы выражается в ее способности
генерировать ПД. Свойство возбудимости сердца следует отличать от автома­
тизма, заключающегося в спонтанной генерации импульсов возбуждения.
Свойством возбудимости обладают как рабочие (сократительные) кардиомиоциты, так и кардиомиоциты проводящей системы. Возбудимость лежит
в основе распространения электрического импульса по сердцу.
Возбудимость сердца описывается законом «все или ничего». Это означает,
что подпороговые раздражители не инициируют ПД, тогда как раздражители
пороговой величины вызывают максимальный по величине и скорости рас­
пространения ПД. Увеличение силы раздражения (надпороговые величины)
не меняет характеристик ПД.
Возбудимость кардиомиоцитов изменяется в отдельные периоды сердечного
цикла. Во время систолы они не возбуждаются, т.е. рефрактерны к раздраже­
нию. Во время диастолы возбудимость сердечных клеток восстанавливается.
Существуют два коротких интервала сердечного цикла, когда возбудимость
миокарда повышена: уязвимый период и период сверхнормальной возбудимости.
Уязвимый период — терминальная часть фазы реполяризации, компонент
относительного рефрактерного периода. В уязвимый период величина порого­
вого МП снижена, а возбудимость повышена. В связи с этим даже сравни-
тельно слабые электрические импульсы и другие раздражители могут вызвать
возбуждение и аритмию. Этот период совпадает с пиком волны Т на ЭКГ
и соответствует третьей фазе реполяризации клетки.
Период сверхнормальной возбудимости следует непосредственно после окон­
чания относительного рефрактерного периода, находится в начале диастолы
и совпадает с волной II на ЭКГ. В этот период даже подпороговые импульсы
могут вызвать ПД.
Причины нарушения возбудимости, проводимости и автоматизма:
—функциональные расстройства нервной системы (неврозы, стресс, вагото-
ния);
— органические поражения нервной системы (опухоли мозга, травмы черепа,
нарушения мозгового кровообращения);
—поражения миокарда (дистрофии, миокардиты, кардиосклероз, кардиомиопатии, инфаркт миокарда);
—нарушения электролитного баланса (изменение содержания в крови ионов
калия, кальция, магния);
—влияние токсичных веществ (окись углерода, бактериальные токсины,
никотин, алкоголь, промышленные токсичные вещества);
—интоксикация ЛС (антиаритмические препараты, р-адреномиметики,
сердечные гликозиды);
—гипоксемия (при сердечной недостаточности, «легочном сердце»).
Механизмы возникновения «комбинированных» аритмий:
—аномальный автоматизм. Комбинированные механизмы нарушений сер­
дечного ритма характеризуются наличием эктопического центра, генери­
рующего собственный ритм, который защищен так называемой блокадой
входа. Блокада входа — зона нарушенной проводимости, препятствующая
подавлению эктопического очага синусовыми импульсами. Таким образом
создаются условия для одновременного сосуществования двух источников
активации сердца: нормального (синусового) и работающего в автономном
режиме альтернативного (парасистолического). Этот механизм проявляет­
ся парасистолией, которую необходимо отличать от экстрасистолии;
— постдеполяризационная и триггерная активность;
—повторный вход (ге-еп&у) и круговое движение импульса;
—нарушения проводимости.
Виды комбинированных аритмий
Нарушения ритма, обусловленные совокупными изменениями свойств воз­
будимости, проводимости и автоматизма, подразделяются на несколько групп:
—экстрасистолии — синусовая, предсердная (наджелудочковая), из пред­
сердно-желудочкового узла (узловая, АВ), желудочковая;
—пароксизмальные тахикардии (предсердная, из предсердно-желудочко­
вого соединения, желудочковая, трепетание и мерцание предсердий);
—трепетание и мерцание желудочков.
Дополнительный материал по аритмиям (включая комбинированные) изло­
жен в приложении «Справочник терминов» (статьи «Аритмия сердца», «Блокада
сердца», «Брадикардия», «Гиперкалиемия», «Кардиоверсия», «Тахикардия»,
«Трепетание», «Фибрилляция», «Экстрасистола», «Экстрасистолии», «Электро­
кардиография» , «Элекгрокардиостимуляция»).
Экстрасистолия
Экстрасистола (наиболее часто регистрируемый вид аритмии) — единич­
ный преждевременный импульс возбуждения и, как правило, единичное
сокращение сердца или отдельных его камер.
Нередко экстрасистолы регистрируются повторно. Если три экстрасистолы
и более следуют одна за другой, говорят об экстрасистолии.
Разновидности экстрасистолии:
— аллоритмия — сочетание (связь) в определенной последовательности
нормальных (своевременных) сокращений с экстрасистолами. Наиболее
частые формы:
о бигеминия (экстрасистола после каждого очередного импульса воз­
буждения;
о тригеминия (экстрасистола после каждых двух импульсов возбуждения);
о квадригеминия (экстрасистола после трех очередных импульсов);
—парасистолия — сосуществование двух или более независимых, одновре­
менно функционирующих очагов генерации импульсов, вызывающих
сокращение всего сердца или отдельных его частей. Один из очагов
определяет основной ритм сердца. Как правило, им является синусно­
предсердный узел. Другой очаг — эктопический (парасистолический).
Он обычно расположен в желудочках.
Пароксизмальная тахикардия
Пароксизмальная тахикардия — приступообразное и внезапное увеличение
частоты имнульсации правильного ритма из эктопического очага сердца.
О пароксизме тахикардии говорят, когда число эктопических импульсов
превышает 3 —5, а частота их колеблется обычно от 160 до 220 в минуту
(при расположении гетеротопного очага в предсердии) или от 140 до 200
(при расположении в желудочках).
Отдельные разновидности пароксизмальных тахикардий охарактеризованы
в статье «Тахикардия» (см. приложение «Справочник терминов»).
Трепетание предсердий и желудочков
Трепетание предсердий и желудочков проявляется очень частыми импульсами
возбуждения и, как правило, сокращениями сердца правильного ритма (пред­
сердий обычно 220-350 в минуту; желудочков — 150—300 в минуту).
Характеризуется отсутствием диастолической паузы и поверхностными,
гемодинамически неэффективными сокращениями миокарда.
При трепетании предсердий, как правило, развивается АВ-блокада. В связи
с этим в желудочки проводится только каждый второй—четвертый предсерд­
ный импульс, поскольку функциональные особенности \В-узла таковы,
что он способен проводить обычно не более 200—250 импульсов в минуту.
Ф ибрилляция (мерцание) пр едсердий и желудочков
Фибрилляция (мерцание) предсердий и желудочков — нерегулярная, бес­
порядочная электрическая активность предсердий и желудочков, сопрово­
ждающ аяся прекращением эффективной насосной функции сердца.
Мерцание предсердий развивается при частоте эктопических импульсов
более 400-500 в минуту, желудочков — более 300-500.
При такой частоте возбуждения клетки миокарда не могут ответить синхрон­
ным, координированным сокращением, охватывающим весь миокард. Отдельные
волокна или микроучастки сердца сокращаются беспорядочно по мере выхода
их из рефрактерного периода.
Нарушения в миокарде, предшествующие аритмиям
Развитию пароксизмальной тахикардии, трепетанию и фибрилляции пред­
шествуют нарушения метаболизма в миокарде (рис. 23.19) и формирование
его биохимической гетерогенности.
Рис. 23.19. Основные биохимические нарушения в миокарде, предшествующие парок­
сизмальной тахикардии, трепетанию и фибрилляции предсердий и/или желудочков
Указанные на рис. 23.19 и многие другие факторы вызывают аритмогенные изменения в миокарде и различные нарушения ритма сердца. Степень
и сочетание этих нарушений при различных видах аритмий различны. К числу
наиболее значимых относят следующие.
• Увеличение внеклеточной концентрации ионов калия.
Причины повышенной концентрации ионов К+ во внеклеточной жидкости
показаны на рис. 23.20.
Аритмогенные эффекты увеличения содержания К+ в интерстициальной
жидкости представлены на рис. 23.21. Указанные эффекты сопровождают­
ся развитием различных видов аритмий, включая пароксизмы тахикардии
и фибрилляцию желудочков.
• Снижение рН в клетках миокарда (развитие метаболического ацидоза).
Развитие ацидоза обусловлено:
—активацией анаэробного гликолиза, вследствие чего накапливается избы­
ток молочной кислоты (МК) в кардиомиоцитах, а также в интерсти­
циальной жидкости и развивается ацидоз; лактат-ацидоз при ишемии
миокарда в результате коронарной недостаточности развивается уже
в течение нескольких секунд после снижения коронарного кровотока;
—торможением процессов аэробного тканевого дыхания (закономерно
наблюдается при коронарной недостаточности в связи с дефицитом
кислорода и субстратов обмена веществ).
Основные причины увеличения внеклеточного содержания ионов калия
ЛГ
Дефицит АТФ
и креатинфосфата
в кардиомиоцитах
1Г
Снижение активности
К*-/Ыа*-АТФазы плазмолеммы
чг
Аномалии мембран
кардиомиоцитов
Рис. 23.20. Основные причины увеличения внеклеточного содержания К+
Рис. 23.21. Аритмогенные эффекты увеличения внеклеточного содержания К+
Аритмогенные эффекты избытка ионов Н+ в миокарде весьма сходны
с эффектами, вызывающими увеличение концентрации ионов К+ в интерстиции (см. выше). Однако выраженность этих эффектов меньше.
• Накопление в кардиомиоцитах избытка цАМФ.
Основные причины нарастания уровня цАМФ в ишемизированном миокарде:
— активация аденилатциклазы (наблюдается при воздействии многих фак­
торов; основными из них являются катехоламины: значительное увели­
чение содержания адреналина и норадреналина в миокарде, например,
при острой коронарной недостаточности или стрессе, сопровождается
активацией аденилатциклазы и повышенным уровнем цАМФ);
—подавление активности фосфодиэстераз, разрушающих цАМФ (наблюда­
ется при ишемии миокарда, миокардитах, кардиомиопатиях).
Аритмогенные эффекты избытка цАМФ возможны благодаря стимуля­
ции под влиянием цАМФ так называемого медленного входящего каль­
циевого тока. ПД при этом развивается за счет медленного транспорта
Са2+ через мембрану клеток миокарда и характеризуется малой скоростью
нарастания деполяризации, что, в свою очередь, обусловливает замед­
ленное проведение возбуждения по сердцу Высокая концентрация вну­
триклеточного цАМФ стимулирует медленный кальциевый ток и создает
тем самым условия для формирования гетеротопных очагов ритмической
активности. О важной аритмогенной роли медленного кальциевого ответа,
стимулируемого цАМФ, свидетельствует факт менее частых аритмий, когда
вход Са2+ в клетку блокируется антагонистами кальция: верапамилом,
нифедипином и др.
• Повышенное содержание ВЖК в клетках миокарда.
Основными причинами накопления ВЖК в кардиомиоцитах являются:
—увеличенное содержание катехоламинов в ткани миокарда (они обладают
выраженной липолитической активностью);
—ишемия миокарда (она характеризуется возросшим содержанием катехо­
ламинов в миокарде и активацией липолиза);
—более частый захват ВЖ К поврежденными кардиомиоцитами (обусловлен
альтерацией мембран кардиомиоцитов и увеличением их проницаемо­
сти, в том числе для ВЖК);
— активация гидролиза мембранных фосфолипидов (например, под действи­
ем катехоламинов и Са2+).
Аритмогенные эффекты вызваны:
—разобщением процессов окисления и фосфорилирования (это приводит
к потенцированию дефицита АТФ и выходу ионов К+ в межклеточную
жидкость);
—подавлением ресинтеза АТФ в процессе гликолиза вызвано тем, что АТФ
гликолитического происхождения используется катионными насосами
при формировании МП, а также при развитии ПД. Дефицит АТФ нару­
шает мембранный электрогенез, что и приводит к появлению аритмий.
Электрофизиологические механизмы
развития аритмий
Выделяют два основных электрофизиологических механизма развития
экстрасистолии, пароксизмальной тахикардии, трепетания и фибрилляции
предсердий и желудочков сердца:
—повторную циркуляцию импульса возбуждения по замкнутому контуру (ге-епЬу ,
возвратный ход импульса возбуждения, циркуляция возбуждения);
— аномальный гетеротопный автоматизм (рис. 23.22).
Рис. 23.22. Основные электрофизиологические механизмы аритмии сердца
Повторная циркуляция возбуждения по замкнутому контуру
Циркуляция возбуждения развивается на базе двух феноменов:
—ретроградного проведения импульса возбуждения;
—продольной диссоциации проведения импульса возбуждения.
Ретроградное проведение импульса возбуждения. Выявлено, что замедление
или блокада проведения импульса возбуждения в одном направлении (антероградное) сочетается с возможностью проведения его в другом (ретроградное).
Такая ситуация складывается обычно в микроучастке на периферии прово­
дящей системы, а также в зонах контактов волокон Пуркинье с рабочими
кардиомиоцитами.
Продольная диссоциация проведения импульса. Этот феномен развивается
в участках с параллельным ходом волокон проводящей системы и наличием
между ними анастомозов. Условиями его возникновения являются блокада
проведения импульса в одном каком-либо волокне и замедленная проводи­
мость в другом.
Типичная ситуация развития циркуляции возбуждения на базе феномена про­
дольной диссоциации заключается в следующем: синусовый импульс не может
распространяться антероградно по волокну А в связи с наличием в нем блока­
ды проведения. Возбуждение движется по волокну Б. Из него по анастомозам
импульс может пройти в дистальный участок волокна А и, распространяясь
в ретроградном направлении через блокированный участок, активировать прок­
симальную часть волокна А. Затем по межклеточным анастомозам возбуждение
вновь попадает в волокно Б, находящееся в состоянии покоя. Этот процесс может
быть однократным или повторяться многократно, обеспечивая длительную цир­
куляцию возбуждения. Описанный феномен характерен для механизма ге-еп!гу
в АВ-узле, пучке Гиса, его ножках и их разветвлениях.
Если импульс возбуждения циркулирует вокруг крупных анатомических пре­
пятствий (например, вокруг зоны ишемии или инфаркта миокарда, рубцовой
ткани, по ткани вокруг отверстий полых вен), то говорят о контуре и феноме­
не макроциркуляции (макро-ге-епйу); если по волокнам проводящей системы
или миоцитам без анатомического препятствия и микромасштаба, то этот кон­
тур и феномен обозначают термином «микроциркуляция» (микро-ге-еяггу).
Аномальный гетеротопный автоматизм
Особенности аномального автоматизма:
— способность сохраняться (а не угнетаться!) при работе водителя ритма
с более частой генерацией импульсов возбуждения. Именно поэтому
аномальный ритм может «подчинять» ритм нормального пейсмекера
сердца, в том числе в условиях кратковременного замедления ритма
нормального пейсмекера (замещающая активность);
—свойство формирования автоматизма имеется и у так называемых рабочих
кардиомиоцитов, в том числе при частичной их деполяризации;
—способность к сохранению или нарастанию аномального автоматизма
сохраняется даже при высокочастотном электрическом раздражении мио­
карда (нормальный автоматизм в этих условиях подавляется!).
Описанные выше механизмы ге-еШгу и аномального автоматизма могут
лежать в основе формирования одиночного импульса и обусловить возникно­
вение экстрасистолы. При наличии условий для повторного возникновения
экстрасистол возможна генерация серии импульсов, приводящих к разви­
тию пароксизмальной тахикардии, трепетания или фибрилляции предсердий
и желудочков.
СЕРДЕЧНАЯ НЕДОСТАТОЧНОСТЬ
Сердечная недостаточность — одна из частых причин утраты трудоспособ­
ности, инвалидизации и смерти пациентов, страдающих заболеваниями ССС.
Сердечная недостаточность — синдром, развивающийся при многих болез­
нях, в том числе поражающих органы и ткани, которые не относятся к ССС.
Сердечная недостаточность — типовая форма патологии сердечной дея­
тельности, при которой сердце не обеспечивает потребности органов
и тканей в кровоснабжении, адекватном их функции и уровню пластиче­
ских процессов в них.
Проявляется сердечная недостаточность меньшей по сравнению с необхо­
димой величиной сердечного выброса и первично-циркуляторной гипоксией
по сравнению с необходимой.
Сущность сердечной недостаточности заключается в том, что сердце
(при данном ОПСС) не может переместить в артериальное русло всю кровь,
притекающую к нему по венам.
Причины сердечной недостаточности
К развитию сердечной недостаточности приводят две основные группы
факторов:
— оказывающих непосредственное повреждающее действие на сердце;
— обусловливающих функциональную перегрузку сердца.
Наиболее значимые факторы, непосредственно повреждающие сердце,
могут иметь физическую, химическую или биологическую природу.
• Факторы физической природы:
—сдавление сердца (экссудатом, кровью, эмфизематозными легкими, опу­
холью);
—воздействие электрического тока (например, при электротравме или дефи­
брилляции сердца);
—механическая травма (при ушибах грудной клетки, проникающих ране­
ниях, хирургических манипуляциях).
• Факторы химические природы:
—нелекарственные химические соединения (например, разобщители окис­
лительного фосфорилирования, соли кальция и тяжелых металлов,
ингибиторы ферментов, гидроперекиси липидов);
—ЛС в неадекватной дозировке (например, антагонисты кальция, сердеч­
ные гликозиды, адреноблокаторы), дефицит кислорода, недостаток
химических соединений, необходимых для обмена веществ (например,
соли различных металлов).
• Факторы биологической природы:
—высокие уровни БАВ (например, катехоламинов, Т4);
—дефицит или отсутствие БАВ, необходимых для метаболизма (например,
ферментов, витаминов и др.);
—длительная ишемия или инфаркт миокарда. Вызывает прекращение сокра­
щений миокарда в зоне повреждения. Это сопровождается функцио­
нальной перегрузкой миокарда вне зоны ишемии или инфаркта;
—кардиомиопатии (поражения миокарда, преимущественно невоспали­
тельной природы, характеризующиеся существенными структурно­
функциональными изменениями в сердце).
Наиболее значимые факторы, вызывающие перегрузку
сердца
Причины перегрузки сердца подразделяются на две подгруппы: увеличи­
вающие преднагрузку или увеличивающие посленагрузку (рис. 23.23).
Рис. 23.23. Основные факторы перегрузки сердца
• Увеличение преднагрузки (объем крови, притекающей к сердцу и увеличи­
вающей давление наполнения желудочков) наблюдается при гиперволе­
мии, полицитемии, гемоконцентрации, клапанных пороках (сопровожда­
ются увеличением остаточного объема крови в желудочках).
• Возрастание постнагрузки (сопротивление изгнанию крови из желудочков
в аорту и легочную артерию. Основным фактором постнагрузки является
ОПСС) происходит при артериальных гипертензиях любого генеза, стено­
зах клапанных отверстий сердца, сужении крупных артериальных стволов
(аорты, легочной артерии).
Виды сердечной недостаточности
Дифференцировка видов сердечной недостаточности базируется на сле­
дующих критериях (рис. 23.24):
—происхождение (миокардиальная и/или перегрузочная);
— скорость развития (острая — ОСН и хроническая — ХСН);
—преимущественное поражение отдела сердца (левожелудочковая и/или
правожелудочковая);
—преимущественно нарушенная фаза сердечного цикла (систолическая и/или
диастолическая).
Виды сердечной недостаточности по происхождению
По этому критерию выделяют следующие формы сердечной недостаточ­
ности:
— миокардиальную;
—перегрузочную;
—смешанную.
Сердечная недостаточность
По происхождению |
| Миокардиальная
1По скорости развития
Перегрузочная
Смешанная
По первичности снижения
сократимости сердца или
притока крови к нему
Первичная
(кардиогенная)
Вторичная
(некардиогенная)
По преимущественно
пораженному отделу сердца
Левожелудочковая
Правожелудочковая
г
Смешанная
Рис. 23.24. Виды сердечной недостаточности
Миокардиальная форма развивается преимущественно в результате непо­
средственного повреждения миокарда.
Перегрузочная форма сердечной недостаточности возникает главным обра­
зом в результате перегрузки сердца (увеличение пред- или постнагрузки).
Смешанная форма сердечной недостаточности — результат прямого повреж­
дения миокарда в сочетании с его перегрузкой.
Виды сердечной недостаточности по скорости развития
По этому критерию выделены острая и хроническая формы.
• ОСН развивается в течение нескольких минут и часов после того, как начи­
нает действовать ее причина. Она является результатом инфаркта миокар­
да, острой недостаточности митрального или аортального клапана, разры­
ва стенок левого желудочка.
• ХСН формируется постепенно, в течение недель, месяцев, годами. Она
является следствием артериальной гипертензии, хронической ДН, дли­
тельной анемии, пороков сердца. Течение ХСН может осложняться ОСН.
Виды сердечной недостаточности
по первичности механизма развития
В зависимости от первичного снижения сократительной функции миокарда
или уменьшения притока венозной крови к сердцу выделены первичная (кар­
диогенная) и вторичная (некардиогенная) формы сердечной недостаточности.
• Первичная (кардиогенная) сердечная недостаточность. Развивается в резуль­
тате преимущественного снижения сократительной функции сердца
при величине притока венозной крови к нему, близкой к нормальной.
Наиболее часто наблюдается при ИБС (может сопровождаться инфарктом
миокарда, кардиосклерозом, дистрофией миокарда), миокардитах (напри­
мер, при воспалительных поражениях мышцы сердца или выраженных
и длительных эндотоксинемиях), кардиомиопатиях.
• Вторичная (некардиогенная) сердечная недостаточность. Возникает вслед­
ствие первичного преимущественного уменьшения венозного притока
к сердцу при близкой к нормальной величине сократительной функции
миокарда. Наиболее часто встречается при острой массивной кровопотере,
нарушении диастолического расслабления сердца и заполнения его камер
кровью (например, при сдавлении сердца жидкостью, накапливающейся
в полости перикарда: кровью, экссудатом), при эпизодах пароксизмаль­
ной тахикардии (что приводит к снижению сердечного выброса и возврату
венозной крови к сердцу), коллапсе (например, при вазодилатационном
или гиповолемическом).
Виды сердечной недостаточности
по преимущ ественно пораженному отделу сердца
В зависимости от преимущественного поражения левого или правого отде­
лов сердца различают левожелудочковую и правожелудочковую сердечную
недостаточность.
• Левожелудочковая сердечная недостаточность может быть вызвана пере­
грузкой левого желудочка (например, при стенозе устья аорты) или сни­
жением его сократительной функции (например, при инфаркте миокарда),
т.е. состояниями, приводящими к уменьшению выброса крови в большой
круг кровообращения, к перерастяжению левого предсердия и застою
крови в малом круге кровообращения.
• Правожелудочковая сердечная недостаточность возникает, например, при меха­
нической перегрузке правого желудочка (в частности, при суженном отвер­
стии клапана легочной артерии) или высоком давлении в легочной артерии
(при легочной гипертензии), т.е. состояниях, сопровождающихся уменьше­
нием выброса крови в малый круг кровообращения, перерастяжением право­
го предсердия и застоем крови в большом круге кровообращения.
• Тотальная сердечная недостаточность характеризуется недостаточностью
и левого, и правого желудочка сердца.
Виды сердечной недостаточности
по преимущ ественной недостаточности фазы сердечного цикла
В зависимости от вида нарушения функций миокарда левого желудоч­
ка (снижение силы и скорости его сокращения или нарушение скорости
расслабления) левожелудочковую сердечную недостаточность подразделяют
на систолическую и диастолическую.
• Диастолическая сердечная недостаточность (СНД) — нарушение расслабле­
ния и наполнения левого желудочка. Оно обусловлено его гипертрофией,
фиброзом или инфильтрацией и приводит к увеличению конечного диа­
столического давления и развитию сердечной недостаточности.
• Систолическая сердечная недостаточность (хроническая) —ХССН — ослож­
няет течение ряда заболеваний. При ней нарушается насосная (нагнета­
ющая) функция сердца, что приводит к уменьшению сердечного выброса.
О бщ ие механизмы развития сердечной недостаточности
• Миокардиальная форма сердечной недостаточности характеризуется сни­
жением развиваемого сердцем напряжения. Это проявляется падением
силы и скорости его сокращения и расслабления.
• Перегрузочная форма сердечной недостаточности формируется на фоне
более или менее длительного периода его гиперфункции, в конце концов
приводящей к уменьшению силы и скорости сокращения и расслабления
сердца.
В обоих случаях (и при перегрузке, и при повреждении сердца) снижение
его сократительной функции сопровождается включением экстра- и интракардиальных механизмов компенсации этого сдвига. Все они, несмотря на известное
своеобразие, в условиях целостного организма взаимосвязаны таким образом,
что активация одного из них существенно влияет на деятельность другого.
Механизмы экстренной компенсации сократительной
функции
Механизмы экстренной компенсации, активирующиеся при снижении
сократительной функции сердца, показаны на рис. 23.25.
Повышение:
-силы сокращения сердца;
-скорости сокращений;
-скорости расслабления миокарда
Рис. 23.25. Экстренные механизмы компенсации сниженной сократительной функ­
ции сердца
К ним относятся:
• Повышение сократимости миокарда при его растяжении притекающей кро­
вью — механизм Франка—Старлинга. Он обеспечивает:
—увеличение развиваемого миокардом напряжения;
—увеличение скорости сокращения и расслабления миокарда;
—увеличение напряжения, развиваемого сердцем (осуществляется в ответ
на нарастающее растяжение миокарда). В связи с этим механизм
Франка—Старлинга называют гетерометрическим, т.е. связанным с воз­
растанием длины мышечного волокна. Молекулярная основа реализа­
ции механизма Франка—Старлинга: ускоренное сокращение и рассла­
бление кардиомиоцитов развиваются в связи с более быстрым выбросом
Са2+ из кальциевых депо (саркоплазматическая сеть) и с последующим
ускоренным закачиванием Са2+ (Са2+-АТФаза) в цистерны саркоплазматической сети.
• Увеличение силы сокращений миокарда в ответ на повышенную нагрузку.
Происходит при неизменной длине миоцитов. Такой механизм называют
гомеометрическим, так как он происходит без значительного изменения
длины мышечных волокон.
• Возрастание сократимости сердца в связи с увеличением ЧСС.
• Повышение сократимости сердца в результате возрастания симпатико-адреналовых влияний. Действие катехоламинов на кардиомиоциты через
РI -адрснорсцепторы влечет за собой ряд последующих изменений:
—стимуляция р-адренорецептора катехоламинами через О-белок активи­
рует аденилатциклазу с образованием цАМФ;
—активация цАМФ-зависимой протеинкиназы сопровождается фосфорилированием белка р27 сарколеммы, в связи с чем увеличивается вход
кальция в саркоплазму через потенциалзависимые Са2+-каналы и уси­
ливается кальцийиндуцированная мобилизация Са2+ в цитозоль через
активированные рецепторы рианодина;
—повышенная концентрация Са2+ в саркоплазме обусловливает его свя­
зывание с тропонином С, что снимает ингибирующее действие тропомиозина на взаимодействие актина с миозином; в результате этого
образуется большее количество актомиозиновых связей и усиливаются
сокращения миокарда.
Компенсаторная гиперфункция серд ца
Функционирование названных выше механизмов обеспечивает экстренную
компенсацию сократительной функции перегруженного или поврежденного
миокарда. Это сопровождается значительным и более или менее длительным
усилением интенсивности функционирования сердца — его компенсаторной
гиперфункцией.
Компенсаторная гипертрофия сердца
Гиперфункция миокарда обусловливает экспрессию отдельных генов кар­
диомиоцитов. Она проявляется усиленной интенсивностью синтеза нуклеи­
новых кислот и белков. Ускорение синтеза нуклеиновых кислот и белков
миокарда приводит к нарастанию его массы — гипертрофии. Биологическое
значение компенсаторной гипертрофии сердца заключается в том, что увели­
ченная функция органа выполняется его возросшей массой.
М еханизмы деком пенсации гипертрофированного сердца
Потенциальные возможности гипертрофированного миокарда увеличивать
силу и скорость сокращения небеспредельны. Если на сердце продолжает
действовать повышенная нагрузка или оно дополнительно повреждается, сила
и скорость его сокращений падают, а их энергетическая «стоимость» возрас­
тает — развивается декомпенсация гипертрофированного сердца. Механизмы
декомпенсации гипертрофированного сердца перечислены на рис. 23.26.
Компенсаторная гипертрофия миокарда I
Нарушение сбалансированности роста структур миокарда и их последствия
Отставание роста Отставание биогенеза
микрососудов
митохондрий от
от нарастания
нарастания массы
массы миокарда
миофибрилл
Отставание
активности
АТФазы миозинов
от необходимой
Отставание скорости
синтеза структур
кардиомиоцитов
от должной
Последствия:
■относительная
коронарная
недостаточность
- нарушение
энергетического
обеспечения
кардиомиоцитов
- снижение
сократимости
миокарда
- нарушение
пластических
процессов;
■дистрофия
миокарда
Рис. 23.26. Основные механизмы декомпенсации гипертрофированного сердца
В основе декомпенсации длительно гипертрофированного миокарда лежит
нарушение сбалансированности роста различных его структур. Эти сдвиги (наря­
ду с другими, см. рис. 23.26) в конечном счете обусловливают уменьшение
силы сердечных сокращений и скорости контрактильного процесса, т.е. раз­
витие сердечной недостаточности.
Клеточно-молекулярные механизмы
сердечной недостаточности
Снижение сократительной функции сердца — итог развития сердечной недо­
статочности самой разной этиологии. Несмотря на различие причин и извест­
ное своеобразие начальных звеньев патогенеза сердечной недостаточности,
ее механизмы на клеточном и молекулярном уровне едины. К числу главных
механизмов снижения сократительной функции сердца относят следующие
(рис. 23.27).
• Недостаточность энергетического обеспечения клеток миокарда. Расстрой­
ство энергоснабжения основных процессов, происходящих в клетках
миокарда (прежде всего его сокращения и расслабления), развивается
вследствие нарушения:
—ресинтеза макроэргов;
—транспорта энергии к эффекторным структурам кардиомиоцитов;
—утилизации этими структурами энергии макроэргических фосфатных
соединений. Эти звенья патогенеза рассмотрены в главе 5 «Повреждение
клетки» (раздел «Общие механизмы повреждения»,
«Расстройства энергетического обеспечения клетки», т. 1).
V___________________________________________________________________________
подраздел
______________________________________________________________'
Снижение:
-с и л ы сокращения сердца;
-ск о р о сти систолического сокращения;
-ск о р о сти диастолического расслабления миокарда
Рис. 23.27. Основные механизмы снижения сократительной функции миокарда
при сердечной недостаточности
Нарушение обеспечения кардиомиоцитов энергией на этапах ее про­
дукции, транспорта и утилизации может быть как стартовым механизмом
снижения сократительной функции сердца, так и существенным фактором
нарастания ее депрессии.
• Повреждение мембран и ферментов кардиомиоцитов. Общие механизмы
повреждения клеточных мембран и ферментов рассмотрены в главе 5
«Повреждение клетки» (см. раздел «Общие механизмы повреждения»,
подраздел «Повреждение мембран и ферментов», т. 1).
• Альтерация мембран и ферментов клеток миокарда — главное, а нередко
и инициальное звено патогенеза сердечной недостаточности.
• Изменение физико-химических свойств и конформации молекул белка
(структурных и ферментов), липидов, фосфолипидов и Л П . Эти измене­
ния сопровождаются значительным обратимым, а часто и необратимым
повреждением структуры и функции мембран и ферментов, в том числе
митохондрий, саркоплазматического ретикулума, миофибрилл, плазма­
тической мембраны, которые обеспечивают условия для сократительной
и ритмической функций сердца.
• Ионный дисбаланс. Нарушение содержания и соотношения между отдельны­
ми ионами внутри и вне клеток рассмотрены в главе 5 «Повреждение клетки»
(см. т. 1). Ниже приведены характерные для развития сердечной недостаточ­
ности особенности ионного дисбаланса. Ионный дисбаланс при сердечной
недостаточности проявляется нарушением соотношения между отдельными
ионами в разных секторах кардиомиоцитов: в органеллах (митохондрии,
саркоплазматический ретикулум, миофибриллы), в цитозоле, по разные сто­
роны плазматической мембраны кардиомиоцитов. В наибольшей степени это
относится к ионам К+, № +, Са2+. Именно эти катионы в основном опреде­
ляют реализацию таких процессов, как возбуждение, электромеханическое
сопряжение, сокращение и расслабление миокарда.
• Нарушения в генетической программе кардиомиоцитов. Эти механизмы
описаны в главе 5 «Повреждение клетки» (см. т. 1). При сердечной
недостаточности происходит активация генов, контролирующих про­
цессы обновления субклеточных структур кардиомиоцитов, а также рост
сосудов микроциркуляторного русла и нервных волокон. Так, при ише­
мическом и стрессорном повреждении сердца подавлена экспрессия
мРН К для Са2+-зависимой АТФазы саркоплазматической сети. Это
потенцирует ингибирование процессов захвата и выброса Са2+ ретикулумом миоцитов. В условиях ишемии и инфаркта миокарда, хронического
эмоционально-болевого стресса подавляется также процесс трансляции
генетической информации, что сопровождается нарушением синтеза
различных белков в клетках миокарда.
• Расстройства нейрогуморальной регуляции сердца. Общая характеристика
нарушений регуляции клеточных функций дана в главе 5 «Повреждение
клетки» (см. т. 1). Ниже рассмотрены важные для развития сердечной
недостаточности изменения симпатической и парасимпатической регуля­
ции сердца.
• Изменение механизмов симпатической рефляции сердечной деятельности
заключается в том, что в ткани сердца уменьшается содержание норадре­
налина — нейромедиатора симпатической нервной системы.
Причины этого:
—снижение синтеза норадреналина в нейронах симпатической нервной
системы (в норме в этих нейронах образуется около 80% медиатора,
содержащегося в миокарде), так как подавляется активность фермента
тирозингидроксилазы и тормозится захват норадреналина нервными
окончаниями;
—снижение адренореактивных свойств сердца, т.е. выраженности ино-,
хроно-, дромо- и батмотропных эффектов норадреналина и адреналина.
• Изменение механизмов парасимпатической регуляции сердца. При сердечной
недостаточности расстройства парасимпатической регуляции выраже­
ны в меньшей мере, чем симпатической, что является результатом более
высокой резистентности этих механизмов к повреждающим факторам.
Известно, что ацетилхолин через М-холинорецепторы снижает частоту
и силу сердечных сокращений (ингибируя образование цАМФ и активи­
руя образование цГМФ, которая активирует цГМФ-зависимую киназу,
подавляющую активность потенциалзависимых Са2+-каналов).
Последствия нарушенных симпатических и парасимпатических влияний
на миокард состоят в меньшей степени управляемости и надежности регуля­
ции сердца. Это приводит к падению темпа и величины мобилизации сокра­
тительной функции сердца, особенно в чрезвычайных условиях.
Проявления сердечной недостаточности
Основные нарушения функции сердца и гемодинамики показаны
на рис. 23.28. Депрессия силы и скорости сокращения, а также расслабле­
ния миокарда при сердечной недостаточности проявляется отклонениями
от нормы показателей функции сердца, центральной и органотканевой гемо­
динамики. Такие отклонения перечислены ниже.
Рис. 23.28. Основные нарушения функции сердца и центральной гемодинамики
при сердечной недостаточности
• Уменьшение ударного и минутного выброса сердца — результат депрессии
сократительной функции миокарда. В подавляющем большинстве слу­
чаев сердечный выброс ниже нормального (как правило, менее 3 л/мин).
Однако при некоторых состояниях сердечный выброс, предшествующий
развитию сердечной недостаточности, бывает выше нормального. Это
наблюдается, например, у пациентов с тиреотоксикозом, хроническими
анемиями, артериовенозными шунтами, после вливания избытка жидко­
сти в сосудистое русло. При развитии у этих пациентов сердечной недоста­
точности величина сердечного выброса остается выше нормы (например,
более 7—8 л/мин). Однако и в этих условиях отмечается недостаточность
кровоснабжения органов и тканей, поскольку сердечный выброс остается
ниже требующейся величины. Подобные состояния известны как сердеч­
ная недостаточность с высоким выбросом крови.
• Увеличение остаточного систолического объема крови в полостях желудочков
сердца — следствие так называемой неполной систолы. Неполное опорож­
нение желудочков сердца — результат избыточного притока крови к нему
(например, при клапанной недостаточности), чрезмерно повышенного
ОПСС (например, при артериальных гипертензиях, стенозе аорты), пря­
мого повреждения миокарда.
• Повышение конечного диастолического давления в желудочках сердца.
Обусловлено увеличением количества крови, скапливающейся в их поло­
сти; нарушением расслабления миокарда, дилатацией полостей сердца
вследствие увеличения в них объема крови и растяжения миокарда.
• Повышение давления крови в тех венозных сосудах и сердечных полостях,
откуда кровь поступает в пораженные отделы сердца. Так, при левожелу­
дочковой сердечной недостаточности повышается давление в левом пред­
сердии, малом круге кровообращения и правом желудочке. При правоже­
лудочковой сердечной недостаточности давление увеличивается в правом
предсердии и венах большого круга кровообращения.
• Снижение скорости систолического сокращения и диастолического рас­
слабления миокарда. Проявляется, главным образом, более длительным
периодом изометрического напряжения и систолы сердца в целом.
Формы сердечной недостаточности
Острая сердечная недостаточность
ОСН — внезапнее нарушение насосной функции сердца, приводящее
к невозможности адекватного кровообращения, несмотря на включение
компенсаторных механизмов.
Этиология острой сердечной недостаточности
ОСН чаще развивается вследствие заболеваний, приводящих к быстрому
и значительному снижению сердечного выброса (наиболее часто при инфар­
кте миокарда). Однако возможна ОСН и при высоком сердечном выбросе.
Основные причины ОСН перечислены ниже.
• ОСН с низким сердечным выбросом:
—инфаркт миокарда (большой объем поврежденного миокарда, разрыв
стенок сердца, острая недостаточность митрального клапана);
—декомпенсация ХСН (неадекватное лечение, аритмии, тяжелое сопут­
ствующее заболевание);
—аритмии (наджелудочковые и желудочковые тахикардии, брадикардии,
экстрасистолии, блокады проведения возбуждения);
—препятствия току крови (стенозы устья аорты и митрального отверстия,
гипертрофическая кардиомиопатия, опухоли, тромбы);
—клапанная недостаточность (митрального или аортального клапана);
— миокардиты;
—массивная тромбоэмболия легочной артерии;
— «легочное сердце»;
— артериальная гипертензия;
—тампонада сердца;
—травма сердца.
• ОСН с относительно высоким сердечным выбросом:
— анемия;
—тиреотоксикоз;
— острый гломерулонефрит с артериальной гипертензией;
— артериовенозное шунтирование.
ОСН имеет три основных клинических проявления:
1) сердечная астма;
2) острый кардиогенный отек легких;
3) кардиогенный шок.
В каждом конкретном случае указывается определенная форма ОСН (сер­
дечная астма, кардиогенный шок или отек легких); не следует употреблять
общий термин «острая сердечная недостаточность».
Сердечная астма (удушье, пароксизмальная ночная одышка) возникает
в результате застоя крови в малом круге кровообращения при положении
больного лежа как проявление интерстициального отека легких и резкого
увеличения давления крови в сосудах малого круга кровообращения.
Острый кардиогенный отек легких развивается при увеличении давления
заклинивания легочных капилляров более 25 мм рт.ст. Острый отек легких
подразделяют на:
—интерстициальный, проявляющийся быстрым отеком паренхимы легких
без выхода транссудата в просвет альвеол (проявляется одышкой и каш­
лем без мокроты). При прогрессировании, например при сердечной
астме, формируется альвеолярный отек. К основным звеньям патогенеза
интерстициального отека легких относят:
о повышение давления в просвете легочных капилляров;
Оувеличение лимфообразования и объема внесосудистой жидкости;
о возрастание сопротивления мелких бронхов;
о уменьшение растяжимости легочной ткани;
— альвеолярный отек легких, характеризующийся накоплением транссудата
в просвете альвеол. Длительное увеличение внутрисосудистого давления
приводит к нарушению целостности альвеолярно-капиллярной мем­
браны и выходу в полость альвеол жидкости, макромолекул и эритро­
цитов. У больных появляется кашель с отделением пенистой мокроты,
удушье, в легких выслушиваются вначале сухие, а затем влажные хрипы.
В последующем развиваются гипоксия, ацидоз и гиперкапния, приво­
дящие к остановке дыхания.
Указанные выше две разновидности отека легких рассматриваются как две
стадии одного процесса.
При остром отеке легких необходимы экстренные мероприятия, поскольку
смертность при нем составляет 15—20%.
Кардиогенный шок развивается в результате острого снижения сердечно­
го выброса. Как правило, он возникает при обширном инфаркте миокарда
на фоне множественного поражения венечных артерий. Подробно патофи­
зиология кардиогенного шока описана в статье «Шок кардиогенный» при­
ложения «Справочник терминов».
Хроническая сердечная систолическая недостаточность
ХССН — синдром, осложняющий течение ряда заболеваний. Характеризуется
наличием одышки, вначале при физической нагрузке, а затем и в покое;
быстрой утомляемостью; периферическими отеками и объективными при­
знаками нарушения функций сердца в покое (например, аускультативными
или эхокардиографическими).
Этиология и патогенез хронической систолической сердечной недостаточности
Основные причины, приводящие к развитию ХССН, перечислены ниже.
• Причины ХССН с низким сердечным выбросом:
—поражение миокарда при:
ОИ БС (постинфарктный кардиосклероз, хроническая миокардиальная
ишемия);
о кардиомиопатиях;
о токсических воздействиях на сердце (например, алкоголя, доксорубицина);
о инфильтративных заболеваниях (саркоидозе, амилоидозе);
о эндокринных формах патологии;
о нарушениях питания (например, дефицит витамина В,).
—перегрузка миокарда:
о при артериальных гипертензиях;
о пороках сердца.
— аритмии:
о наджелудочковые и желудочковые тахикардии;
о фибрилляция предсердий.
• Причины ХССН с относительно высоким сердечным выбросом:
—анемии;
—тиреотоксикоз;
— артериовенозное шунтирование.
Под влиянием указанных причин нарушается насосная функция сердца. Это
приводит к уменьшению сердечного выброса. В результате развивается гипо­
перфузия органов и тканей.
Наибольшее значение имеют:
—уменьшение кровоснабжения сердца, ведущее к активации симпатико-
адреналовой системы и учащению ритма сердца;
—снижение перфузии почек, обусловливающее стимуляцию гипертензивной ренин-ангиотензин-альдостероновой системы;
—снижение перфузии периферических мышц (и, как следствие, развитие
гипоксии), обеспечивающее накопление в них недоокисленных продук­
тов метаболизма.
Основные звенья патогенеза ХССН показаны на рис. 23.29.
Стадии хронической систолической сердечной недостаточности
Выделяют три стадии ХССН.
• Стадия I (начальная) — скрытая сердечная недостаточность. Проявляется
при физической нагрузке (одышкой, тахикардией, быстрой утомляемо­
стью).
• Стадия II (выраженная). Выявляется застоем в большом и малом круге кро­
вообращения, нарушениями функций органов и обмена веществ в покое.
• Стадия III (конечная, дистрофическая). Характеризуется тяжелыми нару­
шениями гемодинамики, стойкими изменениями обмена веществ и функ­
ций органов, необратимыми изменениями структуры тканей и органов.
Сердечная недостаточность диастолическая
СНД характеризуется нарушением расслабления и наполнения левого желу­
дочка. Обусловлена гипертрофией миокарда, его фиброзом или инфильтраци­
ей. СНД приводит к увеличению конечного диастолического давления в левом
желудочке и развитию сердечной недостаточности.
Снижение сократительной функции миокарда
Падение сердечного выброса
Стимуляция
симпатической
нервной
системы
Периферическая
вазоконстрикция
Накопление лактата,
атрофия
I
Увеличение
► концентрации ренина,
ангиотензина II
Бы страя
утом ляем ость
-
I
Увеличение
периферического
сопротивления
I
Возрастание
постнагрузки
на сердце
Увеличение концентрации
альдостерона
Задержка ионов
№ , воды
АДГ Т
Отеки
I
Возрастание объема
плазмы
Увеличение
преднагрузки
на сердце
Рис. 23.29. Патогенез хронической систолической сердечной недостаточности
Этиология и патогенез сердечной недостаточности диастолической
К возникновению СНД наиболее часто приводят:
— И Б С (с инфарктом миокарда или без него);
— гипертрофическая кардиомиопатия;
— амилоидоз сердца;
— артериальная гипертензия;
— клапанные пороки сердца;
-С Д ;
— констриктивный перикардит.
В результате снижения податливости и нарушения наполнения левого желудоч­
ка в нем повышается конечное диастолическое давление, что вызывает снижение
сердечного выброса. Это приводит к увеличению давления в левом предсердии
и малом круге кровообращения. В последующем может возникнуть правоже­
лудочковая сердечная недостаточность.
Принципы нормализации функции сердца
при его недостаточности
Лечебные мероприятия при сердечной недостаточности проводятся ком­
плексно. Они направлены на прекращение (снижение степени) патогенного
действия причинного фактора (этиотропная терапия), разрыв звеньев ее раз­
вития (патогенетическая терапия), потенцирование адаптивных процессов
(саногенетическая терапия).
В табл. 23.1 приведены принципы и цели патогенетической терапии сер­
дечной недостаточности, а также применяемые при ней группы фармаколо­
гических препаратов.
Таблица 23.1. Принципы и цели нормализации функции сердца при его недостаточ­
ности
Цели
Примеры ЛС
Этиотропный принцип
Уменьшить постнагрузку (снизить
Вазодилататоры, а-адреноблокаторы, адретонус резистивных сосудов)
нолитики, АПФ
Уменьшить преднагрузку (снизить
Венозные вазодилататоры, диуретики
возврат венозной крови к сердцу)
Патогенетический принцип
Повысить сократительную функ­
Адреномиметики, сердечные гликозиды,
цию сердца
ингибиторы фосфодиэстеразы
Уменьшить нарушения энергообес­
Антигипоксанты,антиоксиданты,коронапечения кардиомиоцитов
родилататоры
Защитить мембраны и фермен­
ЛС с мембранопротекторным эффектом
ты кардиомиоцитов от факторов
повреждения
Уменьшить дисбаланс ионов
и воды в миокарде
Скорректировать адрено- и холинергическое влияние на сердце,
его адрено- и холинореактивные
свойства
Регуляторы транспорта ионов через мембра­
ны (калийсберегающие вещества, блокаторы
кальциевых каналов и др.), препараты магния
Симпатолитики, ЛС с положительным инотропным действием (при достаточном мио­
кардиальном резерве), холиномиметики
При своевременно начатом лечении и рациональном его проведении воз­
можна длительная нормализация сердечной деятельности и системной гемо­
динамики.
Нарушения системного
артериального давления
Изменения системного АД подразделяют на гвпертензивные и гипотензивные
состояния.
• Гинертснзивные состояния характеризуются повышением ЛД сверх нормы.
К ним относятся гипертензивные реакции и артериальные гипертензии.
• Гипотензивные состояния проявляются снижением АД по соавнению с нор­
мой. Включают гипотензивные реакции и артериальные гипотензии.
Терминология
Для обозначения различных состояний и реакций, характеризующихся
изменениями АД, применяют специальные термины и понятия.
Важно различать значения терминологических элементов «-тония» и «-тензия».
Терминологический элемент «-тония» применяется для характеристики тону­
са мышц, в том числе ГМК сосудистой стенки.
Гипертония означает избыточное напряжение мышц, проявляющееся увели­
чением их сопротивления растяжению.
Гипотония подразумевает снижение напряжения мышц, проявляющееся
уменьшением их сопротивления растяжению.
Терминологический элемент «-тензия» используют для обозначения давле­
ния жидкостей в полостях и сосудах, в том числе кровеносных.
Гипертензия означает повышение, а гипотензия — снижение давления в поло­
стях организма, его полых органах и сосудах.
Адекватным для обозначения гипер- или гипотензивных состояний явля­
ется использование именно элемента «-тензия», поскольку уровень АД зави­
сит не только от тонуса ГМК сосудов (при некоторых видах гипертензии он
не повышен, а может быть даже ниже нормы), но и от величины минутного
выброса сердца и ОЦК.
Тем не менее термины «гипертоническая болезнь» и «гипертонический
криз» применяют для обозначения эссенциальной артериальной гипертен­
зии и осложнения (нередко фатального) артериальной гипертензии соот­
ветственно.
Л С (независимо от механизма их действия на тонус сосудов, сердечный
выброс, ОЦК) называют гипотензивными (ЛС, снижающие АД) и гипертензивными (ЛС, вызывающие повышение АД).
Необходимо различать понятия «гипер- и гипотензивная реакция» и «арте­
риальная гипер- или гипотензия».
Гипер- или гипотензивная реакция — адаптивная и преходящая (временная)
реакция ССС (после нее АД нормализуется в связи с прекращением действия
агента, вызвавшего реакцию), регулируемая физиологическими механизмами.
Артериальная гипер- или гипотензия носит стойкий характер, обычно
не устраняется после прекращения действия причинного фактора и сопро­
вождается повреждением органов и тканей, а также снижением адаптивных
возможностей организма.
АРТЕРИАЛЬНЫЕ ГИПЕРТЕНЗИИ
Артериальная гипертензия — состояние, при котором систолическое АД
составляет 140 мм рт.ст. и белее и/или диастолическое АД 90 мм рт.ст. и более.
(При условии, что эти значения получены в результате как минимум трех
измерений, произведенных в различное время в спокойной обстановке,
а пациент за сутки до измерения не принимал ЛС, изменяющих АД.)
Классификация артериальной гипертензии
Эксперты ВОЗ и Международного общества гипертензии предложили
классификацию артериальной гипертензии по уровню АД (табл. 23.2).
Таблица 23.2. Классификация артериальной гипертензии
Категория
Оптимальное
Нормальное
Высокое нормальное
I степень (мягкая)
подгруппа: пограничная
II степень (умеренная)
III степень (выраженная)
Изолированная систолическая
подгруппа: пограничная
Систолическое АД,
мм рт.ст.
<120
<130
Диастолическое АД,
мм рт.ст.
<80
<85
130-139
8 5 -8 9
9 0 -9 9
9 0 -9 4
140-159
140-149
160-179
>180
>140
100-109
>110
<90
140-149
<90
Примечание. При определении категории следует использовать наибольшее значе­
ние АД, например 140/100 мм рт.ст. — степень II артериальной гипертензии.
Если удается выявить причины артериальной гипертензии, то ее считают
вторичной (симптоматической).
При отсутствии явной причины гипертензии она называется первичной,
эссенциальной, идиопатической, а в России — гипертонической болезнью (ГБ).
Изолированная систолическая артериальная гипертензия диагностируется
при повышении систолического АД более 140 мм рт.ст. и диастолического
АД менее 90 мм рт.ст.
Артериальную гипертензию считают злокачественной при уровне диастоли­
ческого АД более 120 мм рт.ст.
Стремительное и катастрофическое по последствиям развитие артериаль­
ной гипертензии наблюдается при гестозах.
Распространенность артериальных гипертензий
Артериальной гипертензией страдает в среднем 25% взрослого населения.
С возрастом распространенность увеличивается и достигает 65% у людей
старше 65 лет.
В пожилом возрасте преобладает изолированная систолическая артери­
альная гипертензия, которая в возрасте до 50 лет встречается менее чем у 5%
населения.
До 50-летнего возраста артериальная гипертензия чаще бывает у мужчин,
а после 50 лет — у женщин.
Среди всех форм артериальной гипертензии на долю мягкой и умеренной
приходится около 70—80%, в остальных случаях наблюдают выраженную арте­
риальную гипертензию.
Риск поражения сердца и сосудов
По мере накопления эпидемиологических данных о естественном течении
заболевания стал очевидным факт постоянного повышения риска сердечно­
сосудистой заболеваемости и смертности в связи с повышения АД. Однако
четко разграничить нормальный и патологический уровень АД оказалось
невозможно, так как риск осложнений повышается при увеличении АД даже
в пределах его нормального диапазона. При этом абсолютное большинство
сердечно-сосудистых осложнений регистрируется у людей с небольшим повы­
шением АД.
У больных артериальной гипертензией прогноз зависит не только от уровня
АД. Наличие сопутствующих факторов риска, степень вовлечения в процесс
органов-мишеней, а также наличие ассоциированных клинических состоя­
ний имеет не меньшее значение, чем степень повышения АД. В связи с этим
в современную классификацию введена стратификация больных в зависи­
мости от степени риска. Она основывается на традиционной оценке пора­
жения органов-мишеней и сердечно-сосудистых осложнений. Это позволяет
качественно оценить индивидуальный прогноз (чем выше риск, тем хуже
прогноз) и выделить группы для преимущественной социально-медицинской
поддержки.
Оценка индивидуального риска
Для количественной оценки риска используют методики расчета риска
ИБС в течение последующих 10 лет, предложенные Европейским обще­
ством кардиологов, Европейским обществом по атеросклерозу и Европейским
обществом по гипертензии. Общий риск сердечно-сосудистых осложнений
рассчитывается с учетом риска ИБС (риск ИБС умножают на коэффициент
4/3; например, если риск ИБС составляет 30%, то риск сердечно-сосудистых
осложнений равен 40%).
Клинические проявления сердечно-сосудистых заболеваний и поражения
органов-мишеней расцениваются как более значимые прогностические фак­
торы по сравнению с традиционными факторами риска (см. ниже в разделе
«Этиология и патогенез артериальных гипертензий»). Такой подход позволяет
врачам использовать упрошенный метод, определяя уровень риска для каж­
дого отдельного пациента, дает четкое представление о долговременном про­
гнозе и облегчает принятие решения о сроках начала, характере антигипертензивной терапии и целевом уровне АД.
Органы-мишени при артериальной гипертензии
Одно из последствий длительного повышения АД — поражение внутрен­
них органов, так называемых органов-мишеней. К ним относят сердце, голов­
ной мозг, почки, сосуды. При артериальной гипертензии могут наблюдаться
следующие поражения органов:
—сердце:
о гипертрофия левого желудочка;
о стенокардия;
о инфаркт миокарда;
о сердечная недостаточность;
о внезапная сердечная смерть;
—головной мозг:
о тромбозы;
о кровоизлияния;
о гипертоническая энцефалопатия;
о церебральные лакуны;
— почки:
о микроальбуминурия;
о протеинурия;
о хроническая почечная недостаточность (ХПН);
—сосуды — вовлечение в процесс сосудов:
о сетчатки глаз;
о сонных артерий;
о аорты (аневризма).
У нелеченых пациентов с артериальной гипертензией 4/5 летальных исхо­
дов обусловлено сердечными причинами, из них 43% — ХСН, 36% — недо­
статочность венечных артерий. Цереброваскулярные и почечные причины
летального исхода более редки: 14 и 7% соответственно.
Виды артериальной гипертензии по патогенезу
По критерию инициального звена патогенеза, изменению сердечного
выброса, типу повышения АД и характеру клинического течения артериаль­
ные гипертензии подразделяются на несколько видов (рис. 23.30).
• По инициальному звену патогенеза артериальной гипертензии выделяют
общие и местные артериальные гипертензии.
— Общие (системные) артериальные гипертензии по механизму развития
подразделяются на нейрогенные, эндокринные, метаболические, гемические и смешанные:
Рис. 23.30. Виды артериальных гипертензий
о нейрогенные артериальные гипертензии. Среди них выделяют центро­
генные и рефлекторные (рефлексогенные);
о эндокринные (эндокриногенные, гормональные). Они развиваются вслед­
ствие эндокринопатий надпочечников, шитовидной железы и гипофи­
за;
о гипоксические (метаболитогенные, метаболические, ишемические).
Выделяют ишемические (почечно-ишемическая, цереброишемическая), венозно-застойные и гипоксические (без первичного наруше­
ния гемодинамики в органах и тканях);
о гемические («кровяные»). Развиваются вследствие увеличения объема
и/или вязкости крови;
- Местные (регионарные) артериальные гипертензии. Являются, как прави­
ло, результатом значительного сужения участка артериального русла.
• По изменению сердечного выброса артериальные гипертензии подразделя­
ются на гиперкинетические и гипокинетические.
- При гиперкинетических гипертензиях повышен сердечный выброс
(при нормальном или пониженном ОПСС).
- При гипокинетических артериальных гипертензиях сердечный выброс
понижен (при увеличенном ОПСС).
- Эукинетические артериальные гипертензии характеризуются нормальным
сердечным выбросом и повышенным ОПСС.
• По типу повышения АД различают систолические, диастолические и смешан­
ные (систолодиастолические) артериальные гипертензии.
• По характеру клинического течения выделяют артериальные гипертензии
злокачественного и доброкачественного течения.
—«Доброкачественные» гипертензии характеризуются медленным развити­
ем; повышением как систолического, так и диастолического АД; они,
как правило, эукинетические.
—«Злокачественные» артериальные гипертензии быстро прогрессируют,
протекают с преимущественным повышением диастолического АД;
они, как правило, гипокинетические, реже — гиперкинетические (осо­
бенно на начальном этапе).
Этиология и патогенез артериальных гипертензий
Системный уровень АД в организме определяется:
—сердечным выбросом;
— общим периферическим сосудистым сопротивлением (ОПСС);
— объемом циркулирующей крови (ОЦК) и ее вязкостью.
Увеличение сердечного выброса, ОПСС, ОЦК и вязкости крови вызывает
повышение АД, и наоборот.
Причинами развития артериальных гипертензий являются две группы воз­
действий (см. рис. 23.39):
—внутренние гуморальные и нейрогенные факторы (ренин-ангиотензинальдостероновый комплекс, симпатоадреналовая система, баро- и хемо­
рецепторы и др.);
—внешние факторы (например, чрезмерное потребление поваренной соли,
алкоголя, избыточная масса тела).
Системным вазопрессорным эффектом обладают ангиотензин II, вазопрессин, эндотелии, а вазодепрессорным — натрийуретические пептиды, компо­
ненты калликреин-кининовой системы, оксид азота, простагландины (ПГ12,
простациклин).
К главным факторам риска возникновения артериальных гипертензий относят:
—отягощенный семейный анамнез (нарушения липидного обмена у самого
пациента и/или у его родителей, СД у пациента и его родителей, заболе­
вания почек у родителей и др.);
— ожирение;
— злоупотребление алкоголем;
—избыточное употребление поваренной соли;
—повторные и затяжные негативные стрессорные состояния;
—гиподинамия;
—курение;
—ряд других факторов.
Артериальные гипертензии (включая ГБ) существенно различаются по звеньям
патогенеза, и прежде всего их инициального, стартового звена.
Патогенез общ их артериальных гипертензий
Общие (системные) артериальные гипертензии подразделяются в зави­
симости от инициального звена патогенеза на нейрогенные, эндокринные,
метаболические, гемические и смешанные.
Нейрогенные артериальные гипертензии
Эти гипертензии характеризуются либо повышением гипертензивных ней­
рогенных влияний, либо ослаблением гипотензивных нейрогенных эффектов,
либо (чаще) сочетанием того и другого. Эти гипертензии составляют примерно
половину всех артериальных гипертензий. Их подразделяют на рефлекторные
(рефлексогенные) и центрогенные (рис. 23.31).
Рис. 23.31. Виды нейрогенных артериальных гипертензий
Центрогенные артериальные гипертензии
Корковые и подкорковые нейроны, участвующие в регуляции АД, пред­
ставляют собой сложную функциональную систему, состоящую из прессорногипертензивной и депрессорно-гипотензивной подсистем.
В норме доминируют прессорно-гипертензивные механизмы.
Главной структурой, регулирующей системное АД, является кардиовазомоторный (сосудодвигательный) центр. Его эфферентные влияния изменяют
как тонус сосудов, так и функцию сердца.
Наиболее частые причины центрогенных нейрогенных гипертензий:
—расстройства ВНД (невротические состояния);
—органические поражения структур мозга, регулирующих системную гемоди­
намику.
Основные звенья патогенеза центрогенных нейрогенных артериальных
гипертензий изображены на рис. 23.32.
Патогенез артериальных гипертензий, вызванных нарушением ВНД (неврозом)
Повторные и затяжные стресс-реакции с негативной эмоциональной окраской
вызывают цепь взаимозависимых прогрессирующих изменений. Наиболее
важны:
—перенапряжение и срыв основных корковых нервных процессов (возбужде­
ния и активного коркового торможения), нарушение их сбалансирован­
ности и подвижности, приводящее к невротическому состоянию;
—развитие невроза (инициального патогенетического звена артериальных
гипертензий центрогенного характера) обусловливает формирование
корково-подкоркового комплекса возбуждения (доминанты возбужде­
ния); этот комплекс включает симпатические ядра заднего гипоталаму­
са, адренергические структуры ретикулярной формации и кардиовазомоторного центра;
—усиление влияний симпатической нервной системы на органы и ткани;
характеризуется высвобождением избытка катехоламинов и повышением
под их влиянием тонуса стенок артериальных и венозных сосудов, а также
увеличением ударного и минутного выброса крови из сердца, что и при­
водит к повышению как систолического, так и диастолического АД;
— активация (в связи с возбуждением подкорковых нервных центров) и дру­
гих центральных «гипертензивных» систем. Основной среди них является
система «гипоталамус — гипофиз — надпочечники»; это сопровождается
увеличением продукции и концентрации в крови гормонов с гипертензивным действием (АДГ, АКТГ и кортикостероидов, включая минералои глюкокортикоиды, а также катехоламинов, тиреоидных гормонов);
—потенцирование степени и длительности сужения артериол и венул, увели­
чения ОЦК, повышения сердечного выброса.
Рис. 23.32. Основные звенья патогенеза центрогенных нейрогенных артериальных
гипертензий
В целом, указанный выше комплекс патогенных изменений ведет к стой­
кому и значительному повышению АД — развивается артериальная гипертен­
зия. Важно, что описанные выше звенья патогенеза центрогенной артериаль­
ной гипертензии характерны и для инициальных этапов ГБ (эссенциальной
гипертензии).
Патогенез артериальных гипертензий, вызванных органическим повреждением
структур мозга, которые участвуют в регуляции системного АД.
Основные причины гипертензии такого генеза:
—травмы (например, при сотрясении или ушибах мозга);
—энцефалиты;
—опухоли мозга или его оболочек, приводящие к его сдавлению;
—кровоизлияние в желудочки мозга;
—очаговые ишемические поражения мозга.
Общий патогенез гипертензий в результате органического повреждения мозга
включает:
—прямое повреждение структур, участвующих в регуляции уровня АД (сим­
патических ядер гипоталамуса, ретикулярной формации, кардиовазомоторного центра);
— активацию симпатоадреналовой системы и комплекса «гипоталамусгипофиз—надпочечники»;
—повышение в крови содержания прессорных гормонов: катехоламинов,
тиреоидных, АДГ, кортикостероидов. Указанные гормоны обусловливают
увеличение тонуса стенок резистивных сосудов и как следствие — ОПСС,
рост ОЦК и сердечного выброса — развивается артериальная гипертензия.
Цереброишемическая артериальная гипертензия
Причина ее — ишемия головного мозга, особенно продолговатого, где рас­
положен сосудодвигательный центр. Наиболее часто это наблюдается при:
—церебральном атеросклерозе (преимущественно ветвей внутренних сон­
ных артерий);
—тромбозе кровоснабжающих мозг сосудов;
— опухолях, сдавливающих мозг и его сосуды.
Механизм развития цереброишемической артериальной гипертензии
Мозг весьма чувствителен к снижению содержания кислорода в крови.
В организме сформировалась система защиты мозга, активирующаяся при его
ишемии и обеспечивающая повышение системного АД. Это ведет к увеличе­
нию перфузионного давления во всех органах, включая мозг.
При длительном нарушении мозгового кровообращения происходит стойкое
и продолжительное повышение системного АД в результате цепи следующих
патогенных процессов:
—активации (при значительном снижении перфузионного давления в сосу­
дах мозга) симпатико-адреналовой системы, что приводит к существенно­
му и стойкому увеличению концентрации катехоламинов в крови;
—значительного увеличения сердечного выброса (за счет положительного
хроно- и инотропного эффекта) и сужения артериол, что влечет за собой
значительное повышение ОПСС и системного АД, т.е. артериальную
гипертензию;
—потенцирования гипертензии в связи с увеличением в крови р С 0 2 (извест­
но, что повышенный уровень р С 0 2 активирует нейроны вазомоторного
центра и увеличивает возбудимость нейронов других структур мозга; это
еще более активирует симпатическую нервную систему, что сопровожда­
ется нарастанием гиперкатехоламинемии и ростом АД).
Рефлекторные нейрогенные артериальные гипертензии
Рефлекторные (рефлексогенные) нейрогенные артериальные гипертензии
могут развиваться на основе условных и безусловных рефлексов.
Условнорефлекторные артериальные гипертензии
Известны случаи развития рефлекторной гипертензии в форме так назы­
ваемой артериальной гипертензии «белого халата». Для нее характерно повы­
шение АД в лечебном учреждении, в то время как за его пределами АД
нормальное. При проведении суточного мониторирования АД находят нор­
мальные величины среднесуточного АД: менее 125/80 мм рт.ст.
Причина рефлекторных гипертензий — повторное сочетание индифферент­
ных (условных) сигналов (например, информации о предстоящем публичном
выступлении, важном соревновании или событии) с действием агентов, вызы­
вающих повышение АД (например, кофеина, адреномиметиков, психостиму­
ляторов, алкоголя или наркотиков).
После определенного числа сочетаний увеличение АД регистрируется уже
только на индифферентный сигнал. Через некоторое время может развиться
стойкое повышение АД.
Так, описан случай артериальной гипертензии у человека, принимавшего
перед чтением лекций и докладов кофеин (для психологического допинга).
Вскоре у него регистрировалось повышение АД до 180 и 115 мм рт.ст. уже
при одной мысли о предстоящей лекции. Спустя несколько лет у этого чело­
века развилась артериальная гипертензия.
Безусловнорефлекторные артериальные гипертензии
Безусловнорефлекторные артериальные гипертензии развиваются в резуль­
тате:
—хронического раздражения экстеро- и интерорецепторов, нервных ство­
лов и нервных центров;
—прекращения «депрессорной» афферентной импульсации.
Гипертензии в результате хронического раздражения экстеро- и интероре­
цепторов, нервных стволов или нервных центров наблюдаются при длительно
протекающих болевых (каузалгических) синдромах (например, при поврежде­
нии или воспалении тройничного, лицевого, седалищного и других нервов),
энцефалитах, опухолях мозга (например, в области зрительных бугров, про­
межуточного или продолговатого мозга).
Гипертензии вследствие прекращения афферентной импульсации, оказы­
вающей тормозящее влияние на тоническую активность кардиовазомоторного (прессорного) центра (например, от рецепторов дуги или разветвления
сонной артерии), чаще всего развиваются при выраженном атеросклерозе
или артериосклерозе указанных сосудов. Это приводит к «высвобождению»
кардиовазомоторного центра от депрессорных влияний и к развитию арте­
риальной гипертензии.
Эндокринные артериальные гипертензии
Эти гипертензии составляют около 1% всех артериальных гипертензий
(по данным специализированных клиник, — до 12%) и развиваются в резуль­
тате гипертензивного действия ряда гормонов (рис. 23.33).
Гиперпродукция гормонов с
гипертензивным действием
- катехоламинов;
-вазопрессина;
-А К Т Г ;
- минералокортикоидов;
-эндотелина;
-тиреоидных___________
Повышенная чувствительность
сердца и стенок артерий
к гормонам с гипертензивным
действием
Увеличение:
-о б щ его периферического сосудистого сопротивления;
-объ ем а циркулирующей крови;
-сердечного выброса крови
[Артериальная гипертензия]
Рис. 23.33. Общие звенья патогенеза при эндокринных артериальных гипертензиях
Артериальная гипертензия при эндокринопатиях надпочечников
Не менее половины всех пациентов с эндокринными гипертензиями при­
ходится на надпочечниковые артериальные гипертензии.
Надпочечники — главный эндокринный орган, обеспечивающий регу­
ляцию системного АД. Все гормоны надпочечников в норме имеют более
или менее выраженное отношение к регуляции АД, а в патологии участвуют
в формировании и закреплении повышенного АД.
Надпочечниковые артериальные гипертензии подразделяются на две груп­
пы — «катехоламиновые» и «кортикостероидные».
«Катехоламиновые» гипертензии — катехоламины одновременно увеличи­
вают тонус сосудов и стимулируют работу сердца. Гипертензии развиваются
в связи со значительным увеличением в крови уровня катехоламинов —
и адреналина, и норадреналина, вырабатываемых хромаффинными клетками.
Норадреналин стимулирует в основном а-адренорецепторы и в меньшей мере —
р-адренорецепторы. Это приводит к повышению АД за счет сосудосужива­
ющего эффекта. Адреналин воздействует как на а - , так и на Р-адренорецепторы.
В связи с этим наблюдается вазоконстрикция (и артериол, и венул), возрас­
тание работы сердца (за счет положительного хроно- и инотропного эффекта)
и выброс крови из депо в сосудистое русло. В совокупности эти эффекты
и обусловливают развитие артериальной гипертензии.
В 99% всех случаев такой гипертензии у пациентов обнаруживают феохромоцитому (проявления феохромоцитомы разнообразны и неспецифичны:
артериальную гипертензию выявляют в 90% случаев, головную боль — в 80%,
ортостатическую артериальную гипотензию — в 60%, потливость — в 65%,
сердцебиение и тахикардию — в 60%, бледность — в 45%, тремор конечно­
стей — в 35%. В 50% случаев артериальная гипертензия может быть посто­
янной, и в 50% — сочетаться с гипертензивными кризами. Криз обычно воз­
никает без связи с внешними факторами. Часто наблюдается гипергликемия
(в результате стимуляции гликогенолиза).
«Кортикостероидные» артериальные гипертензии. Их разделяют на минералокортикоидные и глюкокортикоидные.
Минералокортикоидные артериальные гипертензии развиваются в резуль­
тате избыточного синтеза минералокортикоида альдостерона (при первич­
ном и вторичном гиперальдостеронизме). Кортизол, 11-дезоксикортизол,
11-дезоксикортикостерон, кортикостерон, хотя и имеют определенную
минералокортикоидную активность, в основном оказывают глюкокортикоидный эффект (их суммарная доля в развитии артериальной гипертензии
мала). Гиперальдостеронизм любого генеза сопровождается повышением АД.
Патогенез артериальных гипертензий при гиперальдостеронизме показан
на рис. 23.34.
Глюкокортикоидные артериальные гипертензии являются результатом
гиперпродукции глюкокортикоидов с минералокортикоидной активностью
(в основном 17-гидрокортизона, гидрокортизона — на его долю приходит­
ся 80%; остальные 20% — кортизон, кортикостерон, 11-дезоксикортизол и
11-дезоксикортикостерон). Артериальные гипертензии глюкокортикоидного
генеза развиваются при болезни и синдроме Иценко-Кушинга (см. в при­
ложении «Справочник терминов»).
Артериальная гипертензия при эндокринопатиях щитовидной железы.
Встречается как при гипертиреозе, так и при гипотиреозе.
Гипертиреоз сопровождается увеличением ЧСС и сердечного выброса,
преимущественно изолированной систолической артериальной гипертензией
с низким (или нормальным) диастолическим АД. Считают, что увеличение
диастолического АД при гипертиреозе — признак другого заболевания, сопро­
вождающегося артериальной гипертензией, или симптом ГБ.
Гипотиреоз нередко сочетается с повышенным диастолическим АД. Другие
проявления со стороны ССС — уменьшение ЧСС и сердечного выброса.
В обоих случаях для уточнения диагноза необходимо определение функций
щитовидной железы.
Патогенез «гипертиреоидных» артериальных гипертензий. В основе развития
гипертензии лежит кардиотонический эффект Т3 и Т4. Он характеризуется
значительным увеличением минутного выброса сердца. Это достигается бла­
годаря выраженной тахикардии (в связи с положительным хронотропным
эффектом) и увеличению ударного выброса (за счет положительного инотропного эффекта тиреоидных гормонов).
Артериальная гипертензия при расстройствах эндокринной функции гипоталамо-гипофизарной системы.
Наибольшее клиническое значение имеет артериальная гипертензия,
развивающаяся при значительном и длительном увеличении в крови АДГ
и АКТГ.
Гиперпродукция АДГ. Патогенез АДГ-зависимой артериальной гипертензии:
— избыток АДГ стимулирует реабсорбцию жидкости почками из первичной
мочи. Это происходит при взаимодействии АДГ с его рецепторами, свя­
занными с водными каналами — аквапоринами. Как следствие, увели­
чивается ОЦК (развивается гиперволемия). Это само по себе уже может
привести к увеличению уровня АД;
— гиперволемия вызывает также повышение сердечного выброса. Повышен­
ный ОЦК, растягивая миокард, увеличивает (согласно закону Ф ранка-
Почечные эффекты
| Стимуляция реабсорбции ионов Ыа1
Гиперосмия крови |
| Активация синтеза и инкреции АДГ
Внепочечные эффекты
^Транспорт избытка № *в клетки
Набухание клеток,
в том числе эндотелия
и миоцитов стенок сосудов
Повышение тонуса
миоцитов стенок
сосудов и сердца
Увеличение чувствительности
стенок сосудов и миокарда
к гипертензивным агентам
_А_
Реабсорбция избытка жидкости
Гиперволемия
Увеличение:
-о б щ его периферического сосудистого сопротивления;
-объ ем а циркулирующей крови;
-сердечного выброса крови
_А_
Артериальная гипертензия |
Рис. 23.34. Общие звенья патогенеза артериальных гипертензий при гиперальдостеронизме
160___________________________________________________________________________________
Увеличение содержания альдостерона в крови
Старлинга) силу его сокращений и как следствие — величину выброса
сердца и АД;
—стимуляция АДГ его рецепторов на ГМ К стенок артериол приводит к суже­
нию их просвета, повышению ОПСС и уровня АД.
Гиперпродукция АКТТ. При гиперпродукции АКТГ аденогипофизом развива­
ется болезнь Иценко—Кушинга (см. в приложении «Справочник терминов»).
Артериальная гипертензия при паранеопластических эндокринопатиях
Артериальные гипертензии часто развиваются при паранеопластических
синдромах (см. «Синдром паранеопластический эндокринный» в приложении
«Справочник терминов»).
Артериальны е гипертензии, вызванные гипоксией тканей
и органов
Артериальные гипертензии, развивающиеся в результате гипоксии органов
(особенно мозга и почек), нередко встречаются в клинической практике. Они
обозначаются как «гипоксические» (или метаболические, органоишемиче­
ские). К ним относят артериальные гипертензии, в основе патогенеза которых
лежат нарушения метаболизма веществ с гипо- и гипертензивным действи­
ем. Возникают эти гипертензии в результате расстройств кровообращения
и последующей гипоксии различных внутренних органов.
Причина гипоксических (метаболических) артериальных гипертензий — избы­
ток гипо- и гипертензивных метаболитов.
Метаболические гипертензии возникают вследствие увеличения эффектов
на клетки- и органы-мишени гипертензивных (прессорных) метаболитов
и/или уменьшения эффектов гипотензивных (депрессорных) метаболитов.
Метаболиты с гипертензивными и гипотензивными эффектами приведены
на рис. 23.35.
Основные звенья патогенеза гипоксических артериальных гипертензий
представлены на рис. 23.36.
Клинические разновидности гипоксических артериальных гипертензий
Наиболее частыми клиническими разновидностями гипоксических (метабо­
лических) артериальных гипертензий являются цереброишемическая гипертензия
(см. выше) и почечные (вазоренальная и ренопаренхиматозная) гипертензии.
Вазоренальная артериальная гипертензия
Вазоренальная (или реноваскулярная, почечно-ишемическая) артериаль­
ная гипертензия — симптоматическая артериальная гипертензия, вызванная
ишемией почки (почек) вследствие уменьшения просвета почечных артерий.
Распространенность почечно-ишемической гипертензии составляет 1-2%
(по данным специализированных клиник, — до 4—16%) среди всех видов
артериальных гипертензий. Из них 60-70% возникает при атеросклерозе
почечных артерий, 30-40% — при фибромускулярной дисплазии, менее 1% —
при аневризме почечной артерии, тромбозе почечных артерий, почечных
артериовенозных фистулах, тромбозе почечных вен).
Патогенез почечно-ишемических артериальных гипертензий
Основные звенья патогенеза вазоренальной артериальной гипертензии
показаны на рис. 23.37.
162
Рис. 23.35. Метаболиты с гипертензивным и гипотензивным действием, участвующие в развитии артериальных гипер- и гипотензий
Патоф изиология, клиническая патоф изиология
Рис. 23.36. Общие звенья патогенеза гипоксических артериальных гипертензий
Рис. 23.37. Основные звенья патогенеза вазоренальной (почечно-ишемической) арте­
риальной гипертензии
Ключевое звено патогенеза вазоренальной гипертензии — активация ренинангиотензиновой системы в результате гипоперфузии почки и нарушения энер­
гообеспечения ее клеток, в том числе ЮГА. Это ведет к спазму ГМ К стенок
артериол и нарастанию ОПСС (под влиянием ангиотензина II), увеличению
синтеза альдостерона и задержке в организме под его влиянием ионов натрия
и воды, увеличению ОЦК, а также — к стимуляции симпатической нервной
системы.
Более подробно отдельные звенья патогенеза вазоренальной гипертен­
зии рассмотрены в разделе «Почечные отеки» главы 12, а также в статьях
«Ангиотензины», «Ренин», «Вазопрессин (АДГ)», «Синдром неадекватной
секреции АДГ», «Ингибитор АПФ», «Система ренин-ангиотензин-альдостероновая», «Альдостерон».
Ренопаренхиматозная артериальная гипертензия
Ренопаренхиматозная (или ренопривная, от лат. геп — почка, р т о —
лишать чего-либо) артериальная гипертензия — симптоматическая (вторичная)
артериальная гипертензия, вызванная врожденным или приобретенным заболева­
нием почек, которое приводит к существенному уменьшению их массы.
Распространенность. Ренопаренхиматозная артериальная гипертензия
составляет 2—3% (по данным специализированных клиник, — 4—5%) среди
всех видов артериальных гипертензий.
Причина ренопривной гипертензии — уменьшение массы паренхимы почек,
вырабатывающей БАБ с гипотензивным действием (ПГ групп Е и I с сосудо­
расширяющим эффектом, брадикинин и каллидин). Чаще всего это наблю­
дается при двусторонних (гломерулонефриты, диабетическая нефропатия,
тубулоинтерстициальный нефрит, поликистоз) и односторонних поражениях
почек (пиелонефрит, опухоль, травма, одиночная киста почки, ее гипоплазия,
туберкулез). Наиболее частая причина — диффузный гломерулонефрит.
Патогенез. В патогенезе ренопаренхиматозной артериальной гипертензии
ключевое значение имеют:
—гиперволемия;
—гипернатриемия (из-за уменьшения количества функционирующих
нефронов и активации ренин-ангиотензиновой системы);
—увеличение ОПСС при нормальном или сниженном сердечном выбросе.
Последовательные этапы патогенеза ренопривной артериальной гипертен­
зии изображены на рис. 23.38.
| Уменьшение массы паренхимы почек |
Рис. 23.38. Основные звенья патогенеза ренопаренхиматозной артериальной гипертензии
Проявляется ренопривная артериальная гипертензия симптомами артери­
альной гипертензии и основного заболевания почек. Основными признаками
этой артериальной гипертензии считают:
—наличие заболеваний почек в анамнезе;
—изменения в анализах мочи (протеинурия более 2 г/сут, цилиндрурия,
гематурия, лейкоцитурия, высокая концентрация креатинина крови);
—выявляемые с помощью УЗИ признаки поражения почек.
Обычно изменения в анализах мочи предшествуют повышению АД.
Гемические артериальные гипертензии
Изменения состояния крови (увеличение ее ОЦК и/или вязкости) неред­
ко служат причиной артериальной гипертензии. Так, при полицитемиях
(истинной и вторичных), гиперпротеинемии и других подобных состояниях
в 25-50% случаев регистрируется стойкое повышение АД.
Смеш анны е артериальные гипертензии
Помимо указанных выше, артериальные гипертензии могут развиваться
в результате одновременного включения нескольких механизмов. Например,
артериальные гипертензии при повреждении мозга или развитии аллергиче­
ских реакций формируются с участием нейрогенного, эндокринного и почеч­
ного патологических факторов.
Лекарственные артериальные гипертензии
В патогенезе артериальных гипертензий, вызванных ЛС, основное значе­
ние имеют:
—длительная вазоконстрикция вследствие стимуляции симпатико-адреналовой системы или прямого воздействия вазопрессорных веществ на
ГМК кровеносных сосудов;
—увеличение О Ц К и вязкости крови;
— активация ренин-ангиотензиновой системы;
— задержка в организме избытка ионов натрия и воды;
— активация ЛС центральных вазопрессорных механизмов.
Адреномиметики. Капли в нос и лекарства от насморка, содержащие адре-
номиметические или симпатомиметические средства (например, эфедрин,
псевдоэфедрин, фенилэфрин), нередко повышают АД.
Контрацептивы для приема внутрь. Возможные механизмы гипертензивного
действия пероральных контрацептивов, содержащих эстрогены:
— стимуляция ренин-ангиотензиновой системы;
—задержка жидкости.
Артериальная гипертензия при приеме контрацептивов развивается при­
мерно у 5% женщин.
Н ПВС вызывают артериальную гипертензию в результате подавления син­
теза ПГ, обладающих вазодилатирующим эффектом, а также в связи с задерж­
кой жидкости.
Трициклические антидепрессанты могут вызвать повышение АД, стимулируя
симпатическую нервную систему.
Глюкокортикоиды повышают АД, так как увеличивают чувствительность
артериол к ангиотензину II и норадреналину, а также в результате задержки
жидкости.
Алкогольная артериальная гипертензия
В 5—25% (!) случаев причиной артериальной гипертензии является хрони­
ческое употребление алкоголя. Точный механизм гипертензивного действия
алкоголя не известен. Обсуждается значение стимуляции симпатической
нервной системы, повышенной продукции глюкокортикоидов, гиперинсу-
линемии, усиленный захват ионов кальция клетками и повышение ОПСС
под влиянием алкоголя.
Артериальная гипертензия у пожилых людей
К возрастной категории пожилых относят людей старше 65 лет.
Распространенность артериальной гипертензии в этой возрастной группе
достигает 50%. Артериальная гипертензия у пожилых может быть изолирован­
ной систолической или одновременно систолической и диастолической.
Патогенетически, кроме других факторов, влияющих на повышение АД,
у пожилых людей большое значение имеет уменьшение эластичности стенок
аорты, что выражается в увеличении систолического АД и снижении диа­
столического АД. Эпидемиологические исследования показали, что по срав­
нению с повышением диастолического АД увеличение систолического АД
имеет большее значение для прогнозирования риска сердечно-сосудистых
осложнений и требует лечения.
Артериальная гипертензия при берем енности
Частота артериальных гипертензий при беременности может достигать
10%, частота эссенциальной артериальной гипертензии — 3%.
Достоверно об эссенциальной артериальной гипертензии у беременной
можно говорить при ее наличии до беременности, при выраженных клини­
ческих проявлениях гипертензивного состояния (головные боли, носовые
кровотечения), положительном семейном анамнезе по ГБ.
В зависимости от ее стадии нарастает риск осложнений для беременной
и плода. Возможные осложнения при этом — преэклампсия и эклампсия,
преждевременная отслойка плаценты, перинатальная гибель плода (более
4% случаев), инфаркт миокарда и кровоизлияния в головной мозг у бере­
менной.
Преэклампсия
Преэклампсия — гестоз с клинической картиной, характерной для нарушения
мозгового кровообращения (головная боль, головокружение, нарушения зрения,
тошнота, рвота, боли в подложечной области), развивающийся во второй поло­
вине беременности.
Основная причина преэклампсии — спазм периферических сосудов.
Этиология и патогенез преэклампсии
Во время нормально протекающей беременности вещества с вазоконстрикторным действием разрушаются ферментами и/или нейтрализуются
веществами плаценты. У беременных с преэклампсией выявлена недостаточ­
ность ферментов.
Локально образующиеся вазоконстрикторные агенты вызывают спазм
артериол стенок сосудов и уменьшение плацентарного кровотока. В резуль­
тате снижается кровоснабжение плода и происходит задержка его развития.
Вещества с вазоконстрикторным действием, циркулирующие в сосудистой
системе беременной, вызывают у нее артериальную гипертензию и снижение
почечного кровотока (что приводит к гипоксии клубочков почки, протеинурии, задержке воды, отекам).
Факторы риска преэклампсии
Возраст беременной. Вероятность преэклампсии увеличивается с возрас­
том.
Первые роды. Преэклампсия характерна главным образом для первородя­
щих, особенно для женщин экстремального детородного возраста (т.е. под­
ростков и женщин старше 35 лет).
Генетические аспекты: дефекты гена ангиотензиногена Л О Т (*106150, 1ц42—
Я43), недостаточность гидроксиацил-КоА-дегидрогеназы, недостаточность
фактора свертывания V, цистиноз, недостаточность метилентетрагидрофолатредуктазы, дефицит синтазы 3 (эндотелиальной) оксида азота.
Проявления преэклампсии
• Периодическое нарастающее повышение АД.
• Увеличение массы тела: слишком быстрое увеличение массы тела (более
900 г/нед) — нередко первый признак преэклампсии; прибавка массы тела
обычно начинается внезапно и связана с патологической задержкой жид­
кости. Возможны отеки.
• Протеинурия. При преэклампсии протеинурия может быть незначитель­
ной и обычно развивается вскоре после артериальной гипертензии.
• Головная боль. При тяжелой преэклампсии сильная головная боль может
предшествовать появлению судорог. Типичная локализация боли — область
лба. В большинстве случаев ненаркотические анальгетики не улучшают
состояния.
• Усиление сухожильных рефлексов. Предвещает развитие судорог.
• Боли в эпигастральной области. Вслед за появлением этой боли очень
быстро могут последовать судороги (развитие эклампсии).
• Нарушения зрения. Варьируют от слабого «затуманивания» зрения до сле­
поты и обусловлены спазмом артериол, ишемией и отеком сетчатки, ино­
гда ее отслойкой.
Осложнения преэклампсии: гипертонический криз, острый некроз печени,
острый отек легких, преждевременная отслойка плаценты, гибель плода,
эклампсия.
Эклампсия
Эклампсия — максимальная степень тяжести гестоза. Обычно она развивает­
ся в III триместре беременности или в течение 24 ч после родоразрешения.
Основное клиническое проявление — судороги с потерей сознания, не свя­
занные с какой-либо другой церебральной патологией (например, эпилепси­
ей или кровоизлиянием в головной мозг). Эклампсия сопровождается нару­
шениями сознания, артериальной гипертензией, отеками, протеинурией,
возможны ДВС, тромбоцитопения, нарушение функций печени, почечная
недостаточность.
Прогноз эклампсии
У 25% женщин с эклампсией при следующей беременности возникает
артериальная гипертензия, у 5% — артериальная гипертензия тяжелого тече­
ния, у 2% — эклампсия. Рожавших женщин с эклампсией относят к группе
высокого риска развития эссенциальной артериальной гипертензии.
Осложнения артериальной гипертензии
К наиболее частым и опасным осложнениям артериальной гипертензии
относят:
—инфаркт миокарда;
—инсульт мозга;
—сердечную недостаточность;
—почечную недостаточность;
—гипертензивную энцефалопатию;
—ретинопатию;
—расслаивающуюся аневризму аорты;
—гипертензивный (гипертонический) криз.
Г и п ер то н и ч еск и й к р и з
Гипертонический (гипертензивный) криз — острое повышение систоличе­
ского и/или диастолического АЦ, сопровождающееся ухудшением мозго­
вого, коронарного или ночечного кровообращения, а также выраженной
вегетативной симптоматикой.
Гипертонический криз, как правило, развивается у нелеченых больных,
у людей, прекративших прием антигипертензивных средств, а также может
быть первым проявлением ГБ или симптоматической артериальной гипер­
тензии у пациентов, не получавших адекватного лечения.
Причины гипертензивного криза:
—неправильный прием ЛС (особенно симпатомиметиков, глюкокортикоидов, пероральных контрацептивов, анорексантов);
—употребление наркотических веществ (например, кокаина, амфетамина);
—отмена гипотензивных препаратов;
—прекращение приема ЛС, угнетающих ЦНС;
—эклампсия и преэклампсия;
—феохромоцитома;
—заболевания почек;
—послеоперационная артериальная гипертензия и тому подобные состояния.
Факторы риска гипертензивного криза:
—артериальная гипертензия в анамнезе;
—алкоголизм, наркомания;
—злоупотребление ЛС.
Проявления гипертензивного криза:
—повышение АД, которое может сопровождаться развитием энцефалопа­
тии, субарахноидальных кровоизлияний, инсульта, инфаркта миокарда,
острой левожелудочковой недостаточности (с отеком легких), расслое­
нием аорты, острой почечной недостаточностью (ОПН);
—нарушения зрения в виде снижения остроты и выпадения полей зрения;
—сильная головная боль, головокружение;
—загрудинные боли (в связи с ишемией миокарда, аорталгией), сердцебие­
ние, одышка.
Экстремальный подъем АД может привести к множественным кровоизлия­
ниям в органы и к отеку тканей.
Основные нозологические формы
артериальных гипертензий
Симптоматическая (вторичная) и эссенциальная (первичная ГБ) артериаль­
ные гипертензии составляют не менее 95% всех артериальных гипертензий.
Сим птом атические артериальные гипертензии
Симптоматические гипертензии — вторичные артериальные гипертензии,
возникающие вследствие поражения органов и систем, участвующих в регу­
ляции АД.
Частота симптоматических гипертензий (по разным оценкам) составляет
10—25% всех артериальных гипертензий.
Этиология симптоматических артериальных гипертензий
Наиболее часто к симптоматическим артериальным гипертензиям приво­
дят следующие формы патологии и факторы.
• Поражения почек: острые и хронические гломерулонефриты и пиелонеф­
риты, обструктивные нефропатии, поликистоз почек, заболевания соеди­
нительной ткани почек, диабетическая нефропатия, гидронефроз, врож­
денная гипоплазия почек, травмы почек, ренинсекретирующие опухоли,
первичная задержка соли (например, синдром Лидцла).
• Эндокринные заболевания (например, акромегалия, гипертиреоз, гиперкальциемия, синдром Иценко—Кушинга, альдостеронизм, врожденная гиперпла­
зия надпочечников, феохромоцитома, вненадпочечниковая хромаффинома).
• Неврологическая патология (повышение внутричерепного давления, опу­
холи мозга, энцефалиты, респираторный ацидоз, ночное апноэ, острая
порфирия, отравление свинцом, синдром Гийена—Барре).
• Сосудистая патология (коарктация аорты и ее основных ветвей, стеноз
почечной артерии).
• Систолические сердечно-сосудистые артериальные гипертензии (недостаточ­
ность аортального клапана, склероз аорты, артериовенозные фистулы,
открытый артериальный проток).
• Л С и другие экзогенные вещества с гипертензивным действием (например,
пероральные контрацептивы, глюкокортикоиды, антидепрессанты, симпатомиметики, алкалоиды спорыньи, препараты лития, НПВС, цикло­
спорин, эритропоэтин, алкоголь, кокаин, пищевые продукты с тирамином
в сочетании с ингибиторами моноаминоксидазы).
Проявления симптоматических гипертензий
Клиническая картина симптоматических артериальных гипертензий не­
специфична и определяется повреждением органов-мишеней.
• Поражение Ц Н С (головная боль, часто при пробуждении и, как правило,
в затылочной области, головокружение, нарушения зрения, преходящее
нарушение мозгового кровообращения или инсульт, кровоизлияния в сет­
чатку или отек соска зрительного нерва, двигательные расстройства и рас­
стройства чувствительности).
• Повреждения сердца (сердцебиение, боли в грудной клетке, одышка, кли­
нические проявления ИБС, дисфункция левого желудочка или сердечная
недостаточность).
• Нефропатии (жажда, полиурия, никтурия, гематурия).
• Поражение периферических артерий (холодные конечности, перемежа­
ющаяся хромота).
Осложнения симптоматических артериальных гипертензий
Исследование, проведенное Американской ассоциацией кардиологов,
показало, что риск инфаркта миокарда и инсульта значительно уменьшается
при снижении диастолического АД до 83 мм рт.ст. (а не до 90, как рекомен­
довали ранее).
Гипертоническая болезнь
Диагноз ГБ (эссенциальной, первичной артериальной гипертензии) устанавли­
вают, исключая вторичные (симптоматические) артериальные гипертензии.
Понятие «эссенциальная» означает, что стойко повышенное АД при ГБ состав­
ляет сущность (главное содержание) этой артериальной гипертензии. Каких-либо
изменений в других органах, которые могли бы привести к артериальной
гипертензии, при обычном обследовании не находят.
Частота эссенциальной артериальной гипертензии составляет 95% всех арте­
риальных гипертензий (при тщательном обследовании пациентов в специали­
зированных стационарах эта величина снижается до 75%).
Этиология гипертонической болезни
Основная причина ГБ — повторный, как правило, затяжной психоэмоцио­
нальный стресс, носящий отрицательный эмоциональный характер.
Главные факторы риска (условия, способствующие развитию ГБ) показаны
на рис. 23.39. К наиболее значимым из них относят:
—генетически детерминированные нарушения структуры и функции мем­
бран клеток как возбудимого, так и невозбудимого типа в отношении
транспорта № + и Са2+; при этом избыток
обусловливает (помимо
прочего) два важных эффекта — усиление транспорта жидкости ь клетки
и их набухание (набухание клеток стенок сосудов ведет к их утолщению,
сужению просвета, повышению ригидности и снижению способности
к вазодилатации);
—повышение чувствительности миоцитов стенок сосудов и сердца к вазоконстрикторным агентам;
—расстройства функций мембранных рецепторов, воспринимающих воздей­
ствие нейромедиаторов и других БАВ, которые регулируют АД, что создает
условия для доминирования эффектов гипертензивных факторов;
—нарушения экспрессии генов, контролирующих синтез сосудорасширяющих
агентов (оксид азота, простациклин, ПГЕ) клетками эндотелия;
—действие факторов окружающей среды. Наибольшее значение имеют:
о профессиональные вредности, например постоянный шум, необходи­
мость напрягать внимание;
о условия проживания, включая коммунальные;
о интоксикация, особенно алкоголем, никотином, наркотиками;
о травмы мозга;
—индивидуальные характеристики организма. К существенному повыше­
нию АД приводят:
о возраст — с возрастом, особенно после 40 лет, доминируют опосредо­
ванные диэнцефально-гипоталамической областью мозга гипертензивные реакции на различные экзо- и эндогенные воздействия;
о повышенная масса тела;
о высокий уровень холестерина в сыворотке крови;
о избыточная продукция ренина;
о особенности реакции ССС на раздражители, заключающиеся в доми­
нировании гипертензивных реакций на различные воздействия.
Увеличенная активность СНС
соли
нарушения
Рис. 23.39. Факторы, участвующие в развитии гипертонической болезни: ГБ —
гипертоническая болезнь, РАС — ренин-ангиотензин-альдостероновая система,
СНС — симпатическая нервная система
Виды гипертонической болезни
В России принята классификация видов ГБ, предложенная экспертами ВОЗ.
I стадия — повышение АД более 160/95 мм рт.ст. без органических измене­
ний в ССС.
II стадия — повышение АД более 160/95 мм рт.ст. в сочетании с изменениями
органов-мишеней (сердце, почки, головной мозг, сосуды глазного дна), обу­
словленные артериальной гипертензией, но без нарушения их функций.
III стадия — артериальная гипертензия, сочетающаяся с поражением органовмишеней (сердце, почки, головной мозг, глазное дно) с нарушением их функций.
Формы эссенциальной артериальной гипертензии
• Пограничная эссенциальная артериальная гипертензия (ЭАГ). Наблюдается
у лиц молодого и среднего возраста, характеризуется колебаниями АД
от нормы до 140/90-159/94 мм рт.ст. Нормализация АД происходит спон­
танно. Признаки поражения органов-мишеней, типичные для эссенци­
альной артериальной гипертензии, отсутствуют. Встречается примерно
у 20-25% лиц; у 20-25% из них затем развивается ЭАГ, у 30% погранич­
ная артериальная гипертензия сохраняется многие годы или всю жизнь,
у остальных АД со временем нормализуется.
• Гиперадренергическая ЭАГ. Характеризуется синусовой тахикардией, неус­
тойчивым АД с преобладанием систолического компонента, потливостью,
гиперемией лица, чувством тревоги, пульсирующими головными болями.
Проявляется в начальном периоде заболевания (у 15% больных сохраняет­
ся и в дальнейшем).
• Гипергидратационная ЭАГ (натрий-, объемзависимая). Проявляется отеч­
ностью лица, параорбитальных областей; колебаниями диуреза с преходя­
щей олигурией; бледными кожными покровами; постоянными распира­
ющими головными болями.
• Злокачественная ЭАГ. Быстро прогрессирующее заболевание с повышени­
ем АД до очень высоких величин с нарушением зрения, развитием энце­
фалопатии, отека легких, почечной недостаточности.
Основные звенья патогенеза гипертонической болезни
Стадия I гипертонической болезни
Инициальный фактор патогенеза ГБ — невротическое состояние. Харак­
теризуется включением центрогенного нейрогенного звена патогенеза ГБ
(рис. 23.40).
|Транзиторная артериальная гипертензия
Рис. 23.40. Основные звенья патогенеза гипертонической болезни (стадия I)
Центрогенный нейрогенный механизм патогенеза ГБ включает:
—формирование корково-подкоркового комплекса устойчивого возбуждения.
Он включает симпатические ядра заднего отдела гипоталамуса, а также
адренергические структуры ретикулярной формации и сосудодвигатель­
ного центра;
—усиление прессорных (гипертензивных) влияний на ССС. Реализуется
по двум взаимозависимым каналам — нейрогенному и гуморальному:
о нейрогенные гипертензивные эффекты заключаются в активации сим­
патических нервных влияний на стенку артериол (развивается длитель­
ное сужение их просвета и повышается ОПСС; при хронически повы­
шенной симпатической стимуляции развивается гипертрофия ГМК
артериол; это потенцирует артериальную гипертензию и приводит
к дальнейшему сужению просвета), венул и вен (характерно сужение
их просвета, увеличение притока венозной крови к сердцу и в связи
с этим — повышение ударного и сердечного выброса) и на сердце
(катехоламины проявляют положительные хроно- и инотропные свой­
ства, что потенцирует увеличение сердечного выброса и АД);
о гуморальные гипертензивные эффекты характеризуются активацией
образования и высвобождения БАВ с гипертензивным действием:
АДГ (вазопрессин), адреналина, АКТГ и кортикостероидов (минерало- и глюкокортикоиды), Т3 и Т4, эндотелина. Это, параллельно
с активацией симпатической нервной системы, обеспечивает повы­
шение ОПСС, веноконстрикцию и увеличение (в связи с этим) воз­
врата венозной крови к сердцу, увеличение ОЦК, усиление сердечного
выброса. Указанные эффекты закрепляют повышенный уровень АД
или увеличивают его дополнительно;
—нарастающая гипертрофия ГМ К артериол и миокарда, развитие атероскле­
роза. Эти изменения являются результатом длительно повышенного
АД. Они обусловливают нарушения кровообращения в органах и тканях
(ишемию, венозную гиперемию), развитие гипоксии (вначале циркуля­
торного, а затем смешанного типа).
I
стадия ГБ (стадия становления, или транзиторная) характеризуется
повторными, преходящими, более или менее длительными эпизодами повы­
шения АД сверх нормы. При этом признаки поражения внутренних органов
отсутствуют.
Стадия II гипертонической болезни
Стадия II характеризуется стабилизацией АД на повышенном уровне.
Основные механизмы этой стадии ГБ изображены на рис. 23.41.
Стабилизацию АД на повышенном уровне в стадии II ГБ обеспечивают реф­
лексогенный, эндокринный, гемический механизмы.
Рефлексогенный (барорецепторный) механизм стабилизации АД на повышен­
ном уровне заключается в нарастающем снижении (или даже прекращении)
афферентной депрессорной импульсации от барорецепторов дуги аорты, синокаротидной и других зон в адрес сосудодвигательного (прессорного) центра.
Эндокринный механизм характеризуется стимуляцией продукции и инкреции в кровь гормонов с гипертензивным действием.
Метаболическое (гипоксическое, органоишемическое) звено патогенеза ГБ
включает почечные гипертензивные механизмы (вазоренальный и ренопаренхиматозный) и органоишемические. Они проявляются избытком БАВ
с гипертензивным действием, уменьшением образования БАВ с гипотензив­
ным эффектом.
Рис. 23.41. Основные звенья патогенеза гипертонической болезни (стадия II)
Гемический механизм закрепления повышенного уровня АД характеризует­
ся развитием (в связи с хронической гипоксией) полицитемии, в основном
за счет значительного эритроцитоза и повышенной вязкости крови (в связи
с полицитемией и диспротеинемией).
В стадии II ГБ регистрируют стабильно повышенное (гипертензивное)
АД, а также признаки поражения тканей и внутренних органов (гипертрофия
сердца, выраженный атеро- и артериосклероз, нефросклероз и др.).
Стадия III гипертонической болезни
Третья стадия ГБ характеризуется повреждением структурных элементов
тканей, грубыми расстройствами функций тканей и органов с развитием
полиорганной недостаточности. Наиболее часто наблюдаются:
—выраженный атеро- и артериосклероз, приводящие к инфарктам в раз­
личных органах (наиболее часто происходит инфаркт миокарда)
и инсультам;
—кардиомиопатии (одна из причин — нарушение сбалансированности
роста структур миокарда);
—склеротическое поражение почек (в виде первично-сморщенной почки;
это название подчеркивает первично гипертензивный генез патологии
почек при ГБ);
—дистрофические и склеротические изменения в тканях и органах (мозг,
эндокринные железы, сетчатка, сердце).
Принципы лечения больных артериальными
гипертензиями
Лечение ГБ базируется на следующих принципах:
— этиотропном (устранение или уменьшение степени действия причины
и факторов риска ГБ);
— патогенетическом (разрыв звеньев патогенеза ГБ);
— симптоматическом (устранение усугубляющих течение ГБ симптомов
и неприятных ощущений).
Указанные принципы лежат в основе программ, алгоритмов и рекомен­
даций по лечению ГБ, сформулированных экспертами ВОЗ, международных
и национальных медицинских организаций.
Эксперты ВОЗ и Международного общества гипертензии считают,
что у людей молодого и среднего возраста, а также у больных СД нужно под­
держивать АД на уровне 130/85 мм рт.ст. У людей пожилого возраста необхо­
димо добиваться снижения АД до уровня 140/90 мм рт.ст.
Вместе с тем необходимо помнить, что чрезмерное снижение АД при зна­
чительной длительности и выраженности заболевания может привести к гипо­
перфузии жизненно важных органов: головного мозга (с развитием гипоксии,
инсульта), сердца (с обострением стенокардии, угрозой инфаркта миокарда),
почек (с возникновением почечной недостаточности).
Цель лечения артериальной гипертензии — не только снижение высокого
АД, но и защита органов-мишеней, устранение факторов риска (отказ от куре­
ния, компенсация СД, снижение концентрации холестерина в крови и избы­
точной массы тела) и в конце концов снижение заболеваемости и смертности
от болезней сердца и сосудов.
В настоящее время для лечения артериальных гипертензий применяют,
как правило, шесть основных групп препаратов:
—блокаторы медленных кальциевых каналов;
—диуретики;
—адреноблокаторы;
—ингибиторы АПФ;
—антагонисты (блокаторы рецепторов) ангиотензина II;
—адреноблокаторы.
Кроме того, широко используют препараты центрального гипотензивного
действия и средства с комбинированными эффектами.
Лекарственную терапию проводят индивидуально по апробированным
схемам и официальным рекомендациям.
АРТЕРИАЛЬНЫЕ ГИПОТЕНЗИИ
Артериальная гипотензия — уменьшение АД ниже нормы: 100/60 мм рт.ст.
у мужчин и 95/60 мм рт.ст. у женщин (границы нормн при хорошем само­
чувствии и полной работоспособности).
Виды артериальной гипотензии
Различают физиологическую и патологическую артериальную гипотензию.
К физиологической артериальной гипотензии относят:
—индивидуальные варианты нормы (так называемое нормальное низкое АД);
— артериальную гипотензию высокой тренированности («спортивная» арте­
риальная гипотензия);
—адаптивную артериальную гипотензию (характерна для жителей высоко­
горья, тропиков, Заполярья).
К патологической артериальной гипотензии относят:
• острые формы гипотензии:
—коллапс (острая недостаточность кровообращения, возникающая вслед­
ствие быстрого снижения функции сердца, падения сосудистого тону­
са и/или уменьшения ОЦК; проявляется коллапс резким снижени­
ем артериального и венозного давления, гипоксией головного мозга
и угнетением жизненно важных функций организма). [Подробнее см.
главу 21 «Патофизиология экстремальных состояний» (см. т. 1) и статью
«Гипотензия ортостатическая» в приложении «Справочник терминов».];
—снижение систолического АД ниже 90 мм рт.ст., сопровождающееся анури­
ей, симптомами нарушений периферического кровообращения и сознания,
например, при шоке. (Подробнее см. главу 21 «Патофизиология экстре­
мальных состояний», т. 1);
• хронические формы артериальной гипотензии:
—хроническая первичная артериальная гипотензия;
—артериальная гипотензия нейроциркуляторная (с нестойким обратимым
течением и выраженная стойкая форма — гипотоническая болезнь);
— артериальная гипотензия ортостатическая идиопатическая (первичная веге­
тативная недостаточность);
—хроническая вторичная (симптоматическая) артериальная гипотензия
с ортостатическим синдромом или без него.
Этиология и патогенез артериальных гипотензий
По инициальному звену механизма развития выделяют нейрогенные, эндо­
кринные и метаболические артериальные гипотензии (рис. 23.42).
Виды артериальных гипотензий
| Центрогенные| | Рефлекторные 11 Надпочечниковые 11 Тиреоидные 11 Гипофизарные I
Рис. 23.42. Виды артериальных гипотензий по начальному (стартовому) звену пато­
генеза
Нейрогенные артериальные гипотензии
Среди нейрогенных артериальных гипотензий выделяют гипотензии:
—центрогенные;
— рефлекторные (рефлексогенные).
Центрогенные артериальные гипотензии
Основные звенья патогенеза центрогенной нейрогенной артериальной
гипотензии представлены на рис. 23.43.
Нейрогенные гипотензии центрогенного генеза являются результатом либо
функционального расстройства ВНД (невроз), либо органического поврежде­
ния мозговых структур, участвующих в регуляции АД.
Рис. 23.43. Основные звенья патогенеза центрогенной нейрогенной артериальной
гипотензии
Артериальные гипотензии вследствие нарушения ВНД (невроз)
Причина их — длительный, повторный стресс, обусловленный необходи­
мостью сдерживать двигательные и эмоциональные проявления. Это влечет
за собой развитие невротического состояния.
Основные звенья патогенеза гипотензии вследствие нарушения ВНД:
—перенапряжение (и срыв) ВНД (невроз) — инициальное звено патогенеза
гипотензии;
—формирование корково-подкоркового комплекса возбуждения; оно распро­
страняется на парасимпатические ядра переднего гипоталамуса и другие
структуры парасимпатической нервной системы (например, на дорсаль­
ное двигательное ядро блуждающего нерва);
— снижение сократительной функции миокарда, сердечного выброса крови
и тонуса резистивных сосудов (как результат активации парасимпатиче­
ских влияний на ССС);
—развитие артериальной гипотензии.
Считают, что именно такой механизм лежит в основе развития гипотони­
ческой болезни. При хроническом ее течении включаются и другие патогене­
тические звенья, что способствует стабилизации АД на пониженном уровне
или даже усугубляет степень его снижения.
Артериальные гипотензии вследствие органических изменений в структурах
мозга
Подобные гипотензии возникают при повреждении центральных (диэнцефально-гипоталамических) и периферических структур, участвующих в регу­
ляции АД.
Наиболее частые причины:
—травмы головного мозга (при его сотрясении или ушибах);
—нарушения мозгового кровообращения (ишемия, венозная гиперемия);
—дегенеративные изменения в веществе головного мозга (дегенерация ней­
ронов экстрапирамидной системы, базальных ядер мозга, заднего ядра
блуждающего нерва);
—нарушение выделения в кровь катехоламинов (при физической нагрузке,
изменениях положения тела из горизонтального в вертикальное —
в этом случае нередко развиваются ортостатические коллапсы и обмо­
роки).
Ключевые звенья патогенеза гипотензий в результате органических изменений
в структурах мозга:
—снижение активности симпатико-адреналовой системы и выраженности ее
влияний на ССС;
—относительное или абсолютное преобладание влияний парасимпатической
нервной системы на сердце и сосуды;
—снижение тонуса стенок артериол, ОПСС, сердечного выброса крови.
В совокупности указанные изменения обусловливают стойкое уменьшение
уровня АД ниже нормы.
Рефлекторные (рефлексогенные, проводниковые) центрогенные артериальные
гипотензии
Причина рефлексогенных артериальных гипотензий — нарушение про­
ведения эфферентных гипертензивных импульсов от сосудодвигательного
центра продолговатого мозга к стенкам сосудов и сердцу. Наиболее часто
это развивается при нейросифилисе, боковом амиотрофическом склерозе,
сирингомиелии, периферических невропатиях различного генеза (например,
диабетического, инфекционного, нейротоксического).
Механизм развития рефлексогенных гипотензий заключается в значительном
уменьшении или прекращении тонических влияний симпатической нервной систе­
мы на стенки сосудов и сердца. Это приводит:
—к снижению ОПСС и, соответственно, диастолического АД;
—уменьшению сократительной функции сердца, величины сердечного
выброса и систолического АД.
В результате развивается артериальная гипотензия.
Эндокринны е артериальные гипотензии
Общие звенья патогенеза артериальных гипотензий эндокринного генеза
представлены на рис. 23.44.
Различают артериальные гипотензии надпочечникового, гипофизарного,
гипотиреоидного генеза.
Снижение синтеза и/или инкреции
гормонов с гипертензивным действием:
-катехоламинов;
-вазопрессина;
-А КТ Г;
- минералокортикоидов;
-эндотелина;
-тиреоидных
Гипосенситизация рецепторов
сосудов и сердца к гормонам
с гипертензивным действием
1
1Г
Снижение:
-общ его периферического сосудистого сопротивления;
-объ ем а циркулирующей крови;
-сердечного выброса крови
I
| Артериальная гипотензия |
Рис. 23.44. Общие звенья патогенеза эндокринных артериальных гипотензий
Артериальные гипотензии надпочечникового происхождения (при надпочечни­
ковой недостаточности)
Причины:
—гипотрофия коры надпочечников;
— опухоль коры надпочечников с разрушением ее;
—кровоизлияние в надпочечник (один или оба);
—туберкулезное поражение надпочечников;
—деструкция надпочечников (например, в результате реакций иммунной
аутоагрессии или травмы, приводящих к повреждению или разрушению
надпочечников).
Патогенез надпочечниковой гипотензии
Дефицит катехоламинов, минерало- и глюкокортикоидов и/или недо­
статочность их эффектов обусловливает снижение тонуса стенок артериол
и ОПСС, О Ц К и сердечного выброса, что и приводит к стойкому снижению АД
по сравнению с нормой.
Артериальные гипотензии при поражении гипофиза
Причина их — гипофункция гипофиза, приводящая к снижению продук­
ции гормонов с гипертензивным эффектом: вазопрессина, АКТГ, ТТГ, СТГ.
Патогенез. Развитие артериальной гипотензии при питуитарной недоста­
точности является результатом недостаточной эффективности вазопрессина,
АКТГ, ТТГ, СТГ. В конечном итоге гипофизарная недостаточность приводит
к снижению тонуса артериол и ОПСС, ОЦК, сердечного выброса.
В совокупности эти изменения обусловливают стойкое снижение и систо­
лического, и диастолического АД.
Артериальная гипотензия при гипотиреоидных состояниях
Причина — дефицит Т3 и Т4 и/или их эффектов.
Механизм развития:
—снижение сердечного выброса;
—стойкая брадикардия (развивается вследствие снижения или отсутствия
положительного хронотропного эффекта тиреоидных гормонов в связи
с их дефицитом, снижения активности симпатико-адреналовой системы);
—снижение тонуса стенок сосудов вследствие их дистрофических измене­
ний и как результат уменьшение ОПСС.
М етаболические артериальные гипотензии
Артериальные гипотензии, вызванные нарушением метаболизма веществ
с гипо- и гипертензивным действием, встречаются редко.
Причины их:
—дистрофические изменения в органах и тканях (например, при хрониче­
ских интоксикациях, инфекциях, голодании); это обусловливает сниже­
ние выработки и/или эффектов метаболитов с гипертензивным действием
(например, эндотелина, ПГР, тромбоксана А2, ангиотензиногена и др.),
падение тонуса миоцитов стенок артериол, снижение сократительной
функции миокарда;
—гипогидратация организма (вызвана уменьшением объема жидкости
в организме в связи со снижением интенсивности метаболизма).
Патогенез метаболических артериальных гипотензий
Основными звеньями патогенеза являются:
—снижение тонуса ГМК стенок сосудов и вследствие этого — ОПСС;
—падение сократительной функции сердца, вследствие чего уменьшается
сердечный выброс крови;
—уменьшение содержания воды в организме, в том числе объема циркули­
рующей жидкости.
В целом указанные факторы обусловливают стойкое снижение АД по срав­
нению с нормой — артериальную гипотензию.
Нарушения регионарного
кровотока
Многочисленные расстройства регионарного (периферического, местного,
органотканевого) кровотока подразделяются на:
—нарушения кровотока в сосудах среднего диаметра;
—расстройства крово- и лимфотока в сосудах микроциркуляторного русла
(рис. 23.45).
Рис. 23.45. Типовые формы патологии регионарного кровообращения
НАРУШЕНИЯ КРОВОТОКА
В СОСУДАХ СРЕДНЕГО ДИАМЕТРА
К нарушениям кровообращения в сосудах среднего диаметра относятся
патологическая артериальная гиперемия, венозная гиперемия, ишемия и стаз.
Артериальная гиперемия
Артериальная гиперемия — типовая форма изменения регионарного кро­
вообращения, характеризующаяся увеличением кровенаполнения и коли­
чества протекающей по сосудам органов и тканей крови в результате
расширения артериол и артерий.
Причины артериальной гиперемии
Причины артериальных гиперемий могут иметь различное происхождение
и природу.
По происхождению выделяют артериальные гиперемии, вызываемые эндо­
генными или экзогенными факторами.
• Экзогенные агенты действуют на орган или ткань извне; к ним относятся
инфекционные (микроорганизмы и/или их эндо- и экзотоксины) и неин­
фекционные факторы различной природы.
• Эндогенные факторы, вызывающие артериальную гиперемию, образуются
в самом организме: например, накопление избытка солей и конкрементов
в тканях почек, печени, подкожной клетчатке; образование избытка БАВ,
вызывающих снижение тонуса ГМК артериол (вазодилатация) — аденозина, ПГ, кининов; образование избытка органических кислот — молочной,
пировиноградной, кетоглутаровой.
По природе причины артериальной гиперемии выделяют факторы:
—физические (например, механическое воздействие, очень высокая тем­
пература, электрический ток);
—химические (например, органические и неорганические кислоты, щело­
чи, спирты, альдегиды);
— биологические (например, физиологически активные вещества, обра­
зующиеся в организме, — аденозин, ацетилхолин, простациклин, оксид
азота).
Виды артериальной гиперемии
Выделяют две разновидности артериальной гиперемии:
—физиологическую;
—патологическую.
Их различают по двум критериям:
—адекватности или неадекватности выраженности и длительности артери­
альной гиперемии по причине, которая ее вызвала, а также изменениям
функции и метаболизма в органах и тканях, в которых эта гиперемия
развилась;
— адаптивности или неадаптивности артериальной гиперемии для ткани,
органа, организма.
Физиологическая артериальная гиперемия адекватна вызвавшему ее воздей­
ствию и имеет адаптивное значение.
Она может быть функциональной и защитно-приспособительной:
—функциональная артериальная гиперемия развивается в органах и тканях
в связи с усилением их функции (например, гиперемия в сокращающейся
мышце или в усиленно работающем органе);
—защитно-приспособительная гиперемия развивается при протекании защит­
ных, компенсаторных и приспособительных реакций и процессов в орга­
низме (например, в очаге воспаления; вокруг трансплантата или зоны
инфаркта). В этих случаях артериальная гиперемия способствует достав­
ке в ткани кислорода, субстратов метаболизма, 1§, фагоцитов, лимфоци­
тов, других клеток и агентов, необходимых для осуществления местных
защитных и восстановительных реакций.
Патологическая артериальная гиперемия не адекватна воздействию, не свя­
зана с усилением функции органа или ткани и играет дизадаптивную — повреж­
дающую роль.
Патологическая артериальная гиперемия сопровождается нарушениями
кровоснабжения, микрогемоциркуляции, транскапиллярного обмена, неред­
ко — кровоизлияниями и кровотечениями.
Примеры:
—патологическая артериальная гиперемия головного мозга при гипертензивном
кризе (может привести к геморрагическому инсульту и/или отеку мозга);
—патологическая артериальная гиперемия органов и тканей, развивающаяся
по нейромиопаралитическому (см. ниже) механизму: например, в артериаль­
ном русле органов брюшной полости после быстрого удаления большого
количества асцитической жидкости она сочетается с «перераспредели­
тельной» ишемией мозга, других органов, а также с развитием коллапса;
артериальная гиперемия в артериях кожи и мышц конечности после снятия
длительно наложенного жгута может вызвать множественные капиллярные
кровоизлияния в них; в зоне хронического воспаления артериальная гипе­
ремия сочетается со стазом; гиперемия в месте длительного (часы) воздей­
ствия тепла — солнечного или грелки — нередко приводит к геморрагиям.
М еханизмы возникновения артериальной гиперемии
Расширение просвета малых артерий и артериол достигается за счет нейроген­
ного, гуморального и нейромиопаралитического механизмов или их сочетания.
Нейрогенный механизм расширения просвета артериол
Выделяют нейротонический и нейропаралитический варианты нейроген­
ного механизма развития артериальной гиперемии:
—нейротонический механизм заключается в преобладании эффектов пара­
симпатических нервных влияний (по сравнению с симпатическими)
на стенки артериальных сосудов;
—нейропаралитический механизм характеризуется снижением или отсут­
ствием («паралич») симпатических нервных влияний на стенки артерий
и артериол.
Гуморальный механизм расширения просвета артериол
Заключается в местном увеличении содержания вазодилататоров — БАВ
с сосудорасширяющим эффектом (аденозин, оксид азота, ПГЕ, ПГ12, кинины
и др.), а также в повышенной чувствительности рецепторов стенок артериаль­
ных сосудов к вазодилататорам.
Нейромиопаралитический механизм
Заключается в истощении запасов катехоламинов в синаптических вези­
кулах симпатических нервных волокон в стенке артериол с последующим
снижением тонуса ГМК артериальных сосудов.
Причины указанных изменений:
—продолжительное действие на ткани или органы различных факторов
физической или химической природы (например, тепла при применении
грелок, согревающих компрессов, горчичников, лечебной грязи, диа­
термических токов);
—прекращение длительного механического давления на стенки артерий
(например, асцитической жидкости, тугого бинта, давящей одежды).
Действие указанных факторов в течение длительного времени существенно
снижает или полностью снимает миогенный и регуляторный (главным образом,
адренергический) тонус стенок артериальных сосудов. В связи с этим они рас­
ширяются, в них увеличивается количество протекающей артериальной крови.
Проявления артериальных гиперемий
Проявления артериальных гиперемий показаны на рис. 23.46. К ним относят:
—увеличение количества и диаметра артериальных сосудов в зоне артериаль­
ной гиперемии (в связи с их расширением);
—покраснение органа, ткани или их участка вследствие:
о повышенного притока артериальной крови;
о расширения просвета артериол и прекапилляров;
о увеличившегося количества функционирующих капилляров;
о «артериализации» венозной крови (т.е. повышения содержания Н Ь02
в венозной крови);
— повышение температуры тканей и органов в зоне гиперемии (в результате
притока более теплой артериальной крови, а также интенсификации
обмена веществ);
—увеличение лимфообразования и лимфооттока (вследствие повышенного
перфузионного давления крови в сосудах микроциркуляторного русла);
—увеличение объема и тургора органа или ткани (в результате возрастания
их крове- и лимфонаполнения);
—изменения в сосудах микроциркуляторного русла:
о увеличение диаметра артериол и прекапилляров;
о возрастание количества функционирующих капилляров (т.е. капилля­
ров, по которым протекает плазма и форменные элементы крови);
о ускорение тока крови по микрососудам;
о уменьшение диаметра осевого «цилиндра» (поток клеток крови по цен­
тральной оси артериолы) и расширение потока плазмы крови с малым
содержанием в ней форменных элементов вокруг этого «цилиндра»
(вследствие увеличения центростремительных сил и отбрасывания
клеток крови к центру просвета сосудов в связи с ускорением тока
крови в условиях артериальной гипертензии).
Рис. 23.46. Основные проявления артериальной гиперемии
Последствия артериальных гиперемий
Последствия артериальной гиперемии приведены на рис. 23.47.
Рис. 23.47. Последствия артериальной гиперемии
Физиологическая артериальная гиперемия обеспечивает:
— активацию специфической функции (функций) органа или ткани;
—потенцирование неспецифических функций и процессов. Например,
при физиологической гиперемии наблюдается:
—активация местного иммунитета (в связи с повышением притока с арте­
риальной кровью 1§, лимфоцитов, фагоцитирующих клеток и других
агентов);
—ускорение пластических процессов;
—увеличение лимфообразования и лимфооттока от тканей; гипертро­
фия и гиперплазия структурных элементов тканей продуктами обмена
веществ и кислородом.
Достижение именно таких эффектов артериальной гиперемии становится
целью при проведении лечебных мероприятий (например, при применении
компрессов, горчичников, при проведении физиотерапевтических проце­
дур; инъекциях сосудорасширяющих ЛС; при хирургических вмешательствах
по пересечению симпатических нервных стволов или иссечению симпати­
ческих ганглиев при некоторых формах стенокардии и др.), направленных
на индуцирование гиперемии. Такие манипуляции выполняются при повреж­
дении органов и тканей, их ишемии, нарушении трофики и пластических
процессов в них, уменьшении активности «местного иммунитета».
Патологическая артериальная гиперемия может вызвать перерастяжение
и микроразрывы в стенках сосудов микроциркуляторного русла, микрои макрокровоизлияния в ткани, кровотечения (наружные и/или внутренние).
Устранение или предупреждение этих негативных последствий — цель
терапии при патологических разновидностях артериальной гиперемии.
Венозная гиперемия
Венозная гиперемия — типовая форма патологии регионарного кровообра­
щения, характеризующаяся увеличением наполнения венозного русла ткани
или органа протекающей по сосудам кровью при уменьшении ее количества.
Венозная гиперемия развивается в результате замедления или прекраще­
ния оттока венозной крови от ткани.
Причины венозной гиперемии
Причиной венозной гиперемии является механическое препятствие оттоку
венозной крови от тканей или органа. Это может быть результатом сужения
просвета венул или вен при:
— их сдавлении (опухолью, отечной тканью, рубцом, жгутом, тугой повязкой);
—их обтурации (тромбом, эмболом, опухолью);
—сердечной недостаточности;
—слабой эластичности венозных стенок, сочетающейся с образованием
в них расширений (варикозы) и сужений.
Проявления венозной гиперемии
Эти проявления показаны на рис. 23.48. К ним относят:
—увеличение числа и диаметра венозных сосудов в зоне гиперемии (в резуль­
тате их растяжения избытком венозной крови);
—отек ткани или органа — происходит вследствие повышенного внутрисосудистого давления в капиллярах, посткапиллярах и венулах; при длительной
венозной гиперемии отек потенцируется за счет включения его осмотиче­
ского, онкотического и мембраногенного патогенетических факторов (см.
«Отек» в главе 12 «Типовые нарушения водного обмена», т. 1);
—мелкоточечные кровоизлияния в ткани, внутренние и наружные кровоте­
чения (в результате перерастяжения и микроразрывов стенок венозных
сосудов — посткапилляров и венул);
— цианоз ткани или органа (вследствие возросшего в них количества веноз­
ной крови и пониженного содержания в ней НЪ02; последнее — резуль­
тат утилизации кислорода тканью из крови в связи с медленным ее
током по капиллярам);
—снижение температуры тканей или органов в зоне венозной гиперемии
(в результате увеличения объема в них венозной крови, более холодной
по сравнению с артериальной, и уменьшившейся интенсивности ткане­
вого метаболизма; это, в свою очередь, является результатом меньшего
притока артериальной крови к тканям в зоне венозной гиперемии);
—изменения в сосудах микроциркуляторного русла:
о увеличение количества и диаметра капилляров, посткапилляров и венул
(в результате растяжения стенок микрососудов избытком венозной
крови);
о возрастание количества функционирующих капилляров на начальном
этапе венозной гиперемии (вследствие оттока венозной крови по ранее
не функционировавшим капиллярным сетям);
о уменьшение количества функционирующих капилляров на более поздних
ее этапах (в связи с прекращением тока крови в результате образования
микротромбов и агрегатов клеток крови в посткапиллярах и венулах);
о замедление (вплоть до прекращения) оттока венозной крови;
о значительное расширение диаметра осевого «цилиндра» и исчезнове­
ние полосы плазматического тока в венулах и венах вследствие низкой
скорости тока крови;
о «маятникообразное» движение крови в венулах и венах «туда-обратно»:
«туда» — от капилляров в венулы и вены (причина — проведение систоли­
ческой волны сердечного выброса крови); «обратно» — от вен к венулам
и капиллярам (причина — «отражение» потока венозной крови от меха­
нического препятствия — тромба, эмбола, суженного участка венулы).
Рис. 23.48. Проявления венозной гиперемии
Патогенные эффекты венозной гиперемии
Венозная гиперемия оказывает повреждающее действие на ткани и органы
за счет ряда патогенных факторов.
Основные патогенные факторы венозной гиперемии:
—гипоксия (вначале циркуляторного типа, а при длительной гиперемии —
смешанного типа);
— отек ткани (в связи с увеличением гемодинамического давления на стен­
ку венул и вен, повышения проницаемости их стенок, гиперосмии
и гиперонкии в зоне венозной гиперемии);
—множественные мелкоточечные кровоизлияния в ткани (в результате перерастяжения и разрывов стенок посткапилляров и венул);
—кровотечения (внутренние и наружные;.
Последствия венозной гиперемии:
— снижение специфических и неспецифических функций органов и тканей;
— гипотрофия и гипоплазия структурных элементов тканей и органов;
— некроз паренхиматозных клеток;
— развитие соединительной ткани (склероз, цирроз) в органах.
Ишемия
Ишемия — типовая форма патологии регионарного кровообращения,
характеризующаяся несоответствием между объемом притекающей арте­
риальной крови и потребностью тканей в ней. При этом потребность в при­
токе крови всегда выше, чем ее реальный приток по артериям.
Причины ишемии
Причины ишемии имеют различное происхождение и природу.
По природе причины ишемии делятся на:
—физические;
—химические;
— биологические.
Физические факторы:
—сдавление артериального сосуда — компрессия его (например, опухолью,
рубцовой тканью, инородным телом, жгутом);
— сужение или закрытие просвета артерии — обтурация ее (например, тром­
бом, эмболом, атеросклеротической бляшкой);
—действие на ткань низкой температуры (приводящее к сокращению ГМК
артериол и снижение притока крови к ткани).
Химические агенты:
—химические соединения, вызывающие сокращение ГМК артериальных
сосудов, сужение их просвета и уменьшение кровотока в ткани: напри­
мер, никотин, ряд ЛС — фенилэфрин (мезатон*), эфедрин, эпинефрин
(адреналин*), АДГ, ангиотензины.
Биологические факторы:
- биологически активные вещества, приводящие к повышению тонуса ГМК
артериол, уменьшению их диаметра и притока к тканям крови по ним
(например, катехоламины, ангиотензин II, АДГ, эндотелии, экзо- и эндо­
токсины микроорганизмов, метаболиты).
По происхождению выделяют ишемии, вызванные:
- эндогенными факторами (физическими, химическими или биологиче­
скими; инфекционными или неинфекционными);
- экзогенными агентами (физическими, химическими, биологическими
факторами внешней среды; инфекционными или неинфекционными).
Механизмы развития ишемии
Механизмы ишемии показаны на рис. 23.49.
Рис. 23.49. Механизмы развития ишемии
Механизмы, обусловливающие меньший приток крови по артериям к ткани
и к ее ишемии, разделяют на:
—нейрогенный;
—гуморальный;
— «механический».
• Нейрогенные механизмы (нейротонический и нейропаралитический) развития
ишемии:
—нейротонический механизм ишемии заключается в преобладании влияний
симпатической нервной системы на стенки артериол над влияниями
парасимпатической системы. Это связано, как правило, с усиленным
выбросом из адренергических терминалей катехоламинов и/или повы­
шенной чувствительностью к ним адренорецепторов стенок артериол.
Таков механизм ишемии, например, при стрессе; при действии на ткани
низкой температуры, механической травмы, химических веществ;
—нейропаралитический механизм ишемии характеризуется устранением
или снижением («паралич») парасимпатических влияний на стенки арте­
риол, уменьшением их просвета и притока к тканям артериальной крови.
Такой механизм развития ишемии наблюдается при торможении или блокаде
проведения нервных импульсов по парасимпатическим волокнам к стен­
кам артериол. В связи с этим замедляется высвобождение ацетилхолина
из нервных волокон стенок артерий, артериол и прекапилляров. Подобная
ситуация имеет место, например, при воспалении или механических трав­
мах парасимпатических нервных окончаний, хирургическом удалении
парасимпатических ганглиев или пересечении парасимпатических нервов.
В этих условиях доминирует симпатическое влияние на рецепторы ГМК
стенок артериол, что обусловливает их сужение и ишемию тканей.
• Гуморальный механизм развития ишемии заключается в том, что в тканях
увеличивается содержание веществ с вазоконстрикторным действием
(например, ангиотензина II, тромбоксана А2, адреналина, ПГГ) и/или повы­
шается чувствительность рецепторов стенок артериол к агентам с сосудо­
суживающим действием, например, если в тканях увеличивается содер­
жание Са2+ или № +. Это приводит к сокращению ГМК стенок артериол,
сужению их и к развитию ишемии.
• Механическое звено патогенеза ишемии представляет собой формирование
механического препятствия притоку крови к тканям по артериальным
сосудам. Причины этого:
—сдавление (компрессия) артериального сосуда опухолью, рубцом, отеч­
ной тканью, жгутом;
—уменьшение (вплоть до полного закрытия — обтурации) просвета артериолы
(например, тромбом, агрегатом форменных элементов крови, эмболом).
Понятие об эмболе и эмболии
Эмбол — образование, находящееся в просвете кровеносного или лимфати­
ческого сосуда либо в полости сердца, в норме в них не встречающееся.
Эмболия — циркуляция эмбола в кровеносном или лимфатическом русле,
в полостях сердца, вследствии чего он закрывает либо сужает просвет сосуда
или клапанное отверстие сердца.
Виды эмболов
• По происхождению:
—эндогенные. Чаще всего это фрагменты тромбов (тромбоэмболы); кусоч­
ки жировой ткани или кости, образующиеся при размозжении органов
или при переломах трубчатых костей; небольшие фрагменты распа­
дающейся опухоли или разрушенной нормальной ткани; конгломераты
микробных клеток, много- и одноклеточные паразиты; пузырьки газов,
в норме растворенных в плазме крови, но образующиеся при быстром
переходе от более высокого барометрического давления к более низко­
му при разгерметизации самолетов или космических аппаратов, а также
при быстром подъеме с больших глубин;
—экзогенные (пузырьки воздуха, попадающие в крупные вены при их ране­
нии либо инородные тела, например ЛС на масляной основе при их вве­
дении в холодном состоянии; осколки или пули).
• По локализации в ССС:
— артериальные (они циркулируют в артериальных сосудах и закупоривают
артерии большого крута кровообращения, попадая в них из легочных
вен или левых камер сердца, либо обтурируют артерии малого круга
кровообращения, попадая в них из правых камер сердца или из вен боль­
шого круга кровообращения);
- венозные (такие эмболы нередко обнаруживаются, например, в мелких
венах и венулах системы воротной вены или венах нижних конечностей);
- внутрисердечные (как правило, это тромбоэмболы. Они попадают в каме­
ры сердца из венозного русла либо образуются в полостях сердца, напри­
мер, при воспалении и травмах эндокарда, дефектах клапанов сердца
и/или их хорд, при инфаркте миокарда).
Механизмы, обусловливающие большую потребность ткани в притоке крови
и развитие ишемии
Факторы, повышающие потребность ткани в кровоснабжении:
- резкое и значительное повышение функции органа или ткани и возраста­
ющая в связи с этим интенсивность метаболизма в них. При этом теку­
щая потребность в притоке артериальной крови превышает его реаль­
ный уровень в данный момент.
Примеры:
- ишемия мышц (включая и миокард) при остро и значительно возрастающей
физической нагрузке;
- ишемия миокарда при резком существенном повышении уровня АД (напри­
мер, в условиях гипертензивного криза, стресса или криза при феохромоцитоме).
В подобных случаях работа сердца значительно возрастает под влиянием
избытка катехоламинов, оказывающих положительный хроно- и инотропный
эффект. При этом, хотя приток крови к миокарду по коронарным артериям
также усиливается, он недостаточен ввиду существенно повышенной функ­
ции сердца. Работа миокарда (и зависящая от этого потребность его в объеме
кровоснабжения) возрастает в значительно большей мере, чем артериальный
кровоток. Развивается ишемия миокарда, проявляющаяся приступом стено­
кардии, а нередко и инфаркт миокарда. При этом одновременно, как правило,
выявляются и признаки сужения артериальных сосудов сердца в связи с раз­
витием коронароатеросклероза.
Проявления ишемии
Проявления ишемии показаны на рис. 23.50.
Последствия ишемии
Основные последствия ишемии представлены на рис. 23.51.
Характер, выраженность и масштаб последствий ишемии зависят от мно­
гих факторов. Наиболее значимые:
- скорость развития ишемии (чем она выше, тем значительнее степень
повреждения тканей);
- диаметр пораженной артерии или артериолы (чем он больше, тем тяжелее
последствия ишемии);
Рис. 23.50. Основные проявления ишемии
Рис. 23.51. Основные последствия ишемии
— «чувствительность» ткани или органа к ишемии (она особенно высока
у ткани мозга, сердца, почек);
—значение ишемизированного органа или ткани для организма (ишемия
таких органов, как мозг, сердце, почки, может повлечь за собой гибель
организма; в отличие от этого, ишемия участка кожи или какой-либо
скелетной мышцы вполне совместима с жизнью);
—степень развития коллатеральных сосудов и скорость включения или акти­
вации коллатерального кровотока в ткани или органе.
Коллатеральный кровоток
Коллатеральный креветок представляет собой систему кровообращения
в сосудах вокруг ишемизированного участка ткани и в нем самом.
Активации (или увеличению) коллатерального кровообращения способ­
ствуют:
— градиент давления крови выше и ниже суженного участка сосуда;
— накопление в зоне ишемии БАВ с сосудорасширяющим действием (адено-
зин, ацетилхолин, ПГ, кинины и др.);
— активация местных парасимпатических влияний (способствующих расши­
рению коллатеральных артериол);
—высокая степень развития сосудистой сети (коллатерали) в пораженном
органе или ткани.
Органы и ткани, в зависимости от степени развития артериальных сосудов
и анастомозов между ними, делятся на три группы:
1) с абсолютно достаточными коллатералями — скелетная мускулатура,
брыжейка кишечника, легкие. В них совокупный просвет коллатеральных
сосудов равен диаметру магистральной артерии или превышает его. В связи
с этим прекращение кровотока по ней не вызывает выраженной ишемии
тканей в зоне кровоснабжения данной артерии;
2) с абсолютно недостаточными коллатералями — миокард, почки, головной
мозг, селезенка. В этих органах суммарный просвет коллатеральных сосу­
дов значительно меньше диаметра магистральной артерии. В связи с этим
окклюзия ее приводит к выраженной ишемии или инфаркту ткани;
3) с относительно достаточными (или, что то же самое, с относительно недо­
статочными) коллатералями — стенки кишечника, желудка, мочевого
пузыря, кожа, надпочечники. В них совокупный просвет коллатераль­
ных сосудов лишь ненамного меньше диаметра магистральной артерии.
Окклюзия крупного артериального ствола в этих органах сопровожда­
ется большей или меньшей степенью их ишемии.
Стаз
Стаз — типовая форма нарушения регионарного кровообращения, харак­
теризующаяся значительным замедлением или прекращением тока крови
и/или лимфы в сосудах органа или ткани.
Причины стаза
Стаз вызывается тремя группами факторов:
—ишемией;
—венозной гиперемией;
—увеличением содержания и/или активности проагрегантов в крови
и лимфе.
Ишемия приводит к стазу вследствие существенного замедления кровотока
и меньшего притока к ткани артериальной крови.
Венозная гиперемия вызывает стаз в результате замедления или прекраще­
ния оттока от ткани венозной крови.
Таким образом, и ишемия, и венозная гиперемия способны вызвать стаз
сами по себе. Кроме того, они создают условия для повышенного образования
и/или активации проагрегантов.
Проагреганты (тромбоксан А2, АДФ, ПГР, катехоламины, АТ к форменным
элементам крови), высокомолекулярные белки, катионы и другие соединения
обусловливают адгезию форменных элементов крови с формированием из них
агрегатов и тромбов, вызывающих стаз (рис. 23.52).
Проагреганты вызывают адгезию, агрегацию, агглютинацию форменных
элементов крови с последующим их лизисом и высвобождением из них допол­
нительного количества БАБ (в том числе проагрегантов, потенцирующих
реакции агрегации и агглютинации).
Рис. 23.52. Механизм развития стаза
Катионы. К+, Са2+, № +, М§2+ высвобождаются из клеток крови, повреж­
денных стенок сосудов и тканей. Адсорбируясь на цитолемме форменных эле­
ментов крови, избыток катионов нейтрализует их отрицательный поверхност­
ный заряд или даже меняет его на обратный. И если неповрежденные клетки
(благодаря отрицательному заряду) «отталкиваются» друг от друга, то повреж­
денные («нейтрализованные») образуют агрегаты. Еще более активно агреги­
руют «перезаряженные» клетки крови. Имея положительный поверхностный
заряд, они сближаются с «нейтрализованными» и особенно с поврежденными
(имеющими отрицательный заряд) клетками, формируя агрегаты, которые
адгезируют на интиме сосудов.
Высокомолекулярные белки (например, у-глобулины, фибриноген) сни­
мают поверхностный заряд неповрежденных клеток (соединяясь с отрица­
тельно заряженной поверхностью клеток с помощью аминогрупп, имеющих
положительный заряд) и потенцируют агрегацию форменных элементов
крови и адгезию их конгломератов к стенке сосуда (происходит в результа­
те фиксации множества белковых мицелл, которые обладают адгезивными
свойствами, на поверхности форменных элементов крови).
На заключительном этапе развития стаза происходит агрегация и/или агглю­
тинация форменных элементов крови. Это вызывает ее сгущение и замедляет
текучесть.
Виды стаза
Все разновидности стаза подразделяются на первичные и вторичные.
Первичный (истинный) стаз
Стартовое патогенетическое звено первичного стаза — активация форменных
элементов крови, в результате чего они выделяют большое количество проагре­
гантов и/или прокоагулянтов.
Следующее патогенетическое звено истинного стаза — агрегация и агглю­
тинация форменных элементов с прикреплением их (при участии фибрина)
к стенке микрососуда. Это и вызывает замедление или остановку кровотока
в сосудах.
Вторичный стаз (ишемический и венозно-застойный)
Ишемический стаз развивается как исход тяжелой ишемии (в связи со снижени­
ем притока артериальной крови, замедлением скорости ее тока, турбулентным его
характером). Это и приводит к агрегации и адгезии клеток крови, сочетающимся
с замедлением и прекращением кровотока в артериолах и капиллярах.
Застойный (венозно-застойный) вариант стаза — результат замедления оттока
венозной крови, сгущения ее, изменения физико-химических свойств, повреж­
дения форменных элементов крови. В последующем клетки крови агглю­
тинируют, адгезируют друг с другом и со стенкой микрососудов, замедляя
и останавливая отток венозной крови.
Проявления стаза
При стазе происходят характерные изменения в сосудах микроциркуляторного русла. К ним относятся:
—уменьшение внутреннего диаметра микрососудов при ишемическом стазе;
—увеличение просвета сосудов микроциркуляторного русла при венозно­
застойном стазе;
—формирование множества агрегатов форменных элементов крови в про­
свете сосудов микроциркуляторного русла и на их стенках;
—образование микрокровоизлияний (чаще при венозно-застойном стазе).
Последствия стаза
Быстрое устранение причины стаза обусловливает восстановление тока крови
в сосудах микроциркуляторного русла, и в тканях не развиваются какие-либо
существенные изменения.
Длительный стаз (особенно ишемический) приводит к развитию дистрофиче­
ских изменений в тканях и нередко к инфаркту.
НАРУШЕНИЕ КРОВО- И ЛИМФООБРАЩЕНИЯ В СОСУДАХ
МИКРОЦИРКУЛЯТОРНОГО РУСЛА
Микроциркуляция крови и лимфы включает триаду процессов:
—упорядоченное движение крови и лимфы по микрососудам;
—трансмуральный перенос плазмы и форменных элементов крови;
—перемещение жидкости во внесосудистом пространстве.
Микроциркуляторное русло
Совокупность артериол, капилляров, венул и артериолокапиллярных шунтов
составляет структурно-функциональную единицу ССС — микроциркуляторное
(терминальное) сосудистое русло.
Терминальное сосудистое русло имеет следующую структуру: от терми­
нальной артериолы отходит метартериола, распадающаяся на сеть капил­
ляров; венозная часть капилляров открывается в посткапиллярные венулы.
К микроциркуляторному руслу относят также мелкие лимфатические сосуды
и межклеточное пространство.
В месте отделения капилляра от артериолы имеется прекапиллярный
сфинктер (скопление циркулярно ориентированных ГМК). Сфинктеры кон­
тролируют локальный объем крови, проходящий через капилляры. Объем
крови, проходящей через терминальное сосудистое русло в целом, определя­
ется тонусом ГМК артериол.
В микроциркуляторном русле имеются артериоловенулярные анастомозы,
связывающие артериолы непосредственно с венулами, и артериовенозные
анастомозы между мелкими артериями и мелкими венами. Стенка анасто­
мозов содержит много ГМК. Обе разновидности анастомозов обеспечивают
юкстакапиллярный кровоток.
Артериоловенулярные анастомозы в большом количестве имеются в неко­
торых участках кожи, где они играют важную роль в терморегуляции (мочка
уха, пальцы).
Причины расстройств микроциркуляции
Многочисленные причины, вызывающие разнообразные нарушения
микроциркуляции, объединяются в три группы.
• Расстройства центрального и регионарного кровообращения. К наиболее зна­
чимым из них относятся:
—сердечная недостаточность;
—патологическая артериальная гиперемия;
—венозная гиперемия;
—ишемия.
• Изменения вязкости и объема крови. Развиваются вследствие гемоконцен­
трации или гемодилюции.
Причины гемоконцентрации:
—гипогидратация организма с развитием полицитемической гиповолемии;
—полицитемия;
—гиперпротеинемия (преимущественно гиперфибриногенемия).
Причины гемодилюции:
—гипергидратация организма с развитием олигоцитемической гиперволе­
мии;
—панцитопения (уменьшение в крови количества всех ее форменных эле­
ментов);
—повышенная агрегация и агглютинация форменных элементов крови (при­
водит к значительному повышению вязкости крови);
—ДВС-синдром.
• Повреждение стенок сосудов микроциркуляторного русла. Наблюдается
при васкулитах, атеросклерозе, воспалении, циррозе органов, в зоне опу­
холевого роста и др.
Типовые нарушения микроциркуляции
Выделяют три группы типовых форм нарушения микроциркуляции:
- внутрисосудистые (интраваскулярные);
- чресстеночные (трансмуральные);
- внесосудистые (экстраваскулярные).
В совокупности расстройства микроциркуляции нередко приводят к капил­
ляротрофической недостаточности.
Внутрисосудистые нарушения микроциркуляции
Типовые формы интраваскулярных (внутрисосудистых) расстройств микро­
циркуляции представлены на рис. 23.53.
Внутрисосудистые нарушения микроциркуляции
г
Замедление тока
крови и/или лимфы
"
Чрезмерное
ускорение
тока крови
1
Нарушения
ламинарности
тока крови
г
Чрезмерное увеличение
внекапиллярного
тока крови
Рис. 23.53. Типовые формы интраваскулярных расстройств микроциркуляции
К ним относят:
• Замедление (вплоть до стаза) тока крови и/или лимфы. Проявления замед­
ления тока крови и/или лимфы сходны с теми, которые наблюдаются
в сосудах микроциркуляторного русла при венозной гиперемии, ишемии
или стазе (см. выше). Причины:
—расстройства гемо- и лимфодинамики (например, при сердечной недоста­
точности, венозной гиперемии, ишемии, лимфорее);
—увеличение вязкости крови (например, в результате гемоконцентрации
при длительной рвоте, диарее, плазморрагии при ожогах, полицитемии,
гиперпротеинемии, ДВС);
—уменьшение просвета микрососудов (вследствие сдавления их опухолью,
отечной тканью, образования в них тромбов, попадания эмбола, набу­
хания или гиперплазии эндотелиальных клеток, образования атероскле­
ротической бляшки и т.п.).
• Чрезмерное ускорение тока крови и/или лимфы. Причины:
—нарушения гемо- и/или лимфодинамики (например, при патологической
артериальной гиперемии или сбросе артериальной крови в венозное
русло через артериоловенулярные шунты);
—уменьшение вязкости крови при гемодилюции, гипопротеинемии, почеч­
ной недостаточности, панцитопении.
• Нарушение ламинарности (турбулентность) тока крови и/или лимфы. Причины:
—повреждение стенок микрососудов и/или нарушение их гладкости (при вас­
кулитах, гиперплазии клеток эндотелия, артериосклерозе, фиброзных
изменениях в сосудистой стенке и т.п.);
—изменения агрегатного состояния крови (например, при формировании
пристеночных микротромбов, нарушающих ламинарный ток крови).
• Чрезмерное увеличение юкстакапиллярного тока крови. Развивается при
открытии артериовенозных и артериоловенулярных шунтов и прояв­
ляется сбросом избытка крови из артерий и артериол в вены и вену­
лы, минуя капиллярную сеть микроциркуляторного русла. Наиболее
частая причина избыточного открытия шунтов — закрытие прекапиллярных сфинктеров. Это происходит в результате спазма ГМК артерий и /
или артериол (например, при значительном повышении уровня кате­
холаминов в крови у пациентов с феохромоцитомой; при чрезмерном
повышении тонуса симпатической нервной системы в условиях стресса;
при гипертензивном кризе). Проявления чрезмерного юкстакапиллярного
кровотока:
—увеличение количества и диаметра функционирующих шунтов;
— признаки ишемии ткани в зоне сброса крови из артериол в венулы;
—турбулентность тока крови в местах ответвлений шунтирующих артерий
и артериол и входа их в вены и венулы (турбулентность кровотока обуслов­
лена тем, что шунты отходят от артерий и артериол и впадают в вены
и венулы, как правило, под значительным углом);
— наличие агрегатов форменных элементов крови и тромбов в местах турбу­
лентности кровотока (вызвано соударением форменных элементов крови
друг с другом, а также с эндотелием, вследствии чего они выделяют
избыток проагрегантов и прокоагулянтов).
Трансмуральные нарушения микроциркуляции
Через стенку микрососуда циркулирует жидкая часть крови и лимфы
(характеризуется понятием «проницаемость сосудистой стенки» для плазмы)
и форменные элементы крови (выражается понятием «эмиграция»),
В зависимости от преобладающих расстройств проницаемости или эми­
грации трансмуральные нарушения микроциркуляции компонентов крови
и лимфы подразделяются на две категории:
—расстройства проницаемости стенок микрососудов для жидкости;
—нарушения эмиграции форменных элементов крови через стенку микрососудов.
Нарушение проницаемости стенки микрососуда для жидкости
При различных патологических состояниях объем перемещения плазмы
крови и/или лимфы через стенку сосуда может чрезмерно (неадекватно) либо
возрастать, либо уменьшаться.
Возрастающий объем жидкости, перемещающейся через стенку сосудов микро­
циркуляторного русла
Основные причины повышенной проницаемости стенок микрососудов
показаны на рис. 23.54.
Главное последствие повышенной проницаемости стенок микрососудов —
потенцирование механизмов циркуляции жидкости:
—фильтрации (транспорт жидкости по градиенту гидростатического давле­
ния);
—трансцитоза (энергозависимый пиноцитоз);
- диффузии (перенос жидкости без затрат энергии);
—осмоса (направленная диффузия жидкости по градиенту осмотического
давления).
Этиологические факторы
V
Развитие ацидоза
''
Неферментный
гидролиз основного
вещества базальной
мембраны сосудов
Активация
гидролаз
и
Округление клеток
эндотелия
1
Ферментный
гидролиз основ­
ного вещества
базальной мем­
браны сосудов
*
Перерастяжение стенок
микрососудов
1.
Расширение
межэндотелиальных
щелей, образование
каналов
1
Растяжение
фенестр,
образование
микроразрывов
в стенках
микрососудов
Повышение проницаемости
стенок микрососудов для жидкости
Рис. 23.54. Основные причины повышенной проницаемости жидкости через стенки
микрососудов
Уменьшение проницаемости стенок сосудов микроциркуляторного русла
Причины снижения проницаемости стенок микрососудов:
- утолщение стенок микрососудов (например, при хронических васкули-
тах);
- уплотнение стенок микрососудов (например, вследствие их кальцифика­
ции).
Наиболее значимым последствием уменьшенной проницаемости стенок
микрососудов является меньшая эффективность механизмов перемещения
через них жидкости:
- фильтрации;
- диффузии;
- трансцитоза;
- осмоса.
Нарушение перемещения форменных элементов крови через стенку микро­
сосуда
Эмиграция лейкоцитов через стенку микрососудов (в ткань и обратно)
осуществляется в норме постоянно. Лейкоциты выполняют в тканях функцию
иммунобиологического надзора (ИБН) за индивидуальным и постоянным
антигенным составом организма. Эритроциты и тромбоциты такой способ­
ностью не обладают.
При различных патологических состояниях эмиграция лейкоцитов через
стенку микрососуда, как правило, возрастает; эритроциты и тромбоциты пере­
мещаются в ткань пассивно с током плазмы крови.
К типовым формам патологии перемещения форменных элементов крови через
стенку микрососуда в ткань относят:
—чрезмерный выход из микрососудов эритроцитов с развитием микрогемор­
рагий (например, при васкулитах или геморрагических синдромах);
—избыточное перемещение в ткань тромбоцитов (обычно вместе с эритро­
цитами).
Экстраваскулярные нарушения микроциркуляции
Внесосудистые (экстраваскулярные) нарушения микроциркуляции сопро­
вождаются увеличением или уменьшением объема межклеточной жидкости,
что приводит к замедлению оттока ее в сосуды микроциркуляторного русла.
Причина чрезмерного замедления оттока интерстициальной жидкости и избы­
точного увеличения ее объема — местные тканевые патологические процессы.
Наиболее часто это:
—воспаление;
— аллергические реакции;
— рост новообразований;
—склерозирование ткани;
—венозная гиперемия;
—стаз.
Последствия замедленного оттока интерстициальной жидкости и избыточного
увеличения ее объема:
—увеличение содержания в интерстициальной жидкости продуктов нормаль­
ного и нарушенного метаболизма (ряд таких метаболитов может оказывать
цитотоксическое и цитолитическое действие);
—дисбаланс ионов (это способствует отеку ткани, нарушает формирование
МП и ПД);
—образование избытка и/или активация БАВ (например, Ф НО-а, про­
коагулянтов, мембраноатакующего комплекса), способных усугубить
повреждение клеток, потенцировать расстройства крово- и лимфообра­
щения, пластических процессов;
—нарушение обмена 0 2, С 0 2, субстратов и продуктов обмена веществ;
—сдавление клеток избытком интерстициальной жидкости.
Причины чрезмерного замедления оттока интерстициальной жидкости, соче­
тающегося с уменьшением ее оттока из интерстициального пространства:
—гипогидратация организма, тканей и органов (например, в результате
длительной диареи, плазморрагии, при интенсивном потоотделении);
—снижение лимфообразования (например, при ишемии ткани или систем­
ной гиповолемии);
—уменьшение эффективности фильтрации жидкости в артериолах и прекапиллярах и/или увеличение реабсорбции ее в посткапиллярах и венулах
(например, при дистрофических и склеротических процессах в тканях).
Последствия чрезмерного уменьшения оттока интерстициальной жидкости
сходны с последствиями, наблюдающимися при увеличении объема интерсти­
циальной жидкости, которое сочетается с замедлением ее оттока (см. выше).
Капилляротрофическая недостаточность
Капилляротрофическая недостаточность — состояние, которое характеризу­
ется следующими нарушениями, приводящими к расстройству обмена веществ
и пластических процессов в тканях и органах:
—крово- и лимфотока в сосудах микроциркуляторного русла;
—транспорта жидкости и/или форменных элементов крови через стенки
микрососудов;
—оттока межклеточной жидкости (рис. 23.55).
Капилляротрофическая недостаточность
__________ Г
Нарушение кровои лимфообращения
в микрососудах
Расстройства транспорта
жидкости и форменных
элементов крови через
стенку микрососудов
Замедление
тока (оттока)
межклеточной
жидкости
Нарушение
обмена веществ
в тканях
и органах
Дистрофии, нарушения пластических процессов,
функции органов, тканей, жизнедеятельности организма
Рис. 23.55. Признаки капилляротрофической недостаточности
Последствия капилляротрофической недостаточности
В результате описанных выше расстройств микроциркуляции крови, лимфы
и межклеточной жидкости в тканях и органах развиваются:
—различные формы дистрофии;
—расстройства пластических процессов (по восстановлению поврежденных
и синтезу новых клеток и внеклеточных структур);
—нарушения функционирования тканей, органов и организма в целом.
Сладж
Сладж — типовая форма патологии агрегатного состояния крови. Характе­
ризуется адгезией, агрегацией и агглютинацией форменных элементов
крови, обусловливающими сепарацию ее на плазму и плотные конгломераты
из эритроцитов, лейкоцитов, тромбоцитов, а также образование тромбов.
Конгломераты и тромбы замедляют кровоток, обтурируют сосуды микроцир­
куляторного русла и приводят к нарушениям микрогемоциркуляции.
Причины сладжа
— Нарушения центральной гемодинамики (например, при сердечной недо­
статочности, венозном застое, ишемии, патологических формах артери­
альной гиперемии).
— Повышение вязкости крови (например, в условиях гемоконцентрации,
гиперпротеинемии, полицитемии).
— Повреждение стенок микрососудов (при местных патологических процес­
сах: воспалении, иммунопатологических состояниях, опухолевом росте
и др.).
Патогенез сладжа
Основные звенья патогенеза сладжа представлены на рис. 23.56.
- Этиологические признаки
1
'’
Актива ция ФЭК,
Устранение поверхностного
Адсорбция мицелл белка
высвобо»едение ими заряда ФЭК, их «перезарядка» на ФЭК, потенцирование
проагр егантов
избытком катионов и молекул
процесса их оседания
белка
на стенках сосудов
1г
1
'
и
Образование конгломератов из ФЭК
±
I Сепарация крови (на плазму и конгломераты ФЭК)
Рис. 23.56. Основные звенья патогенеза сладжа: ФЭК — форменные элементы крови
Последствия сладжа
Сладж является причиной существенных нарушений микрогемоциркуля­
ции и обмена веществ в тканях и органах, которые приводят:
—к внутрисосудистым расстройствам микроциркуляции (замедлению, вплоть
до стаза; турбулентному току крови; открытию артериоловенулярных
шунтов);
—нарушениям трансмурального перемещения форменных элементов крови,
а также оттоку ее от тканей;
—расстройствам метаболизма в тканях и органах с развитием дистрофий
и дисплазий.
В целом, совокупность указанных выше изменений приводит к развитию
капилляротрофической недостаточности.
Отсюда следует важный вывод о том, что феномен сладжа может быть:
—причиной расстройств микроциркуляции (в тех случаях, когда он разви­
вается первично);
—следствием внутрисосудистых нарушений микроциркуляции (при пер­
вичном их развитии).
Глава 24
ТИПОВЫЕ ФОРМЫ ПАТОЛОГИИ СИСТЕМЫ
ВНЕШНЕГО ДЫХАНИЯ
Дыхание — обмен кислорода и углекислого газа — происходит путем
их диффузии. Определяющим фактором этого процесса является разница
в парциальном давлении газов.
Условно выделяют «внутреннее» (тканевое) и «внешнее» (альвеолярное)
дыхание.
Тканевое дыхание — двунаправленная диффузия газов между просветом кро­
веносных капилляров и клетками (термин «тканевое дыхание» имеет и более
широкое значение — утилизация 0 2 в процессе метаболизма клеток).
Внешнее дыхание — двунаправленная диффузия газов между альвеолами
легких и кровью капилляров межальвеолярных перегородок через аэрогематический барьер.
Аппарат внешнего дыхания включает:
—дыхательные пути;
—респираторный отдел легких;
—грудную клетку (включая ее костно-хрящевой каркас и нервно-мышечную
систему);
—нервные центры регуляции дыхания.
Аппарат внешнего дыхания обеспечивает:
— альвеолярную вентиляцию (двунаправленная диффузия кислорода и угле­
кислого через альвеолярно-капиллярную мембрану);
—перфузию ткани легких (кровоснабжение их).
Парциальные или комбинированные расстройства функционирования аппа­
рата внешнего дыхания могут привести к дыхательной недостаточности (ДН).
ДН — состояние, характеризующееся развитием респираторной гипоксии и,
как правило, гиперкапнии в результате нарушения газообменной функции легких.
Оценка функции внешнего дыхания
Для характеристики функции внешнего дыхания применяют показате­
ли, позволяющие оценить функцию воздухоносных путей и респираторного
отдела легких (дополнительные сведения о показателях функции внешнего
дыхания приведены в статьях «Емкость», «Объем» и «Скорость», приложение
«Справочник терминов»).
При исследовании функции легких анализируют, главным образом, состояние
показателей, характеризующих:
—легочные объемы;
— объемную скорость выдоха;
—диффузионную способность альвеолярно-капиллярной мембраны.
Легочные объемы характеризуются статическими и динамическими пока­
зателями:
—показатели статических легочных объемов отражают состояние эластиче­
ских свойств ткани легких и грудной клетки;
—показатели динамических легочных объемов характеризуют проходимость
дыхательных путей.
Объемная скорость выдоха представляет собой максимальную скорость дви­
жения воздушного потока в дыхательных путях при форсированном выдохе.
Скорость воздушного потока зависит от состояния легочных объемов и силы
выдоха. Воздушный поток возрастает при увеличении силы выдоха, особенно
в его начале (этот поток может составлять в течение первой секунды выдоха
более 75% жизненной емкости легких). На объемную скорость выдоха влияют
также эластическая тяга ткани легкого и сопротивление дыхательных путей воз­
душному потоку.
Диффузионная емкость, или диффузионная способность (Дс) аэрогематического барьера, — показатель эффективности транспорта определенного газа
из альвеол в кровь капилляров легких.
Спирометрия (спирография) и ее показатели
Спирометрия (спирография) представляет собой измерение ЖЕЛ и дру­
гих легочных объемов с помощью спирографа (прибор для непрерывной
графической регистрации изменения объемов вдыхаемого и выдыхаемого
воздуха).
Спирограмма регистрируется в момент максимально глубокого вдоха
и спокойного выдоха. После этого вдох и выдох записываются повторно,
но с максимальным усилием.
Показатели спирометрии позволяют дифференцировать обструктивные
и рестриктивные расстройства альвеолярной вентиляции, оценивать степень
ДН и ее динамику при лечении.
Многие показатели спирограммы выражаются в относительных величи­
нах (чаще в процентах) от их средних значений в популяции (учитывается
пол, возраст, рост). Нормальным считают колебания показателя в диапазоне
80-120%.
• Дыхательный объем (ДО) — объем воздуха, поступающего в легкие за один
вдох при спокойном дыхании (норма 500-800 мл). Часть ДО, участвующая
в газообмене, носит название «альвеолярный объем» (АО); остаток (около
30% ДО) — это анатомически мертвое пространство, или вредный объем.
• ЖЕЛ — максимальный объем воздуха, изгоняемого из легких вслед за мак­
симальным вдохом. ЖЕЛ прогрессирующе снижается при рестриктивных
болезнях легких. В связи с этим ЖЕЛ в сочетании с показателем диффу­
зионной емкости легких помогает оценить течение болезни и эффектив­
ность ее лечения у пациентов с рестриктивной патологией.
• Форсированная жизненная емкость (ФЖЕЛ) определяется аналогично
ЖЕЛ, за исключением того, что дыхание при этом производится с макси­
мально возможной силой и скоростью. Форсированный выдох сопрово­
ждается сужением дыхательных путей, замедлением его скорости.
• Объем форсированного выдоха за 1-ю секунду (ОФВ,) — объем воздуха,
изгоняемого с максимальным усилием из легких в течение первой секунды
выдоха после глубокого вдоха, т.е. это часть ФЖЕЛ, выдыхаемая за первую
секунду. ОФВ, отражает состояние крупных дыхательных путей и часто
выражается в процентах от ЖЕЛ (нормальное значение ОФВ, = 75% ЖЕЛ).
• ОФВ,/ФЖ ЕЛ — отношение ОФВ, к ФЖЕЛ (индекс Тиффно), выра­
женное в процентах (в норме больше или равно 70%). Значение ОФВ,/
ФЖЕЛ прямо пропорционально силе выдоха. Этот показатель важен
для выявления обструктивных нарушений дыхания, а также для диагно­
стики рестриктивных его расстройств. Снижение только ОФВ, (ОФВ,/
ФЖЕЛ <70%) свидетельствует об обструкции; снижение обоих показате­
лей (ОФВ,/ФЖЕЛ >70%) указывает на рестриктивную патологию.
• Средняя объемная скорость выдоха (СОС25_75%) — скорость потока форси­
рованного выдоха в его середине (т.е. между 25 и 75% ФЖЕЛ); иначе его
называют максимальным потоком середины выдоха. СОС25_75% отражает
состояние мелких дыхательных путей. Он более информативен, чем ОФВ,,
при выявлении ранних обструктивных нарушений.
• Пик объемной скорости выдоха (мощность выдоха) — максимальная объ­
емная скорость, которую исследуемый может развить при форсированном
выдохе. Это показатель проходимости дыхательных путей на уровне трахеи
и крупных бронхов. Зависит от мышечного усилия пациента.
Другие легочные объемы
Получение других показателей легочных объемов требует применения
не только спирометрии, но и теста с разведением гелия в легочном воздухе
(позволяющего определить объем газа в легких).
• Общая емкость легких (ОЕЛ) — объем воздуха, содержащегося в легких
на высоте максимального вдоха.
• Функциональная остаточная емкость (ФОЕ) — объем воздуха, остающийся
в легких в конце нормального выдоха. Отражает состояние покоя легких
и грудной стенки (объем легких в условиях равновесия эластической тяги
легких, направленной внутрь, и тяги грудной клетки, направленной нару­
жу. ФОЕ представлена двумя компонентами:
- резервным объемом выдоха (РОвьщ) — часть ФОЕ, которая может быть
изгнана из легких при максимально усиленном выдохе;
—остаточным объемом легких (ООЛ) — объем воздуха, остающийся в лег­
ких после максимально усиленного выдоха (в норме 25—30% от ФОЕ).
• Соотношения легочных объемов:
ОЕЛ = ЖЕЛ + ООЛ;
ООЛ = ФОЕ —РОвыд.
Исследование других функций легких
• Диффузионная способность (диффузионная емкость, Дс) легких по окиси
углерода (Дссо) отражает состояние альвеолярно-капиллярной мембра­
ны — аэрогематического барьера. Дссо определяют, измеряя количество
окиси углерода (СО), поступившей из альвеолярного воздуха в кровь
легочных капилляров после того, как пациент вдохнул известное количе­
ство СО (0,1%). Выражают Дссо в мл/мин/мм рт.ст.
• Кривая податливости (растяжимости) легких. Эластичность легких опреде­
ляет соотношение изменений легочных объемов и транспульмонального
давления (разность давления в альвеолах и плевральной полости). Для рас­
тяжения легкого до заданного объема необходимо определенное усилие,
складывающееся из эластической тяги легкого, направленной внутрь,
и эластической тяги грудной стенки, направленной наружу. В норме
в состоянии покоя на момент конца выдоха (т.е. в положении ФОЕ) эла­
стическая тяга легкого полностью сбалансирована эластической тягой
грудной стенки. При полном вдохе (т.е. при ОЕЛ) легкие достигают своей
максимальной эластической тяги. При полном выдохе (т.е. при резервном
объеме выдоха) грудная стенка достигает своей максимальной эластиче­
ской тяги. Зависимость транспульмонального давления от объема легких
отображается в виде кривой растяжимости легких. Податливость, или рас­
тяжимость (С) определяется по наклону кривой давление—объем (Р—V)
над уровнем дыхательного объема:
С = У : Р (в норме 200 мл/см Н20).
Потеря эластической тяги легкого (например, при эмфиземе) увеличивает
растяжимость, смещая кривую податливости влево. Увеличение эластиче­
ской тяги легкого (например, при рестриктивных заболеваниях легких,
таких, как идиопатический легочный фиброз) снижает растяжимость,
смещая кривую податливости вниз и вправо.
• Сопротивление дыхательных путей (СДП). Этот показатель отражает состо­
яние крупных дыхательных путей, так как 80—90% сопротивления воздуш­
ному потоку создают именно они. СДП обычно определяют по динамиче­
ским легочным объемам и объемным скоростям выдоха. Величины СДП
повышены при обструктивных болезнях легких и снижены при рестрик­
тивных легочных заболеваниях.
Спирометрические признаки дыхательной
недостаточ ности
Обструктивные расстройства альвеолярной вентиляции
Объемные скорости. При альвеолярной гиповентиляции вследствие обструк­
ции дыхательных путей показатель ОФВ,/ФЖЕЛ снижается (менее 70%). Этот
показатель тесно связан со временем выдоха. Однако даже при значитель­
ной обструкции периферических дыхательных путей ОФВ,/ФЖЕЛ может
находиться в пределах средней физиологической нормы. В такой ситуации
обструкцию дыхательных путей можно выявить по снижению СОС25_75%
(до 60% или ниже от должной величины).
Легочные объемы. Изменения дыхательных объемов легких могут встречаться
при умеренной и тяжелой обструкции дыхательных путей. Измерение объемов
легких помогает распознать перерастяжение легких вследствие преждевремен­
ного закрытия дыхательных путей (экспираторный коллапс бронхов).
Известно, что во время форсированного выдоха дистальные отделы дыхатель­
ных путей закрываются раньше, чем изгоняется воздух, что приводит к перерастяжению легких, вызывающему увеличение ФОЕ, ООЛ и ООЛ/ЖЕЛ.
При нарушениях проходимости мелких дыхательных путей в них образу­
ются «воздушные ловушки», увеличивающие ООЛ, тогда как ФОЕ и ОФВ,
остаются нормальными.
При эмфиземе легких разрушение стенок альвеол и потеря легкими эла­
стической тяги вызывают увеличение ОЕЛ.
Рестриктивные расстройства альвеолярной вентиляции
Объемные скорости. ОФВ,/ФЖЕЛ и СОС25_75% при рестриктивной форме
гиповентиляции легких могут быть нормальными или повышенными вслед­
ствие увеличенной тяги стенок дыхательных путей.
Легочные объемы. Наиболее информативным признаком при рестриктив­
ных нарушениях вентиляции легких является снижение ЖЕЛ и ОЕЛ.
Ригидность легких при их рестриктивных поражениях увеличивает эласти­
ческую тягу легких и тем самым снижает ФОЕ.
Ригидность грудной стенки (например, при кифосколиозе) уменьшает
легочные объемы, поскольку ограничивает расширение легких.
Растяжимость легких также ограничена вследствие увеличения их эласти­
ческой тяги.
Сопротивление дыхательных путей (СДП) воздушному потоку снижено,
так как благодаря эластическим силам дыхательные пути расширены на уров­
не любых легочных объемов.
Газы артериальной крови
Парциальное давление кислорода (р 0 2) и двуокиси углерода (р С 02),
а также рН — важные параметры для оценки функции легких. Они указыва­
ют на состояние газообмена между легкими и кровью.
р 0 2. При отсутствии патологии р 0 2 снижается с возрастом вследствие
утраты легкими эластичности (в норме оно составляет 90 мм рт.ст. в 20 лет
и около 70 мм рт.ст. к 70 годам).
Уменьшение р 0 2 по сравнению с нормой свидетельствует о гипоксемии,
однако насыщение тканей кислородом при этом существенно не снижается
до тех пор, пока р 0 2 не упадет ниже 60 мм рт.ст.
р С 02. Отражает состояние альвеолярной вентиляции (в норме 35—45 мм рт.
ст.). Гиперкапния (респираторный ацидоз, высокое р С 0 2) свидетельствует
о гиповентиляции.
рН. Сопоставление артериального рН (в норме 7,35—7,45) с рС 02дает возмож­
ность отличить респираторные нарушения КОС от метаболических. Например,
если раС 0 2 и рН обратно пропорциональны (один показатель снижается при уве­
личении другого), расстройство КОС имеет респираторную природу.
Нарушения вентиляционно-перфузионного (У/О) соотношения
Оптимальные вентиляция легких и легочный кровоток обеспечивают
достаточную доставку кислорода к тканям и адекватную элиминацию дву­
окиси углерода из организма.
В среднем соотношение У/О составляет 0,8 (в норме допускается физио­
логический дисбаланс У/О, обусловленный «сбросом» в легких примерно
2% объема артериальной крови в венозную через артериовенозные шунты
без газообмена).
Трактовка последствий вентиляционно-перфузионных нарушений в легких
Низкие значения У/О свидетельствуют о неадекватной вентиляции нор­
мально снабжаемых кровью участков легкого. В результате и происходит
снижение р 0 2 (гипоксемия). Несмотря на окклюзию альвеол или их заполне­
ние жидкостью в отдельных участках легких, степень гипоксемии у пациента
можно снизить за счет дыхания обогащенной кислородом газовой смеси.
Если в каком-либо участке легкого отсутствует альвеолярная вентиляция,
У/О приближается к 0 и происходит шунтирование крови справа налево
(венозная кровь смешивается с артериальной), то такая форма гипоксе­
мии не устраняется оксигенотерапией, поскольку кислород не достигает
альвеолярно-капиллярной мембраны. При этом артериальная кровь, прите­
кающая из хорошо оксигенированных участков легких, не может устранить
низкое содержание кислорода, поскольку НЬ им полностью насыщен.
Высокие значения У/О указывают на адекватную вентиляцию слабо снаб­
жаемых кровью участков легких. Уровень кислородного обмена низок, так
как доступный НЬ способен связать лишь ограниченное количество кислорода.
Если в каком-либо участке легкого отсутствует кровоток и, следователь­
но, в нем не происходит газообмена, то У/О чрезмерно увеличен. Наличие
«мертвого неперфузируемого» альвеолярного пространства приводит к накоп­
лению в организме двуокиси углерода и гипоксии. Гиперкапния стимулирует
дыхательный центр, усиливая дыхание и увеличивая вентиляцию, что может
нормализовать уровень р С 0 2, но (из-за отсутствия перфузии легких кровью)
не увеличивает р 0 2. Развивается острая ДН.
Альвеолярно-артериальный градиент по кислороду — разница между значе­
ниями альвеолярного р 0 2 (рА0 2) и артериального р 0 2 (ра0 2). При уменьшении
альвеолярно-артериального градиента отношение У/О увеличивается. Расчет
градиента кислорода позволяет отличить гиповентиляцию от других причин
гипоксемии и оценить тяжесть поражения легких.
Типовые формы патологии
внешнего дыхания
К типовым формам патологии внешнего дыхания относят нарушения:
- вентиляции альвеол легких воздухом;
- перфузии легких кровью;
- адекватности соотношения вентиляции и перфузии легких (нарушения
вентиляционно-перфузионного соответствия);
—диффузии кислорода и углекислого газа через альвеолярно-капиллярную
мембрану.
НАРУШЕНИЯ ВЕНТИЛЯЦИИ АЛЬВЕОЛ ЛЕГКИХ
Наиболее частая причина нарушений обмена кислорода и углекислого газа
в альвеолах легких — расстройства их вентиляции. Выделяют альвеолярную
гипо- и гипервентиляцию.
Альвеолярная гиповентиляция
Альвеолярная гиповентиляция — типовая форма нарушения внешнего
дыхания, при которой реальный объем вентиляции альвеол за единицу
времени ниже необходимого для организма в данных условиях.
Причины альвеолярной гиповентиляции
Причины альвеолярной гиповентиляции вследствие расстройств подраз­
деляются на две группы:
— нарушающие биомеханику внешнего дыхания
—и/или расстраивающие механизмы его регуляции.
Эти группы причин представлены на рис. 24.1.
Рис. 24.1. Основные причины гиповентиляции легких
Расстройства биомеханики внешнего дыхания
Среди расстройств биомеханики внешнего дыхания различают обструктивные и рестриктивные их разновидности.
Обструктивный тип альвеолярной гиповентиляции
Он заключается в снижении проходимости дыхательных путей. В связи
с этим повышается сопротивление движению воздушного потока, снижается
объем вентиляции соответствующих областей легких, возрастает работа дыха­
тельных мышц, увеличивается энергообеспечение (энергорасход) аппарата
внешнего дыхания.
Даже сравнительно небольшая обструкция бронхов может существенно
повысить их сопротивление воздушному потоку и усилить работу дыхательных
мышц (например, уменьшение диаметра бронха на 1/3 способно увеличить
сопротивление движению воздуха на 300—500%).
Основные причины обструкции дыхательных путей:
- обтурация просвета верхних и/или нижних дыхательных путей инород­
ными телами (например, пищей во время рвоты; западающим языком
-
-
при коме или наркозе, во сне; мокротой, слизью, экссудатом, кровью;
новообразованиями воздухопроводящих путей);
спазм бронхов и/или бронхиол (например, при приступе бронхиальной
астмы); бронхоспазм, как правило, сочетается с отеком слизистой обо­
лочки и образованием вязкой мокроты;
спазм мышц гортани (например, при вдыхании раздражающих веществ
или при невротических состояниях);
сдавление (компрессия) дыхательных путей извне (например, опухолью, уве­
личенными лимфатическими узлами средостения, щитовидной железой);
динамическое сдавление бронхов среднего и мелкого диаметра при повы­
шении внутрилегочного давления во время форсированного выдоха.
Этот феномен, известный как «экспираторная компрессия бронхов»
(или феномен экспираторной компрессии, гипервоздушности легко­
го, экспираторный коллапс бронхов), может наблюдаться при сильном
кашле, у пациентов с эмфиземой легких, при форсированном дыхании
во время физической нагрузки.
Проявления обструкции дыхательных путей
Основные проявления гиповентиляции легких обструктивного типа пред­
ставлены на рис. 24.2.
Рис. 24.2. Основные проявления гиповентиляции легких обструктивного типа
Рестриктивный тип альвеолярной гиповентиляции
Этот тип гиповентиляции характеризуется снижением степени расправления
легких. В связи с этим уменьшается вентиляция легких, увеличивается нагруз­
ка на дыхательную мускулатуру, повышается энергетическая «стоимость»
дыхания.
Основные причины альвеолярной гиповентиляции (внутрилегочные и внелегочные) рестриктивного типа представлены на рис. 24.3.
Рис. 24.3. Основные причины гиповентиляции легких рестриктивного типа
Внутрилегочные (паренхиматозные) причины альвеолярной гиповентиляции
рестриктивного типа.
Они заключаются в снижении растяжимости ткани легких (изменение
объема легких, отнесенное к величине чрезлегочного давления). Наиболее
часто выявляются при:
—фиброзирующих процессах в легочной ткани (например, в результате диф­
фузного воспаления или пневмофиброза);
— обширных и/или множественных ателектазах легких, диффузных опухолях
легких.
Внелегочные причины рестриктивного типа гиповентиляции легких
Они ограничивают расправление легких и диапазон их дыхательных экс­
курсий. Причинами их могут быть:
— сдавление грудной клетки (например, тяжелыми предметами при завалах
грунтом, при разрушении зданий; корсетом, скафандром);
—снижение подвижности суставов грудной клетки и/или окостенение хрящей
ребер («каркасная» гиповентиляция легких); развивается в результате
кифосколиоза, анкилозирующего спондилита;
—воспаление плевры; сильная боль при плеврите заставляет пациента огра­
ничивать объем вдоха;
—фиброз плевры;
— скопление в грудной полости крови, экссудата, транссудата, воздуха.
Рестриктивный тип гиповентиляции легких проявляется:
— снижением общей емкости легких;
—уменьшением остаточного объема легких и ЖЕЛ (этот показатель прямо
отражает степень рестрикции легких).
Н аруш ен и я м е х а н и з м о в р е гу л я ц и и в н е ш н е го д ы х а н и я
Расстройства внешнего дыхания возникают при нарушении его регуляции
на трех уровнях:
—центральном;
—афферентном;
—эфферентном.
Расстройства центральной нервной регуляции внешнего дыхания (на уровне
продолговатого мозга)
Наиболее частые причины центрогенных нарушений дыхания:
—травмы и новообразования в области продолговатого мозга;
—сдавление головного мозга (при его отеке или воспалении, кровоизлияни­
ях в вещество мозга или его желудочки);
— острая выраженная гипоксия различного генеза;
—интоксикации организма (например, этанолом, наркотическими сред­
ствами, эндотоксинами, образующимися при уремии или печеночной
недостаточности);
—деструктивные изменения в ткани мозга (например, при энцефалитах, рас­
сеянном склерозе, сирингомиелии, сифилисе).
Проявления центрогенных нарушений регуляции внешнего дыхания. К клини­
чески значимым формам расстройств дыхания этого типа относятся:
—апнейстическое дыхание — эпизоды временной остановки дыхания,
характеризующиеся удлиненным вдохом за счет судорожного сокра­
щения дыхательных мышц и сравнительно непродолжительным выдо­
хом. Апнейстическое дыхание наблюдается при инфаркте моста мозга,
острой выраженной гипоксии, отравлении барбитуратами;
—дыхание типа «гаспинг» (от англ. %ахр — затрудненное дыхание, удушье).
Наблюдается в агональном состоянии. Характеризуется глубокими судо­
рожными короткими вдохами, большими промежутками между ними,
отсутствием реакций на афферентные воздействия (например, болевые
или повышение содержания углекислоты в крови);
—периодические формы дыхания. Характеризуются периодами усиления
дыхательных движений с последующим их ослаблением и периодами
апноэ. К ним относят дыхание Биота, Чейна—Стокса, Куссмауля (см.
статью «Дыхание» в приложении «Справочник терминов»).
Механизмы развития периодического дыхания:
—периодически нарастающая недостаточность энергообеспечения дыхатель­
ных нейронов;
—расстройство трансмембранного распределения ионов в нейронах дыха­
тельного центра;
—нарушение физико-химического состояния мембран нейронов; в совокупности
с дисионией это приводит к нарушению формирования ими МП и ПД;
—колебание возбудимости нейронов дыхательного центра и вследствие этого
изменения частоты и глубины дыханий.
Расстройства афферентной регуляции дыхания
Характеризуются либо недостаточной, либо избыточной афферентацией от пери­
ферических рецепторов к дыхательным нейронам продолговатого мозга.
Причины недостатка афферентации, активирующей дыхательные нейроны:
— отравление наркотическими средствами или этанолом, вследствие чего огра­
ничивается проведение возбуждающих стимулов к дыхательному центру;
—слабая возбудимость хеморецепторов, воспринимающих содержание кис­
лорода и/или углекислого газа в крови (наблюдается, например, у недо­
ношенных детей или при аномалиях развития мозга);
—снижение тонической активности нейронов ретикулярной формации ствола
мозга (наследуемое или приобретенное, например, при передозировке
наркотических анальгетиков, барбитуратов, транквилизаторов и других
нейро- и психоактивных веществ).
Причины избытка афферентации, активирующей дыхательные нейроны:
—стресс-реакции (сопровождаются активацией стимулирующей импуль­
сации к дыхательному центру от рецепторов сосудов и бронхов);
—энцефалиты;
—кровоизлияние или ишемия в области продолговатого мозга;
—невротические состояния (например, истерии или фобии), чрезмерное
раздражение ноци-, хемо- и механорецепторов при травме органов
дыхания, брюшной полости или ожогах кожи и слизистых оболочек.
Проявления расстройств афферентации дыхательных нейронов:
—частое поверхностное дыхание (тахипноэ);
—гипоксия;
—гиперкапния;
—респираторный ацидоз.
Избыток тормозной афферентации от рецепторов к дыхательным нейронам
Наиболее частые причины:
—сильная боль в области грудной клетки и/или дыхательных путей (напри­
мер, при травме, ожогах, плевритах);
—чрезмерное раздражение слизистой оболочки дыхательных путей (при вды­
хании раздражающих веществ, например нашатырного спирта; при вды­
хании холодного или горячего воздуха во время острого бронхита и /
или трахеита).
Расстройства эфферентного звена регуляции дыхания
Причины их — нарушение проведения импульсации от дыхательных нейронов
продолговатого мозга к дыхательным мышцам:
—по проводящим путям к диафрагме (например, при ишемии или травме
спинного мозга, рассеянном склерозе или полиомиелите). Проявляются
утратой дыхательного автоматизма и переходом на произвольное дыхание
(оно становится неравномерным и прекращается при засыпании (син­
дром «проклятие ундины»);
— по кортико-спинальным путям к дыхательным мышцам (например,
при опухолях, травме или ишемии спинного мозга, сирингомиелии).
Характеризуются утратой произвольного (осознанного) контроля дыхания
и переходом на «автоматизированное» («машинообразное», «стабилизи­
рованное») дыхание;
— по нисходящим спинальным путям к мотонейронам спинного мозга и далее
по нервным окончаниям к дыхательной мускулатуре (например, при травме
или ишемии спинного мозга, полиомиелите, ботулизме, невритах; бло­
каде нервно-мышечной проводимости при миастении или применении
препаратов кураре). Проявляются сокращением амплитуды дыхательных
движений и периодическими эпизодами апноэ.
Альвеолярная гипервентиляция
Альвеолярная гипервентиляция — типовая форма нарушения внешнего дыха­
ния, характеризующаяся превышением реальной вентиляции легких за еди­
ницу времени в сравнении с необходимой организму в данных условиях.
Причины альвеолярной гипервентиляции:
— неадекватный режим ИВЛ (например, при проведении наркоза, пере­
воде пациента на искусственное дыхание при травме мозга или коме).
Развивающуюся при этом гипервентиляцию называют пассивной;
— стресс-реакции;
—невротические состояния (например, истерии или фобии);
— органические поврежзения мозга (например, в результате кровоизлияния,
ишемии, внутричерепных опухолях, ушибе и сотрясении мозга);
— гипертермические состояния (лихорадка, тепловой, солнечный удар и др.);
— экзогенная гипоксия.
Проявления альвеолярной гипервентиляции. Основные из них представлены
на рис. 24.4. К ним относятся:
—гипокапния (она потенцирует торможение утилизации 0 2 тканями,
снижает коронарный и мозговой кровоток за счет уменьшения тонуса
стенок артериол и развития артериальной гипотензии);
—дыхательный алкалоз (как следствие альвеолярной гипервентиляции);
— экономное потребление кислорода тканями и органами (что может при­
вести к тканевой гипоксии);
—дисбаланс ионов в плазме крови и интерстициальной жидкости (характери­
зуется гипернатриемией, гипокалиемией, гипокальциемией, гипомагниемией);
—мышечные судороги (в связи с гипокальциемией и другими проявления­
ми ионного дисбаланса);
— парестезии (как следствие ишемии мозга и ионного дисбаланса).
Рис. 24.4. Основные проявления гипервентиляции легких
РАССТРОЙСТВА КРОВООБРАЩЕНИЯ В ЛЕГКИХ
Причины нарушения перфузии легких:
—гипертензии в сосудах малого круга кровообращения (легочные гипертен­
зии);
—гипотензии в сосудах малого круга кровообращения (легочные гипотензии).
Гипертензия в сосудах малого круга кровообращения
Выделяют три формы легочной гипертензии (рис. 24.5):
—прекапиллярную;
—посткапиллярную;
—смешанную.
Рис. 24.5. Основные виды и причины гипертензии малого круга кровообращения
Прекапиллярная гипертензия
Прекапиллярная гипертензия характеризуется увеличением давления
крови в прекапиллярах и капиллярах выше нормы (более 30 мм рт.ст.
систолического и 12 мм рт.ст. диастолического).
Наиболее частые причины прекапиллярной гипертензии:
— спазм ГМК артериол (например, при стрессе, эмболии легочных сосудов,
выбросе катехоламинов из феохромоцитомы, при ацидозе, остром сни­
жении р 0 2 во вдыхаемом воздухе). Наиболее сильным фактором, вызы­
вающим вазоконстрикцию, является гипоксия; медиаторами вазоспазма
служат катехоламины, эндотелии, тромбоксан А2;
— обтурация микрососудов легких (например, микротромбами, эмболами,
гиперплазированным эндотелием);
—сдавление артериол легких (например, опухолью, увеличенными лимфа­
тическими узлами, повышенным давлением воздуха в альвеолах и брон­
хах при остром приступе кашля).
Посткапиллярная гипертензия
Посткапиллярнал гипертензия характеризуется нарушением оттока крови
из сосудов в левое предсердие и накоплением ее избытка в легких.
Наиболее частые причины посткапиллярной гипертензии:
— стеноз отверстия митрального клапана (например, как результат эндокар­
дита);
—сдавление легочных вен (например, увеличенными лимфатическими
узлами или опухолью);
— недостаточность сократительной функции миокарда левого желудочка
(например, при инфаркте миокарда, ГБ, миокардиодистрофиях).
Смешанная форма легочной гипертензии
Обычно бывает результатом прогрессирования и/или осложнений пре­
нии посткапиллярной гипертензии. Например, затруднение оттока крови
из легочных вен в левое предсердие (характерное для посткапиллярной гипер­
тензии) приводит к рефлекторному снижению просвета артериол легких
(характерное для прекапиллярной гипертензии).
Проявления смешанной легочной гипертензии:
—признаки левожелудочковой и/или правожелудочковой сердечной недоста­
точности (застой крови в венозных сосудах, отеки, асцит и др.);
—уменьшение ЖЕЛ;
— гипоксемия и гиперкапния;
— ацидоз (дыхательный, при хроническом течении — смешанный).
Гипотензия в сосудах малого круга кровообращения
Легочная гипотензия характеризуется стойким снижением давления крови
в сосудах малого круга ниже нормального.
Наиболее частые причины гипотензии в сосудах малого круга кровообращения:
—пороки сердца с шунтированием крови «справа налево». При этом про­
исходит «сброс» венозной крови в артериальную систему (например,
при тетраде Фалло, недостаточности клапанов легочной артерии);
—гиповолемии различного генеза (например, при длительной диарее, шоко­
вых состояниях, в результате хронической кровопотери);
—системная артериальная гипотензия (например, при коллапсах или комах).
НАРУШЕНИЯ ВЕНТИЛЯЦИОННО-ПЕРФУЗИОННОГО
СООТНОШЕНИЯ
В норме соотношение между величинами вентиляции и перфузии сопря­
жены как в отдельных областях, так и в легких в целом: кровоток возможен
в тех участках легкого, в которых осуществляется вентиляция. При этом соот­
ношение перфузии и вентиляции оптимально. Именно в указанных участках
легкого происходит газообмен между воздухом альвеол и кровью, протека­
ющей по межальвеолярным капиллярам. Соотношение выделения легкими С 0 2
к потреблению организмом 0 2 выражают дыхательным коэффициентом, пока­
зывающим интенсивность обмена веществ. В норме он равен в среднем 0,8.
Нарушение сопряжения вентиляции и перфузии легких приводит к раз­
витию ДН. Количественная зависимость между вентиляцией (V) и перфузией
(0) легких выражается показателем У/О, который в норме колеблется в диа­
пазоне 0,8—1,0.
Основные причины дисбаланса вентиляции и перфузии, приводящие либо
к локальной гипоперфузии легких, либо к их локальной гиповентиляции,
изображены на рис. 24.6.
Рис. 24.6. Основные причины нарушения вентиляционно-перфузионных соотношений
Причины, приводящие к локальной гиповентиляции легких (рассмотрены
выше, см. раздел «Альвеолярная гиповентиляция»). Они вызывают регионар­
ное уменьшение поступления воздуха в альвеолы. При этом объем альвео­
лярной вентиляции и объем кровообращения в каком-либо регионе легкого
становится меньше, чем в легких в целом.
Последствия локальной гиповентиляции легких:
—увеличение функционального мертвого пространства;
—снижение оксигенации крови, оттекающей от гиповентилируемого участ­
ка легкого.
Причины, приводящие к локальной гипоперфузии легких:
— обтурация ветвей легочной артерии (например, тромбом или эмболом
при ДВС, жировой эмболии, агрегатами форменных элементов крови
при сепсисе или шоке);
—сдавление ветвей легочной артерии (например, новообразованием, ино­
родным телом, рубцовой тканью);
—спазм мышц стенки какой-либо ветви легочной артерии;
—шунтирование крови в легких (минуя альвеолы). Это происходит, напри­
мер, при наличии сообщений между ветвями легочной артерии и вены.
Указанные выше факторы обусловливают:
—снижение перфузии одного из участков легкого (в результате этого форми­
руется альвеолярное мертвое пространство — вентилируемое, но не кро­
воснабжаемое);
—невостребованность альвеолярной вентиляции (нормальной или даже
повышенной) уровнем перфузии легких;
—уменьшение р 0 2 в оттекающей от легких крови (гипоксемия); р С 0 2
в крови, как правило, остается в норме (нормокапния), поскольку диф­
фузия этого газа не снижена.
НАРУШЕНИЯ ДИФФУЗИИ КИСЛОРОДА И УГЛЕКИСЛОГО
ГАЗА ЧЕРЕЗ АЭРОГЕМАТИЧЕСКИЙ БАРЬЕР
Площадь диффузионной мембраны (аэрогематического барьера) достигает
180—200 м2, а толщина 0,2—2 мкм. Массоперенос кислорода и углекислого
газа оптимален при достаточном градиенте концентрации 0 2 и С 0 2 в альвео­
лярном воздухе и крови, адекватном кровотоке в легких, сохранении вели­
чины площади, нормального структурного и физико-химического состояния
альвеолярно-капиллярной мембраны.
Диффузионную способность легких (БЬ) для кислорода и углекисло­
го газа рассчитывают как отношение объема диффузионного потока газа
(V в мл/мин) и разности парциальных давлений газа (ДР в мм рт.ст.) с разных
сторон мембраны:
01_ [мл/мин/мм рт.ст.]= V : ДР
Эта величина отражает объем газа в миллилитрах, диффундирующего
через альвеолярно-капиллярную мембрану при градиенте его давления в 1 мм
рт.ст. за 1 мин. В норме Б Ь для кислорода составляет примерно 15, а для С 0 2
около 300. Последнее свидетельствует о том, что возможность расстройства
диффузии кислорода весьма велика, а возможность углекислого газа мала.
Дополнительно см. также раздел «Оценка функции внешнего дыхания».
Причины снижения диффузионной способности
Основные причины снижения диффузионной способности альвеолярно­
капиллярной мембраны представлены на рис. 24.7. Наиболее частые из них
перечислены ниже.
• Увеличение толщины альвеолярно-капиллярной мембраны в результате:
—возрастания слоя жидкости на поверхности альвеолярного эпителия
(например, за счет слизи или экссудата при аллергическом альвеолите
или пневмонии);
— отека межмембранного интерстиция (скопление жидкости между двумя
базальными мембранами — эндотелия и эпителия);
—утолщения (увеличение высоты) эндотелия капилляров и эпителия альвеол
(например, в результате их гипертрофии и/или гиперплазии, развития
саркоидоза, многослойности).
• Увеличение плотности альвеолярно-капиллярной мембраны вследствие:
—кальцификации ее (например, компоненты интерстиция);
—возрастания вязкости геля в интерстициальном пространстве;
—увеличения количества и/или толщины коллагеновых, ретикулиновых
и эластичных волокон в межальвеолярных перегородах.
Рис. 24.7. Основные причины снижения диффузионной способности альвеолярно­
капиллярной мембраны
Примеры патологических состояний, снижающих диффузионную способность
аэрогематической мембраны
• Пневмонии (особенно хронически текущие диффузные интерстициаль­
ные).
• Пневмокониозы. Развиваются при вдыхании пыли, содержащей кремнезем
(силикоз), асбест (асбестоз), бериллий (бериллиоз).
• Фиброзирующий альвеолит (диффузный или очаговый).
• Аллергический альвеолит (например, при поллинозах).
• Сердечная недостаточность.
Дыхательная недостаточность
ЩИ — типовая форма патологии системы внешнего дыхания, при которой
эта система не обеспечивает уровня газообмена, необходимого для опти­
мального функционирования организма и нластических процессов в нем.
Характеризуется ДН развитием:
— гипоксемии;
— гиперкапнии (как правило, но не всегда).
Расширенная характеристика понятия ДН включает положение о том,
что к ней относят и такие состояния, при которых газовый состав крови
не выходит за рамки нормального диапазона, но это достигается за счет
гиперфункции аппарата внешнего дыхания. Последнее снижает адаптивные
возможности организма, чревато их срывом и развитием экстремального
состояния.
Причины дыхательной недостаточности
Основные причины ДН приведены на рис. 24.8.
• Легочные (интрапульмональные) причины ДН включают все варианты рас­
стройств (парциальные и смешанные) газообменной функции легких:
—вентиляции;
—перфузии;
—вентиляционно-перфузионных соотношений;
—диффузии газов через альвеолярно-капиллярную мембрану.
• Внелегочные (экстрапульмональные) причины ДН. Их несколько групп:
—расстройства механизмов нейрогенной регуляции внешнего дыхания
(например, при травмах, инсультах, опухолях продолговатого мозга);
—нарушения эффекторного влияния нейронов дыхательного центра через
нервно-мышечные синапсы межреберных мышц и диафрагмы (напри­
мер, при полиомиелите, миастениях, полиневритах);
—расстройство функции дыхательной мускулатуры (например, при миалгиях и миодистрофиях межреберных мышц);
—затруднение дыхательных экскурсий грудной клетки (например, при трав­
мах ребер или позвоночника, анкилозе суставов ребер);
—системная недостаточность кровообращения, в том числе в легких (напри­
мер, при сердечной недостаточности или анемиях).
Рис. 24.8. Основные причины легочной недостаточности
ФОРМЫ ДЫХАТЕЛЬНОЙ НЕДОСТАТОЧНОСТИ
Формы недостаточности системы внешнего дыхания представлены на
рис. 24.9.
Изменения газового состава крови:
-гипоксем ия
- гиперкапния;
- гипоксемия
-гипоксем ия;
- гиперкапния
Рис. 24.9. Формы дыхательной недостаточности и изменения газового состава крови
при них
Выделяют две основные разновидности ДН — гипоксемическую и гиперкапническую.
Гипоксемическая (паренхиматозная, типа I)
форма дыхательной недостаточности
Характеризуется снижением ра0 2 в крови (гипоксемия), оттекающей
от легких.
Основные причины гипоксемической ДН:
—гипоперфузия легких;
—снижение диффузии газов через альвеолярнокапиллярную мембрану
(наиболее частый фактор);
—нарушение вентиляционно-перфузионных соотношений;
— экзогенная гипоксия (гипо- и нормобарическая).
Гипоксемическая форма ДН встречается при тяжелых поражениях парен­
химы легких (например, при генерализованном инфицировании легких, аспи­
рации жидкости, бронхитах и бронхиолитах, вдыхании токсичных газов, отеке
легких, шоке). Этим определяется и одно из ее названий.
Гиперкапническая (гиповентиляционная, типа II)
форма дыхательной недостаточности
Характеризуется развитием гипоксемии и гиперкапнии.
Основные причины гиперкапнической ДН:
— альвеолярная гиповентиляция (основной фактор!);
— нарушение вентиляционно-перфузионных соотношений (в связи с недоста­
точной вентиляцией альвеол).
Гиперкапническая форма ДН наблюдается при бронхитах, бронхопневмо­
ниях, бронхиальной астме, опухолях бронхов.
Смешанная форма дыхательной недостаточности
Характеризуется одновременным развитием первичной гиперкапнии и гипок­
семии.
Основные причины смешанной формы ДН — острые и хронические заболева­
ния легких, ведущие к гиповентиляции обструктивного типа (например, бронхи­
ты, бронхиальная астма, обструктивная эмфизема легких, бронхоэктатическая
болезнь, пневмонии и абсцессы легких).
РЕСПИРАТОРНЫЙ
ДИСТРЕСС-СИНДРОМ
РДС взрослых («влажное легкое») — острая форма ДН, преимущественно
гипоксемического типа.
Название синдрома отражает определенное сходство клинических, мор­
фологических и функциональных изменений с респираторным дистресс-
синдромом новорожденных (однако, в отличие от РДС взрослых, основные
причины последнего — нарушения синтеза сурфактанта и его выделения
на поверхность альвеолоцитов, а также избыточная податливость грудной
клетки).
Причины респираторного дистресс-синдрома
взрослых
Наиболее частые причины РДС взрослых представлены на рис. 24.10.
Рис. 24.10. Основные причины респираторного дистресс-синдрома взрослых
Патогенез респираторного дистресс-синдрома
взрослых
Основные звенья патогенеза РДС взрослых представлены на рис. 24.11.
Важно, что конечным исходом всех разновидностей развития РДС является
гипоксемия.
Проявления респираторного дистресс-синдрома
взрослых
РДС развивается, как правило, через 20-40 ч после действия причинного
фактора и характеризуется прогрессирующим течением.
Наиболее характерные проявления РДС:
— одышка (тахипноэ);
—увеличение МОД;
—уменьшение легочных объемов (общей емкости легких, остаточного объе­
ма легких, ЖЕЛ, функциональной остаточной емкости легких);
—гипоксемия;
—острый дыхательный алкалоз;
—увеличение сердечного выброса (в терминальной стадии синдрома — сни­
жение).
Рис. 24.11. Основные звенья патогенеза дистресс-синдрома взрослых
Глава 25
ТИПОВЫЕ ФОРМЫ ПАТОЛОГИИ
СИСТЕМЫ ПИЩЕВАРЕНИЯ
Органы желудочно-кишечного тракта (ЖКТ) имеют тесную структурно­
функциональную связь и представляют единую физиологическую систему.
Именно поэтому первичное или преимущественное поражение какого-либо
отдела системы пищеварения, как правило, приводит к расстройствам ее
функционирования в целом.
ЭТИОЛОГИЯ ТИПОВЫХ ФОРМ ПАТОЛОГИИ
ЖЕЛУДОЧНО-КИШЕЧНОГО ТРАКТА
Причины патологии ЖКТ подразделяются на две группы:
—повреждающие органы пищеварения непосредственно;
—повреждающие ЖКТ опосредованно.
Факторы, непосредственно повреждающие органы пищеварения, весьма много­
численны. Наиболее клинически значимые из них изображены на рис. 25.1.
Продукты сгорания табака
Токсины
1
*
Лекарства
*
Химические
Добавки к пище Алкоголь
ж
*
Факторы, непосредственно повреждающие органы пищеварения
■Биологические |-
Физические
1
Чрезмерно
Грубая
Инородные Радиация
холодная или
пища
тела
горячая пища
Дефицит
Токсины
или избыток микробов
витаминов
Микробы
Гельминты
Рис. 25.1. Основные причины нарушения пищеварения в желудке и кишечнике —
факторы, непосредственно повреждающие органы ЖКТ
По происхождению они могут быть:
—химическими;
—биологическими;
—физическими.
Факторы, опосредованно повреждающие органы пищеварения, делятся на две
группы.
• Опосредующие свое патогенное действие на систему пищеварения и вызы­
вающие патологию других органов (рис. 25.2). К ним относят факторы,
повреждающие, например, почки, эндокринные железы, печень, органы
системы кровообращения. Примеры:
—при повреждении почек ряд патогенных продуктов метаболизма (аммиак,
производные аммония, индолы, скатолы и др.) выводится из организма
кишечником, что сопровождается повреждением его слизистой оболочки;
—при кортикостеромах высокая концентрация в крови минерало- и глюкокортикоидов приводит к образованию «стероидных» язв на поверхности
слизистой оболочки желудка и/или кишечника;
—при поражении печени нарушается выработка желчи, необходимой
для пищеварения.
Рис. 25.2. Основные причины нарушения пищеварения в желудке и кишечнике —
факторы, опосредованно повреждающие органы ЖКТ
• Опосредующие патогенное воздействие на ЖКТ через расстройство меха­
низмов, регулирующих органы пищеварения. Такими факторами могут быть
дефицит или избыток эффектов регуляторных БАВ либо нервных воздей­
ствий на ЖКТ (см. рис. 25.2). Примеры:
—чрезмерное длительное повышение активности нейронов блуждающего
нерва приводит к избыточной активации секреторной и моторной функ­
ции ЖКТ, что, в свою очередь, может привести к эрозиям слизистой
оболочки желудка и/или кишечника;
—дефицит простациклина в слизистой оболочке желудка может вызвать
ишемию участка его стенки, уменьшить выработку слизи и защитных
бикарбонатов, что приводит к развитию язвы.
Известно, что органы пищеварения, помимо общей соматической и вегета­
тивной иннервации, имеют собственную нервную систему (энтеральная нервная
система) и собственный эндокринный аппарат (энтероэндокринная система).
Энтеральная нервная система — совокупность нервных клеток (интраму­
ральных нейронов) ЖКТ, а также вегетативных нейронов, расположенных
за пределами ЖКТ (экстрамуральные нейроны). Основная функция энтераль­
ной нервной системы — регуляция двигательной и секреторной активности
системы пищеварения.
Энтероэвдокринная система — пул эндокринных (энтероэндокринных) кле­
ток слизистой оболочки и желез ЖКТ. К этой же системе относят некоторые
нейроны энтеральной нервной системы, секретирующие гормоны (в ряде слу­
чаев те же, что и энтероэндокринные клетки). Именно поэтому эндокринную
систему ЖКТ называют еще нейроэндокринной системой.
Энтероэндокринные клетки находятся в слизистой оболочке кишечного
типа среди эпителиальных клеток крипт в кишечнике, железах желудка и две­
надцатиперстной кишки (см. статью «Клетки энтероэндокринные» в при­
ложении «Справочник терминов»). Клетки слюнных и бруннеровых желез
двенадцатиперстной кишки секретируют эпидермальный фактор роста (ЕСР).
При поступлении пищи в просвет ЖКТ различные эндокринные клетки
под действием растяжения стенки, под влиянием самой пищи или изменения
рН в просвете пищеварительного канала начинают выделять гормоны в ткани
и кровь. Деятельность энтероэндокринных клеток находится под контролем
вегетативной нервной системы.
Интрамуральные нейроны пищеварительного тракта выделяют нейропеп­
тид У, относящийся к кальцитониновому гену пептид, вещество Р, гастрин,
гастрин-рилизинг-гормон, нейротензин, метионин-энкефалин, секретин
и другие пептиды.
Функции БАВ в пищеварительном тракте
Адреналин и норадреналин подавляют перистальтику кишечника и моторику
желудка; сужают просвет кровеносных сосудов в стенке пищеварительного
канала.
Ацетилхолин стимулирует все виды секреции в желудке, двенадцатиперст­
ной кишке, поджелудочной железе, а также моторику желудка и перисталь­
тику кишечника.
Брадикинин стимулирует моторику желудка; является вазодилататором.
У1Р стимулирует моторику и секрецию в желудке, перистальтику и секре­
цию в кишечнике. Мощный вазодилататор. Выделяется в ответ на стимуляцию
блуждающего нерва.
Вещество Р вызывает незначительную деполяризацию нейронов в ганглиях
межмышечного сплетения, сокращение ГМК.
Гастрин стимулирует секрецию слизи, бикарбоната, ферментов, соляной
кислоты в желудке, пролиферацию клеток в слизистой оболочке; подавляет
эвакуацию из желудка; активирует перистальтику кишечника и секрецию
инсулина.
Гистамин активирует секрецию в железах желудка и перистальтику.
Глюкагон стимулирует секрецию слизи и бикарбоната, подавляет перисталь­
тику кишечника.
Мотилин стимулирует моторику желудка.
Нейропептид V подавляет моторику желудка и перистальтику кишечника,
усиливает вазоконстрикторный эффект норадреналина во многих сосудах,
включая чревные.
Панкреатический полипептид угнетает секрецию сока в поджелудочной
железе.
ПГЕ стимулирует секрецию слизи и бикарбоната в желудке.
Секретин подавляет перистальтику кишечника, активирует эвакуацию
из желудка, стимулирует секрецию сока в поджелудочной железе.
Серотонин стимулирует перистальтику.
Соматостатин подавляет все процессы в пищеварительном тракте.
Холецистокинин стимулирует перистальтику кишечника, но подавляет мото­
рику желудка; стимулирует поступление желчи в кишечник и секрецию в под­
желудочной железе, усиливает высвобождение инсулина. Холецистокинин
имеет значение для процесса медленной эвакуации содержимого желудка,
расслабления сфинктера Одди.
Е 6Р стимулирует регенерацию клеток эпителия в слизистой оболочке
желудка и кишечника.
Регуляция отдельных процессов в желудочно-кишечном тракте
Образование слизи и бикарбоната в желудке. Стимулируют этот процесс
гастрин, гастрин-рилизинг-гормон, глюкагон, ПГЕ, ЕОР. Подавляет сомато­
статин.
Секреция пепсина и соляной кислоты в желудке. Стимулируют ее ацетилхолин, гистамин, гастрин. Подавляют соматостатин и желудочный ингибиру­
ющий пептид.
Моторика желудка. Стимулируют ацетилхолин, мотилин, У1Р. Подавляют
соматостатин, холецистокинин, адреналин, норадреналин, желудочный инги­
бирующий пептид.
Перистальтика кишечника. Стимулируют ацетилхолин, гистамин, гастрин
(подавляет эвакуацию из желудка), холецистокинин, серотонин, брадикинин,
VIР. Подавляют соматостатин, секретин, адреналин, норадреналин.
Секреция сока поджелудочной железы. Стимулируют ацетилхолин, холеци­
стокинин, секретин. Подавляют панкреатический полипептид, соматостатин.
Желчеотделение. Стимулируют гастрин, холецистокинин.
Факторы риска расстройств пищеварения
К условиям, способствующим воздействию причинных агентов (факторы
риска), относится множество факторов. Наиболее клинически значимые
из них:
—нарушение реактивности организма (расстройства системы пищеварения
часто наблюдаются при гиперреактивных состояниях, например, на фоне
сенсибилизации или длительного эмоционального перенапряжения);
—пол (например, язвенная болезнь желудка и двенадцатиперстной кишки
выявляется преимущественно у мужчин);
—возраст (расстройства пищеварения значительно чаще наблюдаются
в зрелом и пожилом возрасте);
—наследственная предрасположенность (например, часто встречается
семейная предрасположенность к пептическим язвам желудка).
Типовые формы патологии
желудочно-кишечного тракта
Основные расстройства и наиболее клинически значимые типовые формы
патологии ЖКТ приведены на рис. 25.3.
Рис. 25.3. Типовые формы патологии системы пищеварения
РАССТРОЙСТВА ВКУСА
Расстройства вкуса (ощущение горького, сладкого, кислого, соленого
и др.) подразделяются на агевзии, гипогевзии, гипергевзии, парагевзии и дисгевзии (от греч. %еиш — вкус).
Агевзии и гипогевзии
Отсутствие или снижение вкусовых ощущений называется а- или гиногевзияей соответственно.
Причины агевзии и гипогевзии — функциональные расстройства и пораже­
ние структур вкусового анализатора:
— рецепторных (например, при химических ожогах или глосситах);
—нервных стволов, проводящих импульсы от рецепторов к нервным цен­
трам (например, при повреждении, разрыве, невритах, нейродистрофи­
ях язычного и/или языкоглоточного нерва, а также альтерации области
таламуса);
—нейронов коркового анализатора вкусовых ощущений (например, при энце­
фалитах, кровоизлияниях, неврозах, нервно-психических болезнях).
Гипергевзия
Гипергевзия — патологическое усиление вкусовых ощущений. Основные при­
чины:
— гиперсенситизация вкусовых рецепторов (например, при трансмембран­
ном дисбалансе ионов или расстройстве КОС интерстициальной жид­
кости в ткани языка);
—поражение корковых нейронов, участвующих в формировании вкусовых
ощущений (например, при неврозах или психических расстройствах).
Нарушение адекватности вкусовых ощущений
Среди них выделяют парагевзии и дисгевзии.
• Парагевзия — качественное отличие вкусового ощущения от тех, которые
данное вещество вызывает в норме (т.е. ложное ощущение): например, кис­
лое воспринимается как горькое, сладкое — как соленое.
• Дисгевзия — патологическое изменение (извращение) вкусовых ощущений
и склонностей (например, употребление испорченных пищевых продуктов
или опасных для здоровья веществ: травы, бумаги, песка, экскрементов).
Основные причины парагевзий и дисгевзий:
—патология центральных нервных структур, участвующих в формировании
вкусовых ощущений (например, при энцефалитах, шизофрении, невро­
зах).
Возможные последствия расстройств вкуса:
—нарушения аппетита;
—расстройства функций желудка и кишечника (и в целом пищеварения).
НАРУШЕНИЯ АППЕТИТА
К нарушениям аппетита (субъективное ощущение, связанное с потребно­
стью организма в качестве и количестве пищи) относят:
— анорексию;
— гипорексию;
— гиперрексию;
— парарексию.
Все термины происходят от греч. огех1а — аппетит.
Анорексия и гипорексия
Анорексия и гипорексия представляют собой отсутствие или значительнее
снижение аппетита соответственно.
Наиболее частые причины анорексии и гипорексии:
— острые заболевания (например, стоматит, гингивит, пневмонии, энтеро­
колиты, инфаркт миокарда);
—тяжелые затяжные болезни (например, туберкулез, анемии, опухолевый
рост);
—хронические интоксикации экзо- и эндогенными веществами (при почеч­
ной и печеночной недостаточности, алкоголизме, остеомиелите, отрав­
лении препаратами ртути, никотином и др.);
— нейро- и/или психогенные расстройства (например, неврозы, невроген­
ная анорексия, шизофрения).
Патогенез гипо- и анорексий включает нарушения метаболизма и эффек­
тов орексинов, кахектина (ФНО-а), холецистокинина, нейропептида V и ряда
других нейропептидов.
Последствия гипо- и анорексий:
— снижение массы тела, вплоть до истощения (кахексия);
— расстройства пищеварения;
—дистрофии;
—иммунодефицитные состояния (в тяжелых случаях).
Гиперрексия и булимия
Гиперрексия — состояние, характеризующееся патологическим повыше­
нием аппетита.
Булимия (от греч. виз — бык, Птоз — голод) — состояние, проявляющее­
ся повторными эпизодами поедания чрезмерного количества пищи (полифагия)
с одновременным снижением или отсутствием чувства насыщения (акория).
Нередко сопровождается депрессией (подробнее см. статью «Булимия» в при­
ложении «Справочник терминов»),
Парарексия
Парарексия — патологически измененный аппетит, проявляющийся употребле­
нием в пищу несъедобных веществ (например, мела, смолы, угля, золы и др.).
Причина парарексии — нарушение центральных (нейрогенных) механизмов
формирования аппетита (например, после травмы мозга, при росте внутриче­
репной опухоли, кровоизлиянии в мозг, развитии психических болезней).
РАССТРОЙСТВА ПИЩЕВАРЕНИЯ В ПОЛОСТИ РТА
Расстройства пищеварения в полости рта обусловлены нарушениями,
связанными с образованием и выделением слюны (саливация) и пережевы­
ванием пищи.
Нарушения саливации
Характеризуются они гипо- или гиперсаливацией.
Гипосаливация (гипосиалия) — уменьшение, вплоть до прекращения, выработ­
ки слюны слюнными железами и выделения ее в полость рта.
Наиболее частые причины гипосаливации:
—поражение слюнных желез (например, при их воспалении, разрушении
опухолевой тканью, хирургическом удалении, атрофии, воздействии
токсинов или проникающей радиации);
—сдавление протоков слюнных желез извне и/или закрытие их изнутри
(опухолью окружающих тканей, отечной жидкостью, рубцовой тканью;
камнем, густым секретом);
—значительная и длительная гипогидратация организма (приводит к умень­
шению жидкой части слюны);
—нарушение нейрогуморальной регуляции образования слюны (например,
при поражении нейронов гипоталамуса, коры, а также нервных стволов,
иннервирующих железы; при гипертиреоидных состояниях).
Последствия гипосаливации:
—недостаточное смачивание и набухание пищевого комка;
—затруднения пережевывания и глотания пищи в результате ее недостаточ­
ного увлажнения и сухости слизистой оболочки рта (ксеростомия);
—частое развитие стоматитов, гингивитов, глосситов, кариеса зубов. Это
обусловлено дефицитом лизоцима и других бактерицидных веществ
в малом количестве слюны и повреждением сухой слизистой оболочки
плохо смоченными кусочками пищи;
—недостаточная обработка углеводов пищи в связи с дефицитом амилазы
в слюне. Однако это компенсируется амилазами кишечника и к суще­
ственным нарушениям переваривания пищи в целом не приводит.
Гиперсаливация (гиперсиалия) — повышенное образование и выделение слюны
в ротовую полость.
Наиболее частые причины:
—активация нейрогенных парасимпатических влияний на слюнные желе­
зы (например, при повышении возбудимости нейронов блуждающего
нерва под влиянием ЛС, токсинов, при неврозах, энцефалитах);
—острые стоматиты и гингивиты;
—интоксикация организма соединениями ртути, никотином;
—эндотоксинемии (при уремии, комах, токсикозе беременных);
—глистные инвазии.
Последствия гиперсаливации:
—разведение и ощелачивание желудочного содержимого избытком слюны.
Это снижает пептическую активность желудочного сока, бактериостатическую и бактерицидную его способность;
—ускорение эвакуации желудочного содержимого в двенадцатиперстную кишку;
—гипогидратация организма при сплевывании избытка слюны или стекании ее изо рта у тяжелобольных пациентов.
Нарушения пережевывания пищи
Основные причины нарушений жевания:
—заболевания полости рта (стоматиты, гингивиты, глосситы, пародонти­
ты, пародонтоз и др.), сопровождающиеся болевыми ощущениями;
—недостаток или отсутствие зубов;
—патология суставно-мышечного аппарата нижней челюсти (например,
переломы костей, атрофия мышц, их гипертонус);
—привычное недостаточное пережевывание пищи (например, при еде
«на ходу», во время чтения и др.).
Возможные последствия нарушенного жевания пищи:
—механическое повреждение слизистой оболочки желудка плохо пережеван­
ной пищей;
—нарушение желудочной секреции и моторики.
Расстройства глотания
К расстройствам глотания и движения пищи по пищеводу относятся:
—дисфагии (от греч. йу$ — расстройство, рка%е'т — поедать);
— афагия;
—дисфункции пищевода.
Дисфагии и афагии
Дисфагии — состояния, характеризующиеся затрудненным проглаты­
ванием твердой пищи и воды, а также попаданием пищи или жидкости
в носоглотку, гортань и верхние дыхательные пути.
Афагия — состояние, характеризующееся невозможностью проглотить
твердую пищу и жидкость.
Наиболее частые причины:
—сильная боль в полости рта (в результате воспалительных процессов,
изъязвлений слизистой оболочки, повреждений или переломов костей
черепа и др.);
— патология суставов нижней челюсти и/или жевательных мышц (например,
артрозы, артриты, а также спазмы, гипертонус или гипотонус жеватель­
ных мышц, их парезы и параличи);
—поражение нейронов центра глотания и его проводящих путей (наиболее
часто при нарушении мозгового кровообращения);
—нарушение афферентной и эфферентной иннервации жевательных мышц
(например, при повреждении и/или воспалении ветвей блуждающего,
тройничного, языкоглоточного нерва);
—патологические процессы в глотке и пищеводе (например, рубцы, новооб­
разования, язвы);
—психические расстройства (например, афагия при истерическом эпизоде
или сильном стрессе).
Последствия дисфагии и афагии:
—нарушения поступления пищи в желудок и в связи с этим — расстройства
пищеварения и питания;
— аспирация пищи (с угрозой развития бронхоспазма, бронхита, аспираци-
онной пневмонии, абсцесса легкого);
— асфиксия (при попадании большого количества пищи в дыхательные
пути, например, у алкоголиков или при выходе из наркоза).
ДИСФУНКЦИИ ПИЩЕВОДА
Дисфункции пищевода характеризуются затруднением движения пищи
по пищеводу, ее прохождения в желудок и забросом содержимого желудка
в пищевод (рефлюкс). Наиболее часто дисфункции пищевода развиваются
на уровне верхнего и нижнего его сфинктеров.
Дисфункция пищевода на уровне
его верхнего сфинктера и тела
Причины дисфункции пищевода в его верхнем отделе:
—нейрогенные расстройства регуляции моторики пищевода (напри­
мер, при энцефалитах, дистрофических и деструктивных измене­
ниях нейронов блуждающего нерва, интрамуральных пищеводных
сплетений, в условиях патологического стресса). При этом вагусные
влияния усиливают перистальтику пищевода, а интрамуральные спле­
тения могут или активировать (через холинергические мускариновые
ш2-рецепторы), или затормозить (через никотиновые и мускариновые
Ш[-рецепторы) сокращение продольных и циркулярных слоев в мыш­
цах пищевода;
— гуморальные нарушения регуляции тонуса и перистальтики пищевода
(в основном в результате избыточных эффектов У1Р и оксида азота);
—склеротические изменения в стенке пищевода (например, после химиче­
ских или термических ожогов, при дерматомиозите или генерализован­
ной склеродермии, после заживления язв и обширных эрозий);
—спазм стенки пищевода (например, локальный или диффузный эзофагоспазм при невротических состояниях или проглатывании большого
куска твердой пищи).
Последствия дисфункции пищевода на уровне его верхнего сфинктера и тела:
— ахалазия (состояние, проявляющееся длительным спазмом ГМК стенки
тела пищевода, его нижнего сфинктера, утратой перистальтики и недо­
статочным расслаблением сфинктера);
—диффузный спазм пищевода (характеризуется сокращением ГМК всех
отделов стенки пищевода при сохранении — в отличие от ахалазии —
нормального тонуса нижнего пищеводного сфинктера).
Дисфункция пищевода на уровне его нижней части
и нижнего сфинктера
Причины дисфункции пищевода в его нижней части:
—нарушение холинергической иннервации стенки пищевода (например,
при энцефалитах или невритах с поражением тел нейронов и нервных
стволов блуждающего нерва и интрамуральных сплетений);
—снижение или усиление эффектов БАБ, регулирующих тонус мышц пищевода
(мотилин, гастрин, вещество Р и другие, повышающие тонус, или серо­
тонин, секретин, У1Р, соматостатин, дофамин, оксид азота, снижающие
тонус).
Последствия дисфункции пищевода в его нижнем отделе:
— ахалазия кардиального отдела пищевода — состояние, характеризующее­
ся нарушением расслабления нижнего сфинктера пищевода во время
глотания; заключается в замедлении движения пищи по пищеводу после
ее проглатывания и в задержке ее эвакуации в желудок; проявляется
ощущением тяжести и болью в грудной клетке, снижением массы тела;
— гастроэзофагеальный рефлюкс — заброс содержимого желудка в пище­
вод. Частое повторение и длительное сохранение рефлюкса обозначают
термином «гастроэзофагеальный рефлюксный синдром или болезнь».
Для этого состояния характерны:
— отрыжка — неконтролируемое выделение газов и/или пищи (малого
количества) из желудка в пищевод и ротовую полость;
—срыгивание (регургитация) — непроизвольный заброс части желудочного
содержимого в полость рта и носовые ходы; наблюдается у новорожден­
ных и при ахалазии у взрослых;
—изжога — неприятное субъективное ощущение жжения в эпигастральной
области — результат заброса кислого содержимого желудка в пищевод;
—частая аспирация пищи.
НАРУШЕНИЯ ПИЩЕВАРЕНИЯ В Ж ЕЛУДКЕ
В основе нарушений пищеварения в желудке находятся парциальные,
но чаще сочетанные расстройства секреторной, моторной, всасывательной,
барьерной и защитной функций желудка.
Расстройства секреторной функции желудка
Характеристика расстройств секреторной функции желудка дана на рис. 25.4.
Рис. 25.4. Типовые расстройства секреторной функции желудка
В целом ввиду указанных на рис. 25.4 нарушений динамика и/или уровень
секреции различных компонентов желудочного сока не соответствует теку­
щим реальным потребностям в них.
Нарушения динамики и общего объема секреции
желудочного сока
В зависимости от особенностей изменения секреторной функции желудка
выделяют несколько ее типов: тормозной, возбудимый, инертный, астенический.
• Тормозной тип желудочной секреции. Для него характерны:
—увеличенный латентный период секреции (между пищевой стимуляцией
желудка и началом секреции);
—ослабленная интенсивность нарастания и активности секреции;
—укороченная длительность секреции;
—уменьшенный объем секрета. При крайней степени торможения секре­
ции развивается ахилия — практически полное отсутствие желудочного
сока в желудке.
• Возбудимый тип секреции. Он проявляется:
—укороченным латентным периодом начала секреции;
—интенсивным нарастанием секреции;
—увеличенной длительностью процесса секреции;
—повышенным объемом желудочного сока.
• Инертный тип секреции желудка. Характеризуется:
—увеличенным латентным периодом;
—замедленным нарастанием секреции;
—медленным ее прекращением;
—увеличенным объемом желудочного сока.
• Астенический тип желудочной секреции. Проявляется:
—укороченным латентным периодом начала сокоотделения;
—интенсивным началом и быстрым уменьшением секреции;
—малым объемом желудочного сока.
• Хаотический тип секреции желудочного сока. Он характеризуется:
—отсутствием каких-либо закономерностей в динамике и объемах секре­
ции, периодах ее активации и торможения в течение продолжительного
времени (месяцев и лет);
—общее количество сока, как правило, увеличено.
Типовые формы расстройств желудочной секреции
К типовым расстройствам желудочной секреции относятся гиперсекреция,
гипосекреция и ахилия.
Гиперсекреция — увеличение количества желудочного сока, повышение его
кислотности и переваривающей способности.
Основные причины желудочной гиперсекреции:
—увеличение массы секреторных клеток желудка (детерминируется гене­
тически);
—активация влияний блуждающего нерва (например, при невротических
состояниях или конституциональной ваготонии);
—повышение синтеза и/или эффектов гастрина;
—гипертрофия и/или гиперплазия энтерохромаффинных (энтероэндо­
кринных) клеток (например, при гипертрофическом гастрите);
—перерастяжение антрального отдела желудка;
—действие некоторых ЛС (например, ацетилсалициловой кислоты
или глюкокортикоидов).
Последствия гиперсекреции желудочного сока:
—замедление эвакуации пищевой массы из желудка;
—эрозии и изъязвления слизистой оболочки желудка; сопровождающийся
изжогой гастроэзофагеальный рефлюкс;
—нарушения пищеварения в кишечнике.
Птосекреция — уменьшение объема желудочного сока, снижение его кислот­
ности и расщепляющей эффективности.
Основные причины желудочной гипосекреции:
—уменьшение массы секреторных клеток (например, при гипо- и атрофиче­
ской форме хронического гастрита или распадающейся опухоли желудка);
—снижение эффектов блуждающего нерва (например, при неврозах или кон­
ституциональной симпатикотонии);
—менее продуктивное образование гастрина;
—дефицит в организме белков и витаминов;
—действие ЛС, снижающих или устраняющих эффекты блуждающего
нерва (например, блокаторов холинорецепторов или активаторов холинэстераз).
Ахилия — состояние, характеризующееся практически полным отсутствием
желудочной секреции.
Причина ахилии — значительное снижение или прекращение секреторной
функции желудка.
Нарушения моторики желудка
Нарушения моторной функции желудка и последствия расстройств желу­
дочной моторики представлены на рис. 25.5.
Рис. 25.5. Типовые расстройства моторной функции желудка и ее последствия
К расстройствам моторики желудка относят:
—нарушения тонуса ГМК мышечной оболочки желудка (включая мышеч­
ные сфинктеры);
— расстройства перистальтики желудка;
— нарушения эвакуации содержимого желудка.
Нарушения тонуса мышечной оболочки желудка характеризуются:
—избыточным повышением (гипертонус) стенки желудка, либо
—чрезмерным его снижением (гипотонус), либо
—отсутствием мышечного тонуса (атония).
Изменения мышечного тонуса приводят к нарушениям перистолы, свя­
занным с охватыванием пищевых масс стенкой желудка и формированием
порции пищи для внутрижелудочного переваривания, а также с эвакуацией
ее в двенадцатиперстную кишку.
Расстройства тонической активности мышечных сфинктеров желудка про­
являются:
—снижением тонуса сфинктеров (вплоть до их атонии), что обусловливает
длительное открытие («зияние») кардиального и/или пилорического
сфинктера), либо
—повышением тонуса сфинктеров и спазмом их мышц (это приводит
к кардиоспазму и/или пилороспазму).
Нарушения перистальтики желудка характеризуются либо ее ускорением
(гиперкинез), либо замедлением (гипокинез).
Расстройства эвакуации содержимого желудка
Сочетанные и/или раздельные расстройства тонуса и перистальтики стен­
ки желудка вызывают либо ускорение, либо замедление эвакуации пищи
из желудка.
Причины нарушения эвакуации желудочного содержимого:
—нарушения нервной регуляции двигательной функции желудка — с уси­
лением влияний блуждающего нерва усиливается его моторная функ­
ция, а активация эффектов симпатической нервной системы пода­
вляет ее;
—расстройства гуморальной регуляции желудка; например, высокая кон­
центрация в полости желудка соляной кислоты, а также секретин,
холецистокинин тормозят моторику желудка. Напротив, гастрин,
мотилин, сниженное содержание соляной кислоты в желудке стимули­
руют моторику;
—патологические процессы в желудке (эрозии, язвы, рубцы, опухоли могут
ослаблять либо усиливать его моторику в зависимости от их локализа­
ции или выраженности процесса).
Последствия расстройств эвакуации содержимого желудка
В результате нарушений моторики желудка возможно развитие ряда
патологических синдромов: раннего насыщения, изжоги, тошноты, рвоты,
демпинг-синдрома.
Синдром раннего (быстрого) насыщения
Синдром быстрого насыщения — результат снижения тонуса и моторики
антрального отдела желудка. Прием небольшого количества пищи вызывает
чувство тяжести и переполнения желудка. Это создает субъективное ощуще­
ние насыщения.
Изжога
Изжога характеризуется ощущением жжения в области нижней части пище­
вода (результат снижения тонуса кардиального сфинктера желудка, нижнего
сфинктера пищевода и заброса в него кислого желудочного содержимого).
Тошнота
Тошнота представляет собой неприятное, неболезненное субъективное ощу­
щение, предшествующее рвоте. Тошнота развивается при подпороговом воз­
буждении рвотного центра.
Рвота
Рвота — непроизвольный рефлекторный акт, характеризующийся выбро­
сом содержимого желудка (иногда и кишечника) наружу через пищевод, глотку
и полость рта.
Механизмы развития рвоты:
—возбуждение рвотного центра продолговатого мозга;
—усиленная антиперистальтика стенки желудка;
—сокращение мышц диафрагмы и брюшной стенки;
—одновременное расслабление мышц кардиального отдела желудка
и пищевода.
Значение рвоты
• Защитное: при рвоте из желудка извергаются токсичные вещества или ино­
родные тела.
• Патогенное: потеря организмом жидкости, ионов, продуктов питания, осо­
бенно при длительной и/или повторной рвоте.
Демпинг-синдром
Демпинг-синдром — патологическое состояние, развивающееся в результате
быстрой эвакуации желудочного содержимого в тонкую кишку. Развивается,
как правило, после удаления части желудка.
Патогенез демпинг-синдрома. Основные звенья патогенеза демпинг-син­
дрома показаны на рис. 25.6. К ним относят последовательную цепь патоген­
ных изменений в организме:
—гиперосмоляльность содержимого тонкой кишки в результате попадания
в нее концентрированной пищи из желудка;
—интенсивный транспорт жидкости из сосудов в полость кишечника (по гради­
енту осмотического давления), что может привести к учащению стула;
—развитие гиповолемии;
— активация синтеза и выделение в межклеточное пространство БАВ, вызы­
вающих системную вазодилатацию (вследствие эффектов серотонина,
кининов, гистамина и др.) и артериальную гипотензию, включая коллапс;
— быстрое всасывание в кишечнике глюкозы с развитием гипергликемии;
—стимуляция образования и инкреции избытка инсулина. Гиперинсулинемия
активирует массированный транспорт глюкозы в клетки, где она вклю­
чается в метаболические процессы и депонируется в виде гликогена.
К этому времени (обычно через 1,5—2 ч после приема пищи и быстрой
эвакуации ее из желудка в кишечник) пища уже утилизирована и источ­
ник глюкозы недостаточен. В связи с этим развиваются нарастающая
гипогликемия, дисбаланс ионов, ацидоз.
Рис. 25.6. Основные звенья патогенеза демпинг-синдрома
Основные проявления демпинг синдрома:
—прогрессирующая слабость после приема пищи;
—тахикардия, аритмии сердца;
—острая артериальная гипотензия;
—сонливость;
—головокружение, тошнота;
—мышечная дрожь (особенно конечностей);
—нарушения сознания.
Расстройства всасывания в желудке
В норме в желудке всасываются вода, алкоголь, электролиты. При случай­
ном или осознанном приеме могут всасываться токсичные агенты.
При деструктивных изменениях стенки желудка (в том числе при нару­
шениях барьерной функции) во внутреннюю среду организма может попасть
белок, что чревато развитием иммунопатологических процессов: аллергиче­
ских реакций и состояний иммунной аутоагрессии.
Нарушение барьерной и защитной функции
слизистой оболочки желудка
Слизисто-бикарбонатный барьер защищает слизистую оболочку желудка
от действия кислоты, пепсина и других потенциальных повреждающих агентов.
Компоненты защитного барьера:
—защитная слизь, которая постоянно секретируется на поверхность эпите­
лия;
— бикарбонаты (ионы Н С 03_); они секретируются поверхностными слизи­
стыми клетками и обладают нейтрализующим действием по отношению
к НС1;
— рН — слой слизи имеет градиент рН: на поверхности слизистого слоя
слизи рН равен 2, а в примембранной части — более 7, что обеспечивает,
соответственно, бактерицидный эффект и снижает степень агрессивной
кислотности;
—плотные межклеточные контакты; они формируются между поверхност­
ными клетками эпителия. При нарушении их целостности нарушается
функция барьера.
Регуляция состояния защитного барьера слизистой оболочки желудка. Секре­
цию защитных бикарбонатов и слизи усиливают глюкагон, ПГЕ, гастрин,
ЕОР. Чтобы предупредить повреждение и восстановить барьер, применяют
антисекреторные агенты (например, блокаторы гистаминовых рецепторов),
ПГ, гастрин, аналоги сахаров (например, сукральфат).
Разрушение защитного барьера слизистой оболочки желудка
Существуют факторы, нарушающие защитную функцию слизистой обо­
лочки желудка, например НПВС (ацетилсалициловая кислота, индометацин),
этанол, соли желчных кислот.
При неблагоприятных условиях барьер разрушается в течение нескольких
минут — происходит гибель клеток эпителия, возникает отек и кровоизлия­
ния в слизистом слое желудка. Этому способствуют:
—НеИсоЬааег ру1оп — грамотрицательная бактерия, выживающая в кис­
лой среде желудка. Н. ру1оп поражает поверхностный эпителий желудка
и разрушает барьер, способствуя развитию гастрита и язвенного дефекта
в стенке желудка. Этот микроорганизм выделяют у 70% больных язвен­
ной болезнью желудка и у 90% больных язвой двенадцатиперстной
кишки или антральным гастритом;
—снижение кислотности в желудке, что создает условия, способствующие
жизнедеятельности и размножению многих микроорганизмов, напри­
мер холерного вибриона, шигелл, амеб. Так, пациенты с желудочной
ахилией чаще заболевают инфекционными болезнями, передающимися
орально-фекальным путем, подвергаются интоксикациям, имеют более
высокий риск развития новообразований желудка.
РАССТРОЙСТВА ПИЩЕВАРЕНИЯ
В КИШЕЧНИКЕ
Расстройства пищеварения в кишечнике обусловлены нарушением основ­
ных его функций: переваривающей, всасывательной, моторной и барьерно­
защитной.
Нарушения переваривающей функции кишечника
К основным причинам расстройств переваривающей функции кишечника
относят нарушения экзокринной функции поджелудочной железы, выделения
желчи в тонкую кишку, секреции в просвет тонкой кишки слизи и бикар­
боната собственными (бруннеровыми) железами стенки двенадцатиперст­
ной кишки и слизи многочисленными бокаловидными клетками ворсинок
и крипт кишечника.
Нарушения экзокринной функции поджелудочной железы
Нарушения экзокринной функции поджелудочной железы приводят к пан­
креатической гипо- и/или ахилии.
Причины ахилии:
—уменьшение массы поджелудочной железы (например, при некрозе, резек­
ции ее части, поражении опухолью, склерозе);
—нарушение оттока секрета железы по ее протокам в двенадцатиперст­
ную кишку в результате обтурации протоков (камнем, опухолью и др.)
или сдавления протоков (например, новообразованием или рубцом);
—дискинезия протоков железы (вследствие снижения тонуса или, напро­
тив, спазма ГМК протоков);
— нарушение деятельности железы в результате нервных и гуморальных регу­
ляторных расстройств.
Расстройства выделения желчи в тонкую кишку
Рассмотрены в гл. 26 «Типовые формы патологии печени».
Уменьшение секреции бокаловидными клетками и бруннеровыми
железами
Основные причины гипосекреции:
— атрофия или гипотрофия слизистой оболочки кишечника (например,
при хроническом энтерите);
— резекция части тонкой кишки;
—язвенно-эрозивные и некротические изменения в слизистой оболочке кишеч­
ника (например, при острых энтеритах, интоксикации, ишемии стенки
кишки).
Нарушения переваривающей функции кишечника приводят к расстройствам
полостного и пристеночного (мембранного) пищеварения (в том числе к разви­
тию синдрома мальабсорбции).
Эти же расстройства сказываются и на состоянии организма в целом в силу
дефицита субстратного и энергетического обеспечения организма и развития
кишечной аутоинфекции и интоксикации.
Расстройства всасывательной функции кишечника
Основные причины расстройств всасывательной функции кишечника:
—недостаточное полостное и мембранное пищеварение;
—ускорение эвакуации кишечного содержимого (например, при поносе);
—атрофия ворсинок слизистой оболочки кишечника;
—избыточное содержание экссудата на поверхности слизистой оболочки
(например, при острых кишечных инфекциях, хронических энтеритах);
—резекция большого фрагмента тонкой кишки (например, при его опухоле­
вом поражении и/или некрозе);
—расстройства крово- и лимфообращения в стенке кишечника.
Расстройства кишечного всасывания являются значимым компонентом
патогенеза синдрома мальабсорбции.
Нарушение моторной функции кишечника
Типовые формы нарушения моторики кишечника разнообразны. Основные
из них показаны на рис. 25.7.
Рис. 25.7. Типовые формы нарушения моторной функции кишечника и их разновидности
Диарея
Понос (диарея, от греч. сИаггНео — истекаю) — учащенный (более 2—3 раз
в сутки) стул жидкой или кашицеобразной консистенции, сочетающийся
с усилением моторики кишечника.
Основные виды диареи и механизмы их возникновения:
— экссудативная — результат избыточного образования воспалительного
экссудата в слизистой оболочке кишечника (например, при инфекцион­
ных и неинфекционных энтеритах и колитах);
— секреторная — следствие чрезмерной секреции жидкости в просвет
кишечника (например, при холере, вирусных энтероколитах, СПИДе,
гиперпродукции У1Р опухолевой тканью поджелудочной железы);
—гиперосмоляльный — результат значительной гиперосмоляльности
кишечного содержимого (например, если нарушено всасывание ком­
понентов кишечного химуса при мальабсорбции или передозировке
солевых слабительных);
—гиперкинетический — следствие гиперсекреции и повышенной пери­
стальтики кишечника (например, при энтероколитах, синдроме раздра­
женной кишки).
Наиболее серьезные последствия диареи:
—гипогидратация организма, вплоть до эксикоза (крайняя степень гипоги­
дратации организма);
—гиповолемия;
— артериальная гипотензия;
—нарушения электролитного баланса и КОС (различного характера и выра­
женности в зависимости от основного заболевания).
Обстипация
Обстнпацня (запор) — длительная задержка стула или затруднение опо­
рожнения кипшаника (до 3 сут и более).
Наблюдается у 25—30% людей старше 70 лет.
Основные виды и механизмы возникновения запора:
— алиментарный (малообъемный) — результат малого объема кишечного
содержимого (например, при хроническом недоедании, малом потреб­
лении жидкости, недостатке овощей и фруктов в пище, употреблении
легкоусвояемой пищи). Малый объем кишечного содержимого и экскре­
ментов недостаточен для активации рефлекторного процесса дефекации;
—нейрогенный (спастический и атонический запор):
о спастический. Причина его — чрезмерное повышение парасимпатиче­
ских влияний на стенку кишечника может привести к спазму ее муску­
латуры. Это замедляет эвакуацию пищи и опорожнение кишечника;
о атонический. Является результатом ослабленного нейроэффекторного
воздействия на мускулатуру кишечника, что вызывает его гипотонию
и задержку стула;
о ректальный — следствие патологических процессов в прямой кишке
(например, трещина или парапроктит), сопровождающихся болью,
что подавляет рефлекс дефекации;
о механический — результат механической задержки при эвакуации
кишечного содержимого (например, опухолью, рубцом).
Нарушения барьерно-защитной функции кишечника
Стенка кишечника является эффективным механическим, физико-химичес­
ким и иммунологическим барьером для кишечной микрофлоры и токсичных
веществ, образующихся при переваривании пищи и выделяемых микроорга­
низмами.
Нарушение барьерной функции кишечника может привести к инфици­
рованию организма, развитию токсинемии или токсикоинфекции, расстрой­
ствам процесса пищеварения и жизнедеятельности организма в целом.
Патологические синдромы
системы пищеварения
Ниже рассмотрены наиболее характерные и имеющие большое клиническое
значение формы патологии системы пищеварения: язвенная болезнь желудка
и двенадцатиперстной кишки, синдром нарушенного всасывания, синдром раз­
драженной кишки, неспецифический язвенный колит (НЯК) и хронический
колит.
ЯЗВЕННАЯ БОЛЕЗНЬ ЖЕЛУДКА
И ДВЕНАДЦАТИПЕРСТНОЙ КИШКИ
Термины «пептическая язва», «язвенная болезнь», «пептическая язвенная
болезнь» применяют, обозначая группу заболеваний ЖКТ, которые харак­
теризуются участками леструкции слизистой оболочки органов ЖКТ,
образующимися под действием соляной кислоты и пепсина.
Пептические язвы чаще обнаруживают в желудке и проксимальном отде­
ле двенадцатиперстной кишки, иногда в дистальной части пищевода и редко
в тонкой кишке (обычно сочетаются с дивертикулом Меккеля, содержащим
фрагменты слизистой оболочки желудочного типа). Синдром Золлингера—
Эллисона также можно рассматривать как разновидность язвенной болезни.
Основное значение в язвенном процессе имеют повреждение защитного
барьера слизистой оболочки желудка, а также нарушения регуляции кислото­
образующей, кислотонейтрализующей, эвакуаторной функций желудка и две­
надцатиперстной кишки; генетический, бактериальный и другие факторы.
Язвенной болезнью страдают до 5% взрослого населения (при массовых
профилактических осмотрах язвы и рубцовые изменения стенки желудка
и двенадцатиперстной кишки обнаруживают у 10—20% обследованных).
Пик заболеваемости наблюдается в возрасте 40—60 лет. Заболеваемость
выше у городских жителей, чем у сельских.
У мужчин язвенная болезнь развивается чаще, преимущественно в возрас­
те до 50 лет.
Дуоденальные язвы преобладают над желудочными в пропорции 3:1
(в молодом возрасте 10:1).
В возрасте до 6 лет пептическую язву обнаруживают с равной частотой
у девочек и мальчиков (с одинаковой локализацией в двенадцатиперстной
кишке и желудке). У детей старше 6 лет язвы чаще регистрируют у мальчиков
с преимущественной локализацией в двенадцатиперстной кишке.
Рецидивирование наблюдается примерно у 60% пациентов в течение пер­
вого года после заживления язвы двенадцатиперстной кишки и у 80-90%
в течение двух лет.
Смертность при язвенной болезни обусловлена в основном кровотече­
нием (оно наблюдается у 20-25% пациентов) и перфорацией стенки желуд­
ка или двенадцатиперстной кишки с развитием перитонита. Смертность
при перфорации стенки желудка примерно в 3 раза выше, чем при перфора­
ции двенадцатиперстной кишки.
Разновидности язвенной болезни
Выделяют четыре основных клинико-патофизиологических типа язвенной
болезни желудка.
• Тип I: язвы, выявляющиеся в основном в теле желудка, в области «наимень­
шего сопротивления» — 1осиз т т о т геш1епИае — в переходной зоне, рас­
положенной между телом желудка и его антральным отделом.
• Тип II: язвы желудка, возникающие вместе с язвой двенадцатиперстной кишки.
• Тип III: язвы пилорического канала; по течению и проявлениям они сходны
с язвами двенадцатиперстной кишки.
• Тип IV: язвы, локализующиеся вблизи пищеводно-желудочной зоны на малой
кривизне желудка. Несмотря на то что клинически они протекают как язвы
типа I, их выделяют в отдельную группу, так как они склонны к малигни-
зации.
Преимущественно язвы двенадцатиперстной кишки располагаются в начальной
ее части (в луковице); одинаково часто как на передней, так и на задней стенке.
Примерно 5% язв двенадцатиперстной кишки расположено постбульбарно.
Этиология язвенной болезни желудка
и двенадцатиперстной кишки
Причины язвенной болезни желудка и двенадцатиперстной кишки
В настоящее время основной причиной язвенной болезни считают НеНсоЬасТег
ру1оп.
Причинная роль Н. ру1оп в развитии рецидивирующих язв желудка и двенад­
цатиперстной кишки бесспорна. Этот микроорганизм выделяют у 90% больных
язвенной болезнью двенадцатиперстной кишки или антральным гастритом
типа В и у 60-70% пациентов, страдающих язвенной болезнью желудка.
Инфицирование Н. ру1оп разрушает защитный слизисто-бикарбонатный
барьер и повреждает желудочный эпителий. Поражение развивается в резуль­
тате продукции Н. ру1оп гидролитических ферментов (уреаз, фосфолипаз,
протеаз) и широкого спектра цитотоксических веществ (например, мембранотропного или вакуолизирующего), активации синтеза и высвобождения провоспалительных медиаторов, например, фактора некроза опухолей (ФНО-а),
ИЛ, гидролаз лизосом в инфицированной слизистой оболочке желудка. Среди
других возможных причин выделяют:
—алиментарные факторы (нарушение режима и характера питания — дли­
тельное употребление грубой пищи, еда «всухомятку», большие переры­
вы между приемами пищи и т.д.);
—нервно-психический (стрессовый) фактор;
— повышение секреции желудочного сока и снижение активности защитных
агентов (мукопротеидов, бикарбонатов);
— повторный прием алкоголя в избыточных дозах;
— продукты сгорания табака.
Факторы риска язвенной болезни
Язвенная болезнь — результат действия множества взаимопотенцирующих
этиологических факторов. Наиболее значимые факторы риска представлены
на рис. 25.8.
Социальные (нейро- и психогенные) факторы риска язвенной болезни:
—длительные стрессы, умственное переутомление, чрезмерно высокий
(для определенного человека) темп жизни, особенно в профессиональной
сфере. Названные факторы приводят к формированию диффузного
застойного возбуждения в ядрах гипоталамуса, повышают тонические
влияния блуждающего нерва на ЖКТ (стимулируя его секреторную
и моторную активность);
—курение увеличивает риск развития заболевания и уменьшает вероятность
заживления пептических язв. Никотин обусловливает подавление секре­
ции защитных бикарбонатов, обеспечивающих быструю нейтрализацию
соляной кислоты, ускоряет транспорт содержимого желудка с низким
рН в двенадцатиперстную кишку, гиперсекрецию пепсиногена, снижает
тонус сфинктера привратника, что создает условия для заброса кишеч­
ного содержимого, в том числе желчи, в желудок;
- алкоголь непосредственно раздражает слизистую оболочку, стимулирует
желудочную секрецию и разрушает слизисто-бикарбонатный барьер.
Рис. 25.8. Основные этиологические факторы язвенной болезни желудка и кишечника
Алиментарные факторы риска язвенной болезни:
— агенты, повышающие пептическую активность желудочного сока (напри­
мер, частое употребление мясной пищи, которая стимулирует обра­
зование большого количества желудочного сока; преимущественное
употребление рафинированных продуктов питания со сниженной спо­
собностью нейтрализовать соляную кислоту);
—неупорядоченное, нерегулярное и/или однообразное питание (создающее
условия для активации желудочной секреции при недостаточном коли­
честве принятой пищи);
—частое употребление в пищу больших количеств специй, острых приправ,
веществ, раздражающих слизистую оболочку желудка и кишечника (напри­
мер, горчицы, уксуса, майонеза, слишком горячей или холодной пищи,
жидкостей с большим содержанием углекислого газа, крепкого кофе).
Местные факторы риска язвенной болезни:
—желудочная кислотность. Она имеет существенное значение, однако у мно­
гих пациентов находят нормо- или гипоацидность, связанную с увеличе­
нием обратной диффузии ионов водорода (Н+) в стенку желудка. При язве
двенадцатиперстной кишки базальная или стимулированная секреция,
как правило, характеризуется повышенной кислотностью;
—гастрин. У пациентов с язвой желудка содержание гастрина повышено
как натощак, так и после еды; при дуоденальной язве концентрация
гастрина крови натощак в пределах нормы, однако возрастает после
приема пищи;
— рефлюкс желчи в желудок существенно влияет на снижение защитного
барьера слизистой оболочки. Повреждение защитного барьера позволя­
ет кислому желудочному содержимому вступать в контакт с раздражен­
ной слизистой оболочкой и повреждать ее.
Генетические факторы риска язвенной болезни. Об их значении свидетель­
ствуют данные о том, что:
—у ближайших родственников риск возникновения заболевания выше
в 10 раз;
—наблюдается ассоциация болезни с группой крови 0 (I): вероятность раз­
вития язвенной болезни двенадцатиперстной кишки выше на 30—40%;
—заболеваемость язвенной болезнью коррелирует с некоторыми гаплотипами системы Н Ь \ (например, В12, В5, В\у35);
—известны менделирующие заболевания, повышающие риск развития
язвенной болезни желудка;
—семейный полиэндокринный аденоматоз типа I часто сопровождается
развитием гастринсекретирующих опухолей.
Лекарственные средства как фактор риска язвенной болезни
Известно, что ацетилсалициловая кислота (аспирин*) и другие НПВС
подавляют выработку защитных ПГ (ингибируя активность циклооксигеназ).
Стероидные гормоны надпочечников (глюко- и минералокортикоиды) также
подавляют образование слизи и угнетают регенерацию слизистой оболочки
желудка.
Патогенез язвенной болезни
Ключевое звено патогенеза язвенной болезни — нарушение динамического
равновесия между факторами агрессии (их эффекты избыточны!) и факторами
зашиты (их эффекты недостаточны) слизистой оболочки желудка (рис. 25.9).
Рис. 25.9. Роль факторов агрессии и защиты желудочно-кишечного тракта в разви­
тии пептических язв
Выявлено, что в патогенезе язвенной болезни желудка преимущественную
роль играет снижение эффективности факторов защиты, а в развитии пепти­
ческих язв двенадцатиперстной кишки — активация факторов агрессии.
Наиболее вероятный вариант звеньев патогенеза язвенной болезни желудка
представлен на рис. 25.10.
Рис. 25.10. Общие звенья патогенеза язвенной болезни желудка и кишечника
Основные проявления язвенной болезни желудка
• Боль в эпигастральной области (при язвах кардиальной области и задней
стенки желудка она появляется сразу после приема пищи, локализуется
за грудиной, может иррадиировать в левое плечо; при язвах малой кривиз­
ны боли обычно возникают через 15—60 мин после еды).
• Диспепсические расстройства (отрыжка воздухом, пищей, тошнота, изжога,
запор).
• Астеновегетативные явления (снижение работоспособности, слабость, тахи­
кардия, артериальная гипотензия).
• Умеренная локальная болезненность и мышечная зашита в области эпигастрия. Язвы, индуцированные приемом НПВС, часто бывают бессимптом­
ными; они могут дебютировать сразу перфорацией или кровотечением.
Основные проявления язвенной болезни двенадцати­
перстной кишки
• Боль в эпигастрии — преобладающий симптом у 75% больных (боль возни­
кают через 1,5—3 ч после приема пищи (поздняя боль), натощак (голодная)
и ночью (ночная боль). Субъективно боль воспринимается как чувство
жжения в эпигастральной области. Прием пищи улучшает состояние.
• Болезненность в эпигастральной области при пальпации; при этом может
выявляться напряжение мышц брюшного пресса.
• Рвота на высоте боли, приносящая облегчение (уменьшение болей).
• Диспепсические расстройства — отрыжка, изжога (раннее и наиболее частое
проявление), вздутие живота, непереносимость пищи — в 40—70% случаев,
частый запор.
• Астеновегетативные явления (пониженная работоспособность, слабость,
быстрая утомляемость, тахикардия, артериальная гипотензия).
• Сезонность заболевания (обострения весной и осенью).
СИНДРОМ НАРУШЕННОГО ВСАСЫВАНИЯ
Синдром мальабсорбции (синдром нарушенного всасывания компонентов пере­
варенной пищи) — комплекс расстройств, развивающихся в результате нарушений
процессов переваривания пищи и всасывания ее компонентов.
Причины синдрома мальабсорбции
Основные причины мальабсорбции представлены на рис. 25.11.
Проявления синдрома мальабсорбции
Проявления мальабсорбции, их непосредственные причины и механизмы
показаны на рис. 25.12.
Рис. 25.11. Основные причины синдрома мальабсорбции
|Отеки |
Рис. 25.12. Основные проявления синдрома мальабсорбции
Энтеропатии (энтериты)
Энтеропатия (особенно хроническая) — форма патологии, характери­
зующаяся нарушениями кишечного пищеварения и всасывания продуктов
переваренной пищи, — синдром мальабсорбции.
Энтеропатии проявляются признаками воспаления, иммунопатологиче­
ских и дистрофических изменений в слизистой оболочке тонкой кишки.
Виды энтеропатий
Энтеропатии дифференцируются в зависимости от их этиологии, морфо­
логических изменений в кишечнике, функциональных расстройств, клини­
ческих проявлений.
По этиологии энтеропатии подразделяются:
—на инфекционные (развиваются при дизентерии, сальмонеллезе, иерсиниозе, вирусных инфекциях и др.);
—паразитарные (например, при глистных инвазиях, лямблиозе);
—алиментарные (например, при несбалансированном питании и др.);
—энтеропатии физического и химического генеза (например, при воздей­
ствии алкоголя или ионизирующего облучения и др.).
По функциональным признакам вьщеляют энтеропатии, характеризующиеся
преимущественным нарушением:
—мембранного пищеварения (например, при дисахаридазной недостаточ­
ности);
—всасывания определенных веществ (электролиты, железо, вода, витами­
ны, белки, жиры, углеводы);
—моторной функции (гипер- и гипокинетические типы).
В целом, указанные выше изменения и представляют собой суть синдрома
альабсорбции.
По проявлениям различают энтеропатии в зависимости:
— от тяжести синдрома нарушенного всасывания питательных веществ (син­
дром мальабсорбции);
—характера течения (например, острое или хроническое, редко или часто
рецидивирующее);
—фазы патологии (например, рецидив, ремиссия, обострение и др.);
—наличия осложнений (например, солярит, неспецифический мезаденит).
Патогенез энтеропатий
Основные звенья патогенеза энтеропатий связаны с нарушениями полостного
и мембранного пищеварения, а также барьерной функции стенки кишки.
Патогенез энтеропатий характеризуется:
— снижением активности ферментов клеточных мембран;
—нарушением транспорта продуктов гидролиза белков, липидов, углеводов,
ионов и воды;
—расстройством функций других органов пищеварения (например, фермент­
ной активности пищеварительных желез);
—дисбактериозом в кишечнике;
— расстройствами обмена веществ;
—развитием иммунопатологических состояний, которые вторично могут
поддерживать кишечные дисфункции, создавая порочный круг.
В совокупности указанные выше изменения приводят к синдрому нару­
шенного всасывания или рецидивирующей диарее.
Проявления и механизмы развития энтеропатий
Их делят на две группы: внекишечные и кишечные.
Внекишечные проявления энтеропатий
Они обусловлены синдромом мальабсорбции. К внекишечным проявлени­
ям синдрома относятся:
—снижение работоспособности (обусловленное нарушением субстратного
и энергетического обеспечения органов и тканей);
—мышечная слабость (в результате их гипотрофии, дисбаланса ионов
в миоцитах);
—уменьшение массы тела (нередко на 15—20 кг и более);
—трофические изменения кожи и ее придатков — сухость, истончение,
шелушение кожи, выпадение волос, ломкость и утолщение ногтей;
—выраженная гипопротеинемия (вследствие мальабсорбции белка);
— пастозность кожных покровов, системный онкотический (гипопротеинемический) отек (как результат гипопротеинемии);
—гипокальциемия и связанные с ней парестезии, судороги мелких мышц —
симптомы Хвостека (сокращение мышц лица при поколачивании ниже
скуловой дуги) и Труссо (судорога мышц кисти — рука «акушера» —
при наложении жгута на плечо);
—ослабление сухожильных рефлексов, парезы (следствие невропатий и миопатий);
—тахикардия, снижение сегмента 5Т на ЭКГ, уплощение и двухфазность
зубца Т, экстрасистолия (обусловленные в основном гипокалиемией);
—смешанный гиповитаминоз и дисвитаминоз (сопровождаются различной
симптоматикой: гемералопией вследствие недостаточности витамина А,
хейлита — дефицита витамина В2, глоссита — витамина В12, подкожных
кровоизлияний — витамина К; формированием оссалгии и миопатии в результате недостаточности витамина Б ; развитием невропатий
при недостаточности витаминов В,, В2, Е);
—вторичный по отношению к синдрому мальабсорбции иммунодефицит
(сопровождается частыми инфекционными заболеваниями).
Кишечные проявления энтеропатий
Если в процесс вовлекается только начальный отдел тощей кишки, забо­
левание протекает с минимальными кишечными симптомами.
При поражении подвздошной кишки может нарушаться абсорбция желчных
кислот, происходящая в норме в дистальном отделе кишечника. Это влечет
за собой комплекс изменений:
—избыточное поступление желчных кислот в толстую кишку, что вызывает
диарею, поскольку желчные кислоты стимулируют секрецию жидкости,
ионов натрия и хлора в просвет кишечника и активизируют его мотор­
ную функцию;
—нарушение функции илеоцекального клапана, что ведет к забросу в под­
вздошную кишку содержимого толстой кишки и обсеменению ее микро­
флорой;
—нарушение абсорбции витамина В12 с последующим развитием В12-дефицитной анемии;
—боли в средней части живота (схваткообразные, тупые, распирающие;
они возникают обычно через 3—4 ч после еды, что вызвано спазмами
мускулатуры стенки кишечника, избыточным газообразованием, нейрои миопатиями);
—диарею (стул до 5 -6 раз в сутки, обильный; обозначается термином
«полифекалия»);
— стеаторею (каловые массы блестящие, содержат непереваренные жиры;
вызвана дефицитом желчи и липаз в кишечнике).
КОЛИТЫ
К колитам отнесятся хронический колит, синдром раздраженной кишки
и НЯК.
Хронический колит
Хронический колит — заболевание, характеризующееся воспалительно­
дистрофическими изменениями слизистой оболочки толстой кишки
и нарушением ее функций.
Заболевание довольно широко распространено, так как около половины
больных, обращающихся за медицинской помощью по поводу различных
заболеваний органов пищеварения, страдают хроническим колитом. У женщин
заболевание чаще возникает в возрасте 20-60 лет, у мужчин — 40-60 лет.
Этиология хронического колита
Наиболее частые причины колита:
—возбудители кишечных инфекций (дизентерии, сальмонеллеза, иерсиниоза и др.; колит также могут вызывать гельминты, простейшие — амебы,
лямблии, трихомонады, балантидии);
—нарушение питания: однообразная, содержащая большое количество
белков или углеводов, лишенная витаминов пища; частое употребление
трудноперевариваемых и острых блюд;
— частое злоупотребление алкоголем;
—длительное раздражение слизистой оболочки толстой кишки продуктами
неполного расщепления пищи (при врожденной недостаточности фермен­
тов, например, при дисахаридазной недостаточности);
—дисбактериоз (вызывает воспаление толстой кишки);
—интоксикации организма: экзогенные (например, отравление солями ртути,
свинца, фосфора, мышьяка) и/или эндогенные (например, при уремии,
печеночной недостаточности, гипертиреозе, болезни Аддисона);
—длительный бесконтрольный прием некоторых ЛС (слабительных, анти­
биотиков; салицилатов; препаратов наперстянки и др.);
—воздействие радиации (лучевая терапия, массивное рентгеновское облу­
чение);
—нарушение кровообращения в сосудах брыжейки.
Патогенез колитов
К числу ключевых звеньев патогенеза колитов относят:
—повреждение слизистой оболочки толстой кишки причинным фактором (при­
водит к местному воспалению, нарушениям полостного и мембранного
пищеварения, секреторной и всасывательной функций кишечника);
—поражение нервного аппарата стенки кишечника патогенными агентами (усу­
губляет нарушение моторики толстой кишки и трофические расстройства
в ее стенке; обусловливает хронизацию и прогрессирование течения);
— аутосенсибилизация организма экзо- и эндоаллергенами;
—нарушение жизнедеятельности нормальной микрофлоры кишечника с раз­
витием дисбактериоза;
—развитие диспепсии.
Основные проявления хронического колита:
—боли в животе (спастического или ноющего характера, возникают через
7—8 ч после приема пищи; локализация боли зависит от распространен­
ности процесса);
—расстройства стула (диарея, запор).
Синдром раздраженной кишки
Синдром раздраженной кишки — устойчивая совокупность функциональ­
ных расстройств, проявляющаяся болью и/или дискомфортом в брюшной
полости. Они обычно устраняются после дефекации, сопровождаются
изменением частоты и консистенции стула.
В патологический процесс вовлекается преимущественно толстая кишка.
Заболевание широко распространено. По данным мировой статистики,
15—20% всего населения страдает синдромом раздраженной кишки. Женщины
болеют в 2—4 раза чаще, чем мужчины.
Синдром раздраженной кишки сочетается не менее чем с двумя стойкими
признаками нарушения функций кишечника:
—изменениями частоты стула;
—нарушениями акта дефекации (императивные позывы, тенезмы, чувство
неполного опорожнения кишечника, дополнительные усилия при дефе­
кации);
—изменениями консистенции кала;
— выделением слизи с калом;
—метеоризмом.
Эти расстройства продолжаются не менее 12 нед на протяжении последних
12 мес.
Этиология и патогенез син д ром а раздраженной кишки
Причинный фактор, повлекший за собой нарушение функций толстой
кишки, определить удается не всегда.
• В возникновении заболевания большое значение имеет тип личности.
Для пациентов с синдромом характерны истерические, агрессивные реак-
ции, депрессия, навязчивость, канцерофобия, ипохондрические проявле­
ния.
• Важным звеном патогенеза синдрома является дисбаланс БАВ, регулиру­
ющих функции кишечника (серотонин, гистамин, брадикинин, холеци­
стокинин, нейротензин, У1Р, энкефалины и эндорфины).
• Нарушения моторики (как следствие дисбаланса БАВ; они могут быть
как гипер-, так и гиподинамического типа и нередко чередуются).
• Нарушение секреторной функции кишечника. Оно вызывается дисбалансом
БАВ и воздействием бактериальных токсинов (проявляется повышенной
секрецией жидкости и электролитов в просвет кишки).
• Развитие синдрома потенцируется нарушениями питания (наиболее часто —
нерегулярным приемом пищи, преобладанием в ней рафинированных
продуктов). Это усугубляет нарушения моторно-эвакуаторной функции
кишечника, состава микрофлоры; приводит к повышению внутрикишечного давления.
• В патогенезе синдрома раздраженной кишки важную роль играют острые
кишечные инфекции, приводящие к дисбактериозу.
Неспецифический язвенный колит
НЯК — хроническое воспалительное заболевание толстой кишки, характе­
ризующееся язвенно-деструктивными изменениями ее слизистой оболочки.
Распространенность синдрома — 50—230 случаев на 100 ООО населения.
Заболевание возникает во всех возрастных группах, но основной пик при­
ходится на 20-40 лет.
Мужчины и женщины болеют с одинаковой частотой.
Начало заболевания может быть постепенным или острым.
Этиология и патогенез неспециф ического язвенного колита
Этиология изучена еще недостаточно. Доказана генетическая предрас­
положенность к НЯК — описаны семейные случаи язвенного колита; среди
ближайших родственников заболевание возникает в 15 раз чаще, чем в общей
популяции; имеются данные о связи заболевания с Аг НЬА БК2 и В27.
В патогенезе НЯК существенное значение имеют:
—изменения иммуногенной реактивности (сенсибилизация организма);
—формирование аллергических и иммунных аутоагрессивных реакций;
—нервно-психические расстройства;
—развитие воспаления в стенке толстой кишки (при умеренном воспалении
процесс захватывает слизистую оболочку; при тяжелых формах воспа­
ление распространяется на глубокие слои кишечной стенки; слизистая
оболочка отечна и изъязвлена).
Проявления неспециф ического язвенного колита
В клинической картине выделяют три основных синдрома, связанных
с поражением кишки:
—нарушения стула (как правило, диарея);
—геморрагический синдром (наличие мелкоточечных кровоизлияний
на всем протяжении толстой кишки);
— болевой синдром.
При длительном течении НЯК присоединяются общие симптомы:
—тошнота, рвота, анорексия (как проявления хронической интоксикации
организма);
—нарастающая слабость (вследствие интоксикации, нарушения питания,
гипоксии);
—снижение массы тела;
—анемия (в результате интоксикации, полигиповитаминоза, гипоксии,
дефицита железа и других веществ для синтеза гемоглобина);
—лихорадка неинфекционного генеза (в связи с повреждением большого
массива стенки кишечника) и инфекционного (вследствие дисбактериоза
и иммунодефицита).
Особенно тяжело протекает молниеносная форма НЯК. Она почти всегда
характеризуется тотальным поражением толстой кишки, развитием тяжелых
осложнений (токсическая дилатация толстой кишки, перфорация ее стен­
ки, кровотечение в брюшную полость, перитонит, парез кишечника и др.)
и в большинстве случаев требует срочного хирургического вмешательства.
Глава 26
ТИПОВЫЕ ФОРМЫ ПАТОЛОГИИ ПЕЧЕНИ
Печень — один из основных органов, обеспечивающих гомеостаз/гомеокинез организма (рис. 26.1).
Рис. 26.1. Участие печени в процессах гомеостаза/гомеокинеза организма
ФУНКЦИИ ПЕЧЕНИ
Функции печени многообразны. Наибольшее значение имеют следующие.
• Участие в функции пищеварения. Оно связано с образованием в печени
и выделением в кишечник желчных кислот, способствующих эмульгации,
расщеплению и всасыванию жиров и жирорастворимых веществ (напри­
мер, витаминов А, Б , Е, К), а также активации липаз.
• Дезинтокснкационная функция. В печени происходит взаимодействие ток­
сичных веществ с глюкуроновой кислотой и сульфатами, инактивация
аммиака, индола, скатолов, фенолов и других соединений, поступающих
из ЖКТ, а также попадающих в организм извне.
• Кроветворение (происходит у плода).
• Синтез компонентов системы гемостаза. Печень поддерживает оптимальное
состояние (содержание и/или активность) факторов системы гемостаза
и агрегатного состояния крови (например, через образование компонен­
тов свертывающей системы; путем депонирования и выброса крови в сосу­
дистое русло).
• Участие в реакциях системы ИБН. Обеспечивается в связи с наличием
в печени фагоцитирующих клеток фон Купфера, способных также к про­
цессингу и презентации Аг лимфоцитам.
Участие в обмене веществ
В печени протекают многие метаболические процессы (печень — «цен­
тральный орган метаболизма», «биохимическая лаборатория» организма),
в том числе обмен:
—белков (гепатоциты синтезируют все альбумины, 2/3 а-глобулинов, поло­
вину (3-глобулинов; участвуют в де- и переаминировании аминокислот);
—липидов и ЛП (в гепатоцитах образуются и/или трансформируются
ЛПНП, ЛПВП, холестерин и КТ);
—углеводов (в клетках печени протекает гликогенолиз, гликогенез, глюконеогенез);
—витаминов (печень принимает участие в обмене витаминов групп А, В, С,
О, К, РР, фолиевой кислоты);
—минеральных веществ (гепатоциты участвуют в обмене железа, меди,
хрома и др.);
—желчных кислот (они образуются в клетках печени и секретируются
в желчные капилляры).
Различные формы патологии печени могут приводить к недостаточности
функций печени (печеночная недостаточность, включая печеночную кому)
и/или к синдрому желтухи.
НЕДОСТАТОЧНОСТЬ ФУНКЦИЙ ПЕЧЕНИ
Печеночная недостаточность характеризуется стойким снижением или пол­
ным выпадением одной, нескольких или всех функций печени, что приводит
к нарушению жизнедеятельности организма.
Виды печеночной недостаточности
По различным критериям (масштаб повреждения, происхождение, ско­
рость возникновения, обратимость повреждений) выделяют несколько видов
печеночной недостаточности (рис. 26.2).
• По происхождению:
—печеночно-клеточная (печеночная). Является результатом первичного
повреждения гепатоцитов и недостаточности их функции;
—шунтовая (обходная). Обусловлена нарушением тока крови в печени
и в связи с этим ее сбросом (минуя печень) по анастомозам (портокавальным и каво-кавальным) в общий кровоток.
• По скорости возникновения и развития:
— молниеносная (син. — фульминантная). Развивается в течение несколь­
ких часов;
— острая. Развивается в пределах нескольких суток;
—хроническая. Формируется в течение нескольких недель, месяцев
или лет.
• По обратимости повреждения гепатоцитов:
— обратимая. Наблюдается при прекращении воздействия патогенного
агента и устранении последствий этого воздействия;
—необратимая (прогрессирующая). Развивается в результате продолжающе­
гося влияния причинного фактора и/или неустранимости патогенных
изменений, вызванных им. Нередко приводит к гибели пациента.
Рис. 26.2. Виды печеночной недостаточности
Причины печеночной недостаточности
Причинами развития недостаточности печени могут быть факторы двух
категорий:
— собственно печеночные (или гепатогенные): патологические процессы и /
или воздействия, прямо повреждающие клетки печени;
— внепеченочные (или негепатогенные): патологические процессы, проте­
кающие в других органах и тканях, вторично повреждающие печень.
Причины обеих категорий недостаточности изображены на рис. 26.3.
Основные из непосредственно повреждающих печеночную ткань причин пере­
числены ниже.
Дистроф ии печени
Дистрофии и дегенеративные изменения в печени наиболее часто разви­
ваются под действием химических веществ (например, антибиотиков, суль­
фаниламидов, наркотиков; промышленных ядов: бензола, четыреххлористого
углерода, метанола, нитрокрасок, бытовых ядов, этанола и других спиртов,
отравлений грибами).
Рис. 26.3. Основные причины печеночной недостаточности
Гепатиты
Гепатиты — поражения печени воспалительного и/или иммуновоспалитеяьного характера. Наиболее часто гепатиты возникают в результате
вирусной инфекции или гепатотропной интоксикации.
Социальная значимость и экономический ущерб, наносимый вирусными
гепатитами, весьма существенны, что и определяет их как важную проблему
здравоохранения. В частности, гепатитом А ежегодно заболевают более 1 млн
человек, а число носителей вируса гепатита В превышает 1 млрд.
Вирусные гепатиты — группа полиэтиологичных вирусных поражений пече­
ни с различными механизмами и путями передачи возбудителей.
Впервые отделить инфекционный гепатит от прочих поражений пече­
ни предложил выдающийся отечественный терапевт С.П. Боткин (1888).
Клинико-морфологическая картина заболеваний характеризуется преиму­
щественным развитием диффузного воспалительного процесса в печеноч­
ной ткани с соответствующими астеновегетативными и общетоксическими
проявлениями, желтухой, гепатоспленомегалией и рядом возможных внепеченочных поражений (артриты, узелковые периартерииты, гломерулонефриты и т.д.).
Причиной вирусного поражения печени могут быть различные вирусы (напри­
мер, возбудитель желтой лихорадки или герпесвирусы). Однако развитие тяже­
лого, клинически манифестного поражения печени при этих инфекциях либо
непостоянно, либо возникает только у пациентов с иммунодефицитами.
К возбудителям вирусного гепатита относят вирусы различных таксоно­
мических групп; всех их отличает способность вызывать преимущественно
специфические поражения клеток печени.
В настоящее время выделяют восемь типов возбудителей вирусного гепатита,
обозначаемых заглавными латинскими буквами соответственно от А до О,
и вирус ТТУ (характеристика разных вирусных гепатитов и их возбудителей
дана в статье «Гепатиты», см. приложение «Справочник терминов»).
Пути передачи вируса — возбудителя гепатита
• Парентеральный путь передачи возбудителя характерен для вирусных гепа­
титов В, С, О, О. Это происходит при:
—трансфузии крови и ее препаратов, гемодиализе, инъекциях, оперативных
вмешательствах;
—половых контактах с людьми, инфицированными вирусом гепатита В, С, Б;
—рождении детей матерями, инфицированными вирусом гепатита В, С, Б , С;
—тесных бытовых контактах с членом семьи, инфицированным вирусом
гепатита В, С, Б , О.
• Энтеральный (фекально-оральный) путь передачи вируса. Характерен
для вирусных гепатитов А и Е. Они возникают при:
—контакте с больным за 15-60 сут до начала заболевания;
— проживании в эпидемиологически неблагополучном районе;
—групповой заболеваемости с формированием эпидемических очагов в дет­
ских и молодежных коллективах.
Цирроз печени
Цирроз печени — хронически протекающий патологический процесс
в печени, характеризующийся прогрессирующим повреждением и гибелью
гепатоцитов, развитием в ней избытка соединительной ткани (фиброз),
замещающей паренхиму. Проявляется цирроз недостаточностью функций
печени и нарушением кровотока в ней.
Нарушения кровообращ ения в печени
Из нарушений кровообращения наибольшее клиническое значение имеет
развитие портальной гипертензии различного происхождения — стойкого повыше­
ния давления в сосудах системы воротной вены сверх нормы (выше 6 мм рт.ст.).
Длительно текущая портальная гипертензия нередко приводит к дистро­
фии печени и ее недостаточности.
Наиболее частые причины внутрипеченочной портальной гипертензии:
— цирроз печени;
— шистосомоз;
— опухоли печени;
— гемохроматоз.
Портальная гипертензия «предпеченочного» генеза обусловлена блокадой при­
тока крови по портальным сосудам (например, в результате сдавления, окклю­
зии, аневризм, тромбозов ствола воротной или селезеночной вены).
Портальная гипертензия «подпеченочного генеза» вызвана препятствием отто­
ка крови от печени (например, при констриктивном перикардите в результате
кальцификации ткани перикарда, при тромбозе, эмболии, сдавлении нижней
полой вены).
Гипоксия печени
Гипоксия различного генеза (например, циркуляторная при сердечной
недостаточности, тканевая при интоксикациях, субстратная при СД) является
существенным фактором повреждения печеночной ткани, ведущим к ее недо­
статочности.
Паразитарные поражения печени
Многие паразиты тропны к печеночной ткани, поражают ее непосред­
ственно и вызывают печеночную недостаточность (например, при шистосомозе, клонорхозе, описторхозе, фасциолезе).
Н аследуемы е формы патологии печени
Ряд наследуемых заболеваний приводит к нарушению функций и недоста­
точности печени (например, гликогенез типа IV или гемохроматоз).
Эндокринопатии
Повреждение печени наблюдается при эндокринных формах патологии:
например, при гипокортицизме, СД, патологии паращитовидных желез.
Существует множество и других причин развития печеночной недостаточ­
ности.
Общие звенья патогенеза печеночной недостаточности
Воздействие фактора, повреждающего гепатоциты, формирует разветвлен­
ную сеть взаимозависимых и взаимопотенцирующих изменений. Главные
звенья патогенеза печеночной недостаточности представлены на рис. 26.4.
Рис. 26.4. Основные общие звенья патогенеза печеночной недостаточности
Модификация и/или деструкция плазмолеммы, других мембран и цито­
скелета гепатоцитов, развитие иммунопатологических, воспалительных, сво­
боднорадикальных процессов, активация гидролаз сопровождается массиро­
ванным разрушением клеток печени, выходом в интерстиций их содержимого,
включая многочисленные гидролитические ферменты.
Названные факторы дополнительно потенцируют воспалительные, иммуно­
патологические и свободнорадикальные реакции. Это, в свою очередь, делает
процесс поражения печени тотальным и нарастающим по степени.
Проявления печеночной недостаточности
Печеночная недостаточность характеризуется признаками расстройств
обмена веществ и функций печени.
Расстройства обмена вещ еств
Нарушения обмена белка характеризуются:
—нарушением синтеза гепатоцитами альбуминов, которое проявляется гипо-
альбуминемией и диспротеинемией. Гипоальбуминемия способствует
развитию отеков и формированию асцита (в условиях повышенного
давления крови в сосудах воротной вены);
—торможением синтеза белков системы гемостаза (проконвертин, проакцелерин, фибриноген, протромбин, факторы Кристмаса и СтюартаПрауэр, антикоагулянтные белки С и 8), что приводит к гипокоагуляции
белков крови, развитию геморрагического синдрома (кровоизлияние
в ткани, кровотечение);
—меньшей эффективностью реакций дезаминирования аминокислот в соче­
тании с возросшим содержанием аминокислот в крови и моче;
—подавлением в гепатоцитах орнитинового цикла синтеза мочевины из ток­
сичного для организма аммиака; проявляется повышенной концентра­
цией аммиака в крови.
Расстройства липидного обмена сопровождаются:
—нарушением синтеза в печеночных клетках ЛПНП и ЛПОНП (облада­
ющих атерогенными эффектами), а также ЛПВП (оказывающих анти-
атерогенное действие); нередко сопровождается развитием липидной
дистрофии печени (жировой гепатоз);
—повышением в плазме крови уровня холестерина (обладающего проатерогенным свойством).
Нарушения метаболизма углеводов проявляются подавлением гликогене­
за, менее эффективным гликогенолизом, нарушениями образования глюкозы.
Указанные расстройства проявляются низкой резистентностью организма
к нагрузке глюкозой — гипогликемией натощак и гипергликемией вскоре
после приема пищи, особенно углеводной.
Расстройства обмена витаминов характеризуются развитием гипо- и дисвитаминозов за счет:
—нарушения высвобождения из продуктов питания и всасывания в кишечни­
ке жирорастворимых витаминов А, Б , Е, К;
—менее эффективной трансформации провитаминов в витамины (например,
Р-каротина в витамин А);
—замедленного образования коферментов из витаминов (например, тиамин-
пирофосфата из витамина В,, флавинмононуклеотида и динуклеотида
из витамина В2, пиридоксаль-5-фосфата из витамина В6, коэнзима
А из пантотеновой кислоты).
Нарушения обмена минеральных веществ (железа, меди, хрома и др.) наблю­
даются, например, при наследственном гемохроматозе, при котором в ткани
печени накапливается избыток железа, развиваются гепатомегалия, цирроз
и недостаточность функций печени.
Расстройства функций печени
• Недостаточность дезинтокснкационной функции печени. Характеризуется
менее выраженными процессами детоксикации в печени:
—эндогенных токсинов (образующихся и/или накапливающихся в кишеч­
нике — фенолов, скатолов, аммиака, путресцина, кадаверинов и пато­
генных продуктов метаболизма — низкомолекулярных жирных кислот,
сульфатированных аминокислот и др.);
—экзогенных ядовитых веществ (например, токсинов грибов и микроорга­
низмов; ядохимикатов; ЛС).
• Недостаточность антимикробной функции. При печеночной недостаточ­
ности нарушается фагоцитоз различных микроорганизмов клетками фон
Купфера, транспорт 1§А в желчь, где они оказывают бактериостатическое
и бактерицидное действие.
• Нарушение желчеобразования и желчевыделения (с развитием желтух и нару­
шений пищеварения).
Печеночная кома
Финалом прогрессирующей печеночной недостаточности является печеночная
кома. Она характеризуется потерей сознания, подавлением или значительным
снижением выраженности рефлексов и расстройствами жизнедеятельности орга­
низма (включая нарушения дыхания и кровообращения), чреватыми смертью.
Причины и виды печеночной комы
Шунтовая кома («обходная»). Причина шунтовой (обходной) комы — инток­
сикация организма продуктами метаболизма, а также экзогенными вещества­
ми, в норме обезвреживающимися гепатоцитами. Это результат попада­
ния их в общий кровоток, минуя печень, по портокавальным анастомозам.
Последние развиваются в связи с портальной гипертензией.
Паренхиматозная кома. Причина печеночно-клеточной (паренхиматозной)
комы — интоксикация организма в связи с повреждением и гибелью значительной
массы печени (например, при ее травме, некрозе, удалении). В результате этого
нарушаются все функции печени. Наибольшее патогенное значение имеет
утрата дезинтокснкационной функции.
Патогенез печеночных ком
Основные факторы патогенеза печеночных ком представлены на рис. 26.5.
К ним относят:
—гипогликемию; является результатом нарушения гликогенеза и гликоге-
нолиза в гепатоцитах;
— ацидоз (метаболический; в завершающих стадиях дополнительно разви­
вается респираторный и выделительный ацидоз);
—дисбаланс ионов; выявляется в клетках, интерстициальной жидкости,
крови (в крови нарастает [К+], в клетках — [№ +], [Са2+], [Н+]);
—интоксикацию организма. Эндотоксинемия (особенно продуктами бел­
кового и липидного метаболизма, непрямым билирубином) обусловлена
нарушением трансформации билирубина и конъюгации с глюкуроновой
кислотой;
—нарушения центральной, органотканевой гемоциркуляции и микроциркуля­
ции как следствие сердечной недостаточности, нарушения тонуса арте­
риол, развития феномена сладжа;
—полиорганную недостаточность. Раньше всего и наиболее выраженно
нарушаются функции сердца, дыхательного и кардиовазомоторного
центра. Последнее приводит к смешанной гипоксии, прекращению сер­
дечной деятельности, дыхания и к смерти пациента.
Этиологические факторы
1Г
уг
лг
Гипогли­
Дисбаланс
Расстройства
Полиорганная
Ацидоз
Эндотоксинемия
кемия
ионов
кровообращения недостаточность
Кома
Рис. 26.5. Основные факторы патогенеза печеночных коматозных состояний
Ж ЕЛТУХА
Многие формы патологии печени начинаются и/или сопровождаются
желтухой.
Желтуха характеризуется избыточным содержанием в интерстициальной
жидкости и крови компонентов желчи (включая желчные пигменты),
а также желтушным окрашиванием кожи, слизистых оболочек и мочи.
Все виды желтухи объединены одним признаком — гипербилирубинемией,
от которой зависит яркость окраски кожи: от светло-лимонной до оранжево­
желтой, зеленой или оливково-желтой (пожелтение кожи и склер начинается
при концентрации билирубина более 26 ммоль/л).
Метаболизм билирубина
Метаболизм билирубина представлен на рис. 26.6.
Вначале происходит высвобождение гема. Более 80% гема образуется
в результате разрушения эритроцитов и около 20% — миоглобина и цитохромов.
Клетки системы
мононуклеарных
фагоцитов
Плазма крови
Гепатоциты
Рис. 26.6. Основные этапы метаболизма билирубина
Затем протопорфирин гема трансформируется в биливердин; это происходит
под влиянием микросомальных оксидаз гепатоцитов.
Биливердин окисляется в непрямой билирубин, что катализируется цитозоль­
ной биливердинредуктазой.
Непрямой билирубин, циркулирующий в крови, связан с альбуминами и поэто­
му не фильтруется в почках и отсутствует в моче.
Затем происходит диссоциация комплекса неконъюгированный билирубин —
альбумин и транспорт билирубина в гепатоциты. В их цитозоле билирубин
образует комплекс с белками и глутатион-8-трансферазами.
Диглюкуронизация билирубина в гепатоцитах завершается образованием рас­
творимого в воде конъюгированного билирубина. Прямой билирубин не связан
с альбумином. В связи с этим он активно («прямо») взаимодействует с диазоре­
активом Эрлиха, выявляющим этот пигмент.
Конъюгированный билирубин выводится в желчевыводящие пути, после чего
происходит трансформация конъюгированного билирубина:
- в уробилиноген (в верхнем отделе тонкой кишки), всасывающийся в тон­
кой кишке и попадающий по системе воротной вены в печень, где он
разрушается в гепатоцитах;
—в стеркобилиноген (в основном в толстой кишке). Часть стеркобилиногена всасывается в нижнем отделе толстой кишки и с кровью гемор­
роидальных вен попадает в общий кровоток. Стеркобилиноген хорошо
растворим в воде, не связан с белками и поэтому фильтруется в почках
в мочу (придавая ей в норме соломенно-желтый цвет). Другая часть
стеркобилиногена выделяется с экскрементами, окрашивая их.
Виды желтухи
По этиопатогенезу различают:
— механическую;
—паренхиматозную;
—гемолитическую желтуху.
В клинической практике существует множество терминов (в том числе
синонимов), которые относятся к разным заболеваниям, сопровождающим­
ся желтухой (названия и характеристики разных видов желтухи см. в статье
«Желтуха» приложения «Справочник терминов»).
Нарушения обмена желчных пигментов наблюдаются при расстройствах
функций гепатоцитов (развивается паренхиматозная желтуха), внепеченочных формах патологии (например, при выраженном гемолизе эритроцитов
возникает гемолитическая желтуха), а также при нарушениях оттока желчи
от печени (развивается механическая желтуха).
В общем виде выделяют печеночные и внепеченочные группы желтухи
(рис. 26.7).
Рис. 26.7. Виды желтухи по происхождению
Печеночные желтухи (паренхиматозные и энзимопатические) возникают
при первичном повреждении гепатоцитов.
Непеченочные желтухи первично не связаны с повреждением гепатоцитов.
К ним относятся гемолитические (надпеченочные) и механические (подпеченочные) желтухи.
Печеночные желтухи
Причины паренхиматозных печеночных желтух
Различают инфекционные и неинфекционные причины возникновения
паренхиматозных желтух.
• Инфекционные причины. К ним относят вирусы, бактерии, плазмодии.
• Неинфекционные причины паренхиматозных желтух:
— органические и неорганические гепатотоксические вещества (например,
четыреххлористый углерод, этанол, парацетамол и др.);
— гепатотропные факторы (АТ, цитотоксические лимфоциты и макрофаги,
новообразования).
Характер и выраженность нарушений функций печени зависят от степени
альтерации и массы поврежденных гепатоцитов.
В значительной части случаев повреждение, начинаясь с изменения структуры
клеточных мембран и/или подавления активности ферментов, нарастает и может
завершиться деструкцией печеночных клеток.
В любом случае при повреждении паренхимы печени происходят расстрой­
ства желчеобразования и желчевыведения.
Характер расстройств и степень их выраженности на разных этапах (ста­
диях) патологического процесса (следовательно, и выраженности желтухи)
различны.
Стадии паренхиматозной желтухи
I стадия паренхиматозной желтухи (преджелтушная)
Ключевые звенья патогенеза и проявления преджелтушной стадии парен­
химатозной желтухи представлены на рис. 26.8. К ним относят:
— повреждение и снижение активности ферментов, разрушающих уробилиноген (в связи с этим данная стадия проявляется уробилиногенемией
и уробилиногенурией);
—альтерацию мембран гепатоцитов, повышение их проницаемости и выход
в интерстиций и кровь компонентов цитоплазмы, что приводит к ферментемии (в связи с чем в крови увеличивается уровень трансаминаз АЛТ
и АСТ, а также других ферментов гепатоцитов) и гиперкалиемии (вызвана
повреждением и разрушением большого количества клеток печени);
—снижение активности глюкуронилтрансферазы (проявляется уменьшен­
ным образованием прямого билирубина и, как следствие, содержания
стеркобилиногена в крови, моче и экскрементах).
Этиологические факторы
, г
г
Повреждение
мембран
гепатоцитов
Снижение активности
ферментов разрушения
уробилиногена
------Уробилиногенемия |
1г
Снижение
активности
ГТФ
Проявления ------1
Ферментемия|
1р
|Капиемия|
,,
Уробилиногенурия!
Рис. 26.8. Основные звенья патогенеза и проявления I стадии печеночно-клеточной
желтухи: ГТФ — глюкуронилтрансфераза
II стадия паренхиматозной желтухи (желтушная)
Основные звенья патогенеза и проявления желтушной стадии изображены
на рис. 26.9.
Для желтушной стадии характерно дальнейшее усугубление альтерации
мембран гепатоцитов и их ферментов. В связи с этим нарушается работа
«билирубинового конвейера», которая возможна благодаря цитоплазмати­
ческому белку гепатоцитов лигандину и ферменту глюкуронилтрансферазе.
Лигандин способствует транспорту желчных пигментов из участка гепатоцита,
обращенного к кровеносному капилляру, в зону, прилежащую к желчному
капилляру. Расстройство этого механизма в совокупности с повреждением
мембран клеток обусловливает нарушение однонаправленного транспор­
та/трансформации билирубина.
Звенья патогенеза
Усугубление
альтерации
ГТФ
Нарастание
повреждения
мембран
Сдавление
желчных
капилляров
Рис. 26.9. Основные звенья патогенеза и проявления I I стадии печеночно-клеточной
желтухи: ГТФ — глюкуронилтрансфераза
Проявления II стадии паренхиматозной желтухи:
- выход прямого билирубина в кровь, развитие билирубинемии и появление
желтушного окрашивания кожи и слизистых оболочек;
- фильтрация прямого билирубина в мочу почками и его экскреция с мочой:
билирубинурия, сочетающаяся с изменением окраски мочи;
- попадание компонентов желчи в кровь и развитие холемии.
III стадия паренхиматозной желтухи
Характеристика III стадии паренхиматозной желтухи представлена
на рис. 26.10.
Рис. 26.10. Основные звенья патогенеза и проявления I I I стадии печеночно-клеточной
желтухи: ГТФ — глюкуронилтрансфераза
Прогрессирующее подавление активности глюкуронилтрансферазы гепато­
цитов усугубляет расстройство трансмембранного переноса конъюгированного
билирубина в гепатоциты и вызывает торможение глюкуронизации билирубина.
Проявления III стадии паренхиматозной желтухи:
— нарастание уровня непрямого (!) билирубина в крови;
—уменьшение содержания в крови прямого (!) билирубина (как результат
подавления реакции глюкуронизации);
—снижение (в связи с этим) концентрации стеркобилиногена в крови, моче
и экскрементах;
—уменьшение содержания уробилиногена (вплоть до исчезновения) в крови
и как результат этого — в моче. Последнее является результатом малого
поступления прямого билирубина в желчевыводящие пути и кишечник.
Проявляется прогрессирующее усугубление повреждения структур и фер­
ментов гепатоцитов в III стадии паренхиматозной желтухи:
—нарастанием холемии;
— сохранением ферментемии и гиперкалиемии;
—прогрессированием признаков печеночной недостаточности, чреватой раз­
витием комы.
Энзим опатические желтухи
Различают две группы энзимопатических желтух:
—наследуемые (первичные);
—приобретенные (вторичные).
Первичные энзимопатические желтухи
Причины энзимопатий:
—генетически обусловленные дефекты ферментов метаболизма билирубина;
—генетические дефекты белков, обеспечивающих метаболизм пигментного
обмена в гепатоцитах.
Описано несколько нозологических форм энзимопатических желтух.
Наиболее часто в клинической практике встречаются:
—синдром Жильбера (семейная негемолитическая гипербилирубинемия
с желтухой, развивающаяся, как правило, у мальчиков вследствие
дефектов генов 110Т1А1 и С1ЧТ1 хромосомы 2, без признаков пораже­
ния печени, закупорки желчевыводящих путей и гемолиза эритроцитов
при нормальных анализах крови);
—синдром Дабина-Джонсона (результат мутации гена белка, обеспечи­
вающего транспорт органических анионов, в том числе билирубина,
из гепатоцитов в желчь);
—синдром Криглера—Найяра (следствие недостаточности глюкуронозилтрансферазы КФ 2.4.1.174; это приводит к нарушению образования
билирубинглюкуронида — прямого билирубина — из непрямого);
—синдром Ротора (конъюгированная гипербилирубинемия, сочетающаяся
с увеличением экскреции с мочой копропорфирина).
Признаки указанных синдромов приведены на рис. 26.11, этиология и пато­
генез — в статьях «Желтуха» и «Синдром» приложения «Справочник терминов».
Приобретенные (вторичные) энзимопатические желтухи
Общая их причина — сниженная активность и/или дефект ферментов, уча­
ствующих в метаболизме желчных пигментов и синтезе компонентов мембран
гепатоцитов. Это наблюдается при:
— интоксикации организма, особенно гепатотропными ядами (например,
этанолом или четыреххлористым углеродом), некоторыми ЛС, напри­
мер парацетамолом, хлорамфениколом (левомицетин*), веществами
для холецистографии;
—инфекционных поражениях печени (например, вирусами, бактериями,
их эндо- и экзотоксинами);
—повреждении гепатоцитов АТ, цитотоксическими лимфоцитами и макро­
фагами.
I Основные проявления |
- Повышение содержания
- Длительное
неконъюгированного
повышение уровня
неконъюгированного билирубина в крови
(особенно при типе I).
билирубина в крови.
-Снижение уровня
- Снижение уровня
стеркобилиногена
стеркобилиногена
в крови, моче, кале.
в крови, моче, кале
- Значительное
(у отдельных
увеличение
пациентов).
содержания
- Увеличение
моноглюкуронида
содержания
билирубина в желчи.
моноглюкуронида
билирубина в желчи.' - Билирубиновая
энцефалопатия
(при типе I у детей).
- Повышение уровня — Повышение
конъюгированного
содержания
конъюгированного
билирубина в крови.
- Возрастание
билирубина
(моноглюкуронид).
содержания
неконъюгированного —Увеличение
уровня общих
билирубина в крови
копропорфиринов
(за счет
деглюкуронизации
в гепатобилиарной
системе).
- Желудочно-кишечные
расстройства.
Рис. 26.11. Наиболее частые виды энзимопатической желтухи и их признаки
Внепеченочные желтухи
К внепеченочным относят:
—гемолитическую желтуху;
—механическую желтуху.
Гемолитическая желтуха
Причины и проявления гемолитической (надпеченочной) желтухи приведе­
ны на рис. 26.12, а также в статье «Желтуха» (см. приложение «Справочник
терминов»).
Механическая желтуха
Для механической (подпеченочной, застойной, обтурационной) желтухи
характерно стойкое нарушение выведения желчи:
— по желчным капиллярам печени (с развитием внутрипеченочного холеста-
за);
—желчным протокам печени;
желчевыводящим протокам из полости желчного пузыря в двенадцати­
перстную кишку (это вначале приводит к внепеченочному, а при хрони­
ческом течении — и к внутрипеченочному холестазу).
Рис. 26.12. Основные непосредственные причины (а) и проявления (б) надпеченочных (гемолитических) желтух
Причины внутри- и внепеченочного холестаза. К ним относятся факторы:
- закрывающие желчевыводящие пути изнутри (например, конкременты,
опухоли, паразиты, гранулематозная ткань при билиарном циррозе);
- сдавливающие желчные пути снаружи (например, новообразования
головки поджелудочной железы или большого дуоденального сосочка,
рубцовые изменения ткани вокруг желчевыводящих путей, увеличенные
лимфатические узлы);
- нарушающие тонус и снижающие моторику стенок желчевыводящих путей
(дискинезии).
Указанные и другие факторы обусловливают повышение давления в желч­
ных капиллярах, перерастяжение (вплоть до микроразрывов) и повышение
проницаемости стенок желчеотводящих путей, диффузию компонентов желчи
в кровь. В результате этого развивается билиарный гепатит с угрозой цирроза
печени.
Проявления механической желтухи представлены на рис. 26.13. К ним
относятся:
- синдром холемии («желчекровия»);
— синдром ахолии.
Рис. 26.13. Основные проявления подпеченочной (механической) желтухи
Холемия — комплекс расстройств, обусловленных появлением в крови ком­
понентов желчи, главным образом желчных кислот (гликохолевой, таурохолевой
и др.), прямого билирубина и холестерина.
Признаки холемии:
— высокая концентрация конъюгированного билирубина в крови (с развитием
желтухи) и, как следствие, в моче (в сочетании с желчными кислотами).
Это придает моче темный цвет;
—гиперхолестеринемия (избыток холестерина поглощается макрофагами
и накапливается в виде ксантом в коже кистей, предплечий, стоп и /
или ксантелазм в коже вокруг глаз);
—зуд кожи (вследствие раздражения желчными кислотами нервных окон­
чаний);
— артериальная гипотензия (вследствие сниженного базального тонуса
ГМК артериол, менее выраженных адренореактивных свойств рецеп­
торов сосудов и сердца, повышенного тонуса бульбарных ядер блужда­
ющего нерва под действием жирных кислот);
—брадикардия (вследствие прямого тормозного влияния желчных кислот
на клетки синусно-предсердного узла);
— повышенная раздражительность и возбудимость пациентов (в результате
уменьшившейся активности тормозных нейронов коры больших полу­
шарий под действием компонентов желчи);
—депрессия, нарушение сна и бодрствования, повышенная утомляемость
(развивается вследствие хронической холемии).
Ахолия — состояние, характеризующееся значительным уменьшением или пре­
кращением поступления желчи в кишечник, сочетающееся с нарушением полост­
ного и мембранного пищеварения.
Проявления ахолии:
— стеаторея (потеря организмом жиров с экскрементами в результате нару­
шения эмульгирования, переваривания и усвоения жира в кишечнике
в связи с дефицитом желчи);
—дисбактериоз;
— кишечная аутоинфекция и интоксикация (вследствие отсутствия бакте­
рицидного и бактериостатического действия желчи, что способствует
активации процессов гниения и брожения в кишечнике и развитию
метеоризма);
—полигиповитаминоз в основном за счет дефицита жирорастворимых вита­
минов А, Э, Е, К; вследствие дефицита указанных витаминов нарушает­
ся сумеречное зрение, происходит деминерализация костей с развитием
остеомаляции и переломов, уменьшается эффективность системы антиоксидантной защиты тканей, развивается геморрагический синдром;
— обесцвеченный кал (вследствие уменьшения или отсутствия желчи
в кишечнике).
Глава 27
ТИПОВЫЕ ФОРМЫ НАРУШЕНИЙ
ЭКСКРЕТОРНОЙ ФУНКЦИИ ПОЧЕК
Почки выполняют ряд жизненно важных функций. Основные из них пока­
заны на рис. 27.1. Перечень указанных функций (наряду с другими) свидетель­
ствует о том, что почки относятся к числу главных органов, обеспечивающих
процессы гомеостаза/гомеокинеза в организме.
Регуляция парамет­
ров организма
-рН;
-Р
1 ОСМ 5•
-массы циркулирующей
крови;
-артериального давления
[глюкозы];
Синтез и
инкреция БАВ
Мочеобразование,
мочевыделение
-п у те м реализации
процессов:
Офильтрации;
Ореабсорбции;
Осекреции
-
Регуляция
гемопоэза
простагландинов; -посредством
ренина;
синтеза
кининов;
эритропоээритропоэтина;
тина
серотонина;
Рис. 27.1. Участие почек в процессах гомеостаза/гомеокинеза организма
Поражения почек выявляются в среднем у 1,8% населения. У взрослых этот
показатель достигает 9%. При этом более 2/3 обследованных лиц не подозре­
вают о наличии у них почечной патологии.
Патология почек характеризуется рядом важных особенностей:
—высоким уровнем заболеваемости в возрасте до 35—45 лет (пациенты этого
возраста составляют более 60% всех страдающих болезнями почек);
—затяжным характером течения;
— сравнительно низкой эффективностью терапевтических мероприятий;
—частой утратой трудоспособности пациентами;
—значительным и постоянно возрастающим числом пациентов с поражением
почек лекарственного генеза (более 15% всех причин);
—высокой летальностью, в том числе в молодом возрасте.
ПРИЧИНЫ ПАТОЛОГИИ ПОЧЕК
Причины различных форм патологии почек дифференцируют по их при­
роде, происхождению и уровню преимущественной реализации действия.
Виды причин патологии почек по их природе
Природа причинных факторов почечной патологии может быть:
—инфекционной (например, бактерии, вирусы, риккетсии);
—неинфекционной. Среди них выделяют факторы химической, физиче­
ской и биологической природы:
о химические факторы (например, соединения свинца, сулемы, ртути,
мышьяка, некоторые антибиотики, диуретики);
о физические агенты (например, проникающая радиация, продукты
радиоактивного распада, низкая температура, травма почек);
о биологические факторы (например, противопочечные АТ, ]ЧК-лимфоциты, макрофаги; иммунные комплексы; аллергены; избыток или
дефицит катехоламинов, эндопероксидов, ПГ, ПТГ и других БАВ).
Виды причин патологии почек по их происхождению
По происхождению причины почечной патологии подразделяют на:
—первичные (наследственные и врожденные);
—вторичные (приобретенные).
Первичные причины почечной патологии
Их составляют заболевания, вызванные мутациями генов:
—обеспечивающих синтез структурных белков и энзимов, которые участвуют
в функционировании почек;
—дефекты морфогенеза почек.
К заболеваниям этой группы относят ферментопатии, мембранопатии,
поликистоз, дисплазии, нефрогенный несахарный диабет (ННД), псевдогипоальдостеронизм, аминоацидурии, фосфатурию и др.
Вторичные (приобретенные) причины патологии почек
Приобретенные заболевания, не являющиеся результатом действия при­
чинных факторов на геном клеток в период внутриутробного развития,
составляют основную часть патологии почек.
Виды причин патологии почек по уровню их преимущ ественного
действия
По этому критерию различают причины патологии почек:
—преренальные;
—ренальные;
—постренальные.
К преренальным причинам относят:
—нервно-психические расстройства: длительный стресс, психические трав­
мы, состояния, сочетающиеся с сильной болью (в частности, рефлек­
торная болевая анурия);
—эндокринопатии (например, избыток или недостаток АДГ, альдостерона,
тиреоидных гормонов, инсулина, катехоламинов);
—расстройства кровообращения в виде гипотензивных и гипертензивных
состояний.
К ренальным причинам нефропатий относятся факторы:
—прямо повреждающие паренхиму, сосуды, компоненты межклеточного
матрикса почек инфекционного или неинфекционного характера;
—нарушающие кровоток в почках в виде ишемии, венозной гиперемии, стаза;
—вызывающие постнатальные мутации генов, которые обеспечивают функ­
ционирование почек.
Постренальные патологии почек вызываются нарушением оттока мочи
по мочевыводящим путям (это сопровождается повышением внутрипочечного
давления, например, при камнях и опухолях мочевыводящих путей, их отеке,
аденоме простаты, перегибах мочеточника и т.д.).
Названные выше группы причин обусловливают различные расстройства
функции почек. Часть из них описана в других главах и в приложениях учеб­
ника.
В этой главе рассматриваются нарушения мочеобразовательной и мочевы­
делительной (т.е. экскреторной в широком смысле слова) функций почек.
ОБЩИЕ МЕХАНИЗМЫ ВОЗНИКНОВЕНИЯ
И РАЗВИТИЯ ПОЧЕЧНОЙ ПАТОЛОГИИ
Нарушения мочеобразования — результат парциальных или чаще комби­
нированных расстройств, обеспечивающих экскреторную функцию почек:
— фильтрацию (образование первичной мочи в почечных тельцах);
— реабсорбцию (транспорт ионов, жидкости, белков, аминокислот, глюко­
зы и других веществ из просвета почечных канальцев в просвет капилля­
ров вторичной сети);
— секрецию (транспорт ионов, жидкости и ряда других веществ в просвет
канальцев).
На начальных этапах повреждения почек, как правило, происходит акти­
вация какого-либо одного из нижеописанных звеньев патогенеза. По мере
развития патологического процесса подключаются и другие звенья патогенеза.
Именно поэтому в клинической нефрологии трудно выделить какие-либо
специфические, характерные только для одного заболевания механизмы
и клинические проявления.
Многие нефрогенные синдромы и симптомы наблюдаются в различной
степени выраженности и в разных сочетаниях при разнообразных заболева­
ниях и поражениях почек.
Нарушение клубочковой фильтрации
Нарушения клубочковой фильтрации сопровождаются либо снижением,
либо увеличением объема фильтрата.
Снижение объема клубочкового фильтрата
Причины:
—снижение эффективного фильтрационного давления (например, при гипо­
тензивных состояниях: артериальной гипотензии, коллапсе и др., ише­
мии почки или почек, гиповолемических состояниях);
—уменьшение площади клубочкового фильтрата (наблюдается при некрозе
почек или их части, миеломной болезни, хронических гломерулонефритах и других состояниях);
— снижение проницаемости фильтрационного барьера (вследствие утолще­
ния, реорганизации базальной мембраны или других ее изменений;
это происходит при хронических гломерулонефритах, СД, амилоидозе
и других болезнях).
Увеличение объема клубочкового фильтрата
Причины:
— повышение эффективного фильтрационного давления при увеличении тонуса
ГМК-выносящих артериол (под влиянием катехоламинов, ПГ, ангиотензина, АДГ) или уменьшении тонуса ГМК-приносящих артериол (под воздей­
ствием кининов, ПГ и др.), а также вследствие гипоонкии крови (например,
при печеночной недостаточности, голодании, длительной протеинурии);
—увеличение проницаемости фильтрационного барьера (например, вследствие
разрыхления базальной мембраны) под влиянием БАВ — медиаторов вос­
паления или аллергии: гистамина, кининов, гидролитических ферментов).
Нарушения канальцевой реабсорбции
Эффективность канальцевой реабсорбции снижается при:
— ферментопатиях почечного эпителия;
—дефектах систем трансэпителиального переноса веществ (например, ами­
нокислот, альбуминов, глюкозы, лактата, бикарбонатов и др.);
— мембранопатиях эпителия и базальных мембран почечных канальцев.
Важно, что при преимущественном повреждении проксимальных отделов
нефрона нарушается реабсорбция органических соединений (глюкозы, амино­
кислот, белка, мочевины, лактата), а также бикарбонатов, фосфатов, С1_, К +,
а при повреждениях дистальных отделов почечных канальцев расстраиваются
процессы реабсорбции № +, К+, М§2+, Са2+, воды.
Нарушения секреции
Нарушения секреции различных органических и неорганических веществ
развиваются преимущественно при генных дефектах эпителия.
Они приводят к цистинурии, аминоацидурии, фосфатурии, ННД, бикарбонатурии, почечному ацидозу.
Виды почечной патологии
В настоящее время нет общепринятой классификации заболеваний почек,
основанной на едином подходе.
Различными специалистами разработаны и используются классификации,
учитывающие преимущественно морфологические, этиологические, патоге­
нетические, клинические и другие критерии разграничения нефропатий.
Во всех имеющихся в настоящее время классификациях форм почечной
патологии делают акцент на одном или нескольких признаках:
—на преимущественном поражении определенных структур (с выделением,
например, гломерулопатий или тубулопатий);
—на причинах, вызывающих нефропатии;
—на механизмах развития нефропатий;
—на характере лечебных мероприятий («хирургические», «терапевтические»
заболевания почек) и т.д.
Ввиду этих обстоятельств ниже рассматриваются нефропатии и характе­
ризуются отдельные группы типовых форм патологии почек с обязательным
указанием их причин и механизмов развития.
ВИДЫ ПОЧЕЧНОЙ ПАТОЛОГИИ ПО ПРОИСХОЖДЕНИЮ
Виды почечной патологии по их происхождению представлены на рис. 27.2.
Их разделяют на две группы — первичные и вторичные.
Рис. 27.2. Виды патологии почек по происхождению
• Первичные формы патологии почек (наследственные, врожденные, генети­
чески обусловленные формы). К ним относят:
—аномалии развития почек (числа, формы, макро- и микроструктуры);
- тубулопатии (с преимущественным поражением канальцев почек: ННД,
почечный псевдогипоальдостеронизм и др.);
— энзимопатии эпителия канальцев (например, цистинурия, аминоацидурия);
- нефропатии (генерализованные поражения почек: семейная нефропатия
с глухотой или без нее, семейная почечная дистрофия и др.).
• Вторичные (приобретенные, симптоматические) формы патологии почек.
К ним относятся нефропатии:
— инфекционного происхождения — микробного, паразитарного, грибкового,
протозойного (например, нефриты, пиелонефриты, эхинококкоз, акти-
номикоз почек, нефротический синдром, почечная недостаточность);
—иммуноаллергического генеза (нефриты, иммуноаллергические нефропа­
тии и др.);
- обусловленные прямым повреждением почек факторами физической,
химической, биологической природы (например, травмы, радиацион­
ные поражения; токсигенные, лекарственные нефропатии);
- сопутствующие (сателлитные) нефропатии при амилоидозе, эндокринопатиях (например, при СД), нефролитиазе, миграции почки, сердечно­
сосудистых заболеваниях (например, при атеросклерозе, ГБ), иммуноагрессивных болезнях (например, при СКВ);
— онкогенного генеза (опухоли почек различного гистогенеза).
ФОРМЫ ПАТОЛОГИИ,
РАЗВИВАЮЩИЕСЯ ПРИ ПОРАЖЕНИИ ПОЧЕК
Они развиваются при разных формах патологии почек (рис. 27.3).
Типовые формы патологии почек
1Г
Г
Нефриты
1'
Нефротический
Пиелонефриты
синдром
1Г
V
Почечная
недостаточность
(острая, хроническая)
Нефролитиаз
I Уремия
Почечная кома |
Рис. 27.3. Типовые формы патологии почек
Характеристика отдельных форм патологии почек
К отдельным группам патологии почек относят:
- нефрит, нефритический синдром (например, острые постстрептококковый и нестрептококковый гломерулонефриты, хронический гломерулонефрит, быстропрогрессирующий гломерулонефрит);
-
пиелонефрит;
нефротический синдром;
ОПН и ХПН;
нефролитиаз.
Нефриты
Нефриты — группа заболеваний, характеризующаяся диффузным пора­
жением почечной ткани воспалительно-иммунопатологического генеза
с вовлечением в патологический процесс всех отделов нефронов, интер­
стициальной ткани и сосудов печки.
Одной из наиболее распространенных форм патологических процессов
этой категории являются гломерулонефриты.
О стры й гломерулонефрит
Острый гломерулонефрит — заболевание, как правило, инфекционно­
аллергического и/или иммуноаутоагрессивного генеза.
Причины острого гломерулонефрита
Острый гломерулонефрит может быть вызван инфекционными и неинфек­
ционными факторами (рис. 27.4). Наиболее значимые:
- инфекционные агенты: стрептококки (чаще р-гемолитический стрепто­
кокк группы А), пневмококки, менингококки, сальмонеллы, бледная
трепонема, вирусы (вызывающие гепатит, инфекционный мононуклеоз,
оспу и др.), малярийные плазмодии, токсоплазмы;
—неинфекционные факторы. Чаще всего это аутоагрессивные или/и пере­
крестные АТ, циркулирующие в крови комплексы Аг, 1§, факторов ком­
племента, а также чужеродные белки (например, вакцины, сыворотки
или цельной крови, белки опухолевых клеток или поврежденных тка­
ней).
Рис. 27.4. Наиболее частые причины острого гломерулонефрита
Патогенез острого диффузного гломерулонефрита
Одна из наиболее частых форм гломерулонефрита — острый диффуз­
ный гломерулонефрит. Его вызывает гемолитический стрептококк группы А
(штамм 12). На рис. 27.5 и в сопровождающем рисунок тексте показаны
основные звенья патогенеза острого диффузного гломерулонефрита. К ним
относятся:
— образование АТ к Аг стрептококка;
—воздействие антистрептококковых АТ на стрептококки (их деструкция)
и на структуры почечных клубочков (!), особенно их мембраны, имеющие
Аг, которые сходны с Аг гемолитического стрептококка;
—денатурация белков, расцениваемых системой ИБН как чужеродные (!)
для нее;
—прямое повреждение структур нефрона токсинами стрептококка, приводя­
щее к дополнительному образованию аутоАг;
—выработка нефроцитотоксических аутоАТ и лимфоцитов в ответ на появ­
ление в крови аутоАг. Это потенцирует повреждение почек в связи
с развитием реакций иммунной аутоагрессии, аллергии, воспаления
(в ответ на повреждение почечной ткани). Об этом свидетельствует
инфильтрация почек лейкоцитами (включая лимфоциты) и макрофа­
гами, наличие 1§С, факторов комплемента СЗ, С 1я , С4 (выявляемых
иммунофлюоресцентным методом) в петлях капилляров и в мезангии
почечных телец;
—периодическая активация иммуноагрессивного процесса под влиянием
неспецифических повреждающих «разрешающих» (аллергические реак­
ции) факторов (например, охлаждение организма, интоксикации, инфек­
ции, попадание в кровь белоксодержащих препаратов, облучение); обра­
зующиеся при этом иммунные комплексы фиксируются на базальной
мембране клубочков и сосудов микроциркуляторного русла, потенцируя
и расширяя масштаб повреждения почечной ткани, делая его диффузным
(отсюда и название — диффузный гломерулонефрит).
Рис. 27.5. Ключевые звенья патогенеза острого диффузного гломерулонефрита
О том, насколько важна роль инфекционного компонента в патогенезе острого
диффузного гломерулонефрита (ОДГ), свидетельствуют следующие факты:
— возникновение болезни после какой-либо стрептококковой инфекции (анги­
на, тонзиллит, фарингит, скарлатина);
— обнаружение в организме очагов инфекции (в миндалинах, аденоидах, сли­
зистой оболочке гортани и глотки);
— выявление в крови противострептококковых АТ (в частности, антигиалу-
ронидазы, антистрептолизина О);
—воспроизведение модели ОДГ в эксперименте на животных путем введения
им смеси, содержащей убитую культуру гемолитического стрептококка
и гомогенизированную почечную ткань.
Аргументами в пользу иммуноаллергического и/или иммуноаутоагрессивного
компонента патогенеза ОДГ следует считать:
—развитие заболевания через 14—18 сут после перенесенной стрептококковой
инфекции (время, необходимое для образования АТ, их комплексов с Аг,
а также медиаторов аллергии и воздействия их на мембраны клубочков);
—обнаружение в крови нефроцитотоксических аутоАТ;
—выявление неспецифического «разрешающего» фактора заболевания
(охлаждения, интоксикации, инфекции);
— обнаружение в стенках клубочковых капилляров комплексов «Аг + АТ +
комплемент» (например, с помощью иммунофлюоресцентной реакции);
—экспериментальное воспроизведение признаков заболевания после инъек­
ций нефроцитотоксической сыворотки животным.
Хронический диффузный гломерулонефрит
Хронический диффузный гломерулонефрит (ХДГ) — одно из наиболее
частых заболеваний почек.
У 10—20% пациентов ХДГ является исходом острого диффузного гломеру­
лонефрита, а у 80—90% — результатом медленно текущего, клинически слабо
манифестирующего (скрытого) течения болезни.
Причины ХДГ
По происхождению:
• инфекционные агенты (бактерии, вирусы, плазмодии и др.):
—эндогенные микроорганизмы (например, имеющиеся на слизистых обо­
лочках кишечника, глотки или дыхательных путей);
—экзогенные микроорганизмы (попадающие в организм, например, с водой,
пищей, парентерально вводимыми веществами, при переливании крови
и жидкостей);
• неинфекционные патогенные факторы:
—эндогенные агенты (например, Аг опухолей или образующиеся в результа­
те массированного повреждения тканей при ожоговой болезни или син­
дроме длительного раздавливания тканей и т.п.);
—экзогенные вещества (например, ЛС, содержащие литий или золото,
некоторые антибиотики, ненаркотические анальгетики, вакцины, сыво­
ротка крови, алкоголь, органические растворители).
Патогенез ХДГ:
—инициальное звено патогенеза — образование АТ к причинному агенту и /
или к аутоАг, появляющимся при повреждении почечной ткани;
—формирование иммунных комплексов «Аг + АТ + факторы комплемента»,
а также цитотоксических Т-лимфоцитов;
—воздействие этих комплексов и цитотоксических Т-лимфоцитов на компонен­
ты базальных мембран и клеток почечных клубочков, а также их капилляров;
— индукция воспаления и иммунопатологических процессов;
—потенцирование процессов воспаления и иммунопатологических реакций
при повторном образовании в организме или попадании в него чуже­
родных Аг. Вследствие этого нарастает степень и масштаб повреждения
почечной ткани, что делает процесс хроническим, диффузным и потен­
циально необратимым.
Пиелонефриты
Пиелонефриты — группа синдромов (болезней), вызываемых микроор­
ганизмами и характеризующихся развитием воспалительного процесса
в почечных лоханках и интерстиции почки.
Этиология пиелонеф ритов
Причина пиелонефритов — микроорганизмы (в большинстве случаев —
кишечная палочка, клебсиеллы, энтерококки, протеи), попадающие в орга­
низм как из эндогенных очагов инфекции, так и из внешней среды.
Экзогенные микроорганизмы. Они попадают в почку:
—через уретру (например, у женщин при наличии периуретральных коло­
ний бактерий во влагалище, после полового акта или при вагините);
—после инструментальных вмешательств или цистоскопии.
Эндогенные микроорганизмы проникают в почки из очагов инфекции в орга­
низме (например, в миндалинах, кариозных зубах, костях при остеомиелите).
Факторы риска пиелонефритов
Условия, в наибольшей мере способствующие возникновению пиелонеф­
рита, представлены на рис. 27.6. К ним относятся:
—закрытие (обтурация) и/или сдавление (компрессия) мочевыводящих путей
и самих почек (например, камнем; тромбом, формирующимся в резуль­
тате повреждения стенок мочевыводящих путей; опухолями органов
брюшной полости);
— замедление оттока мочи от почек по мочевыводящим путям (например,
при гипотонии их мышечной стенки; сужении мочеточников опухолью
или рубцом; при беременности). Указанные факторы обусловливают
также сдавление и почек, замедление кровотока в них, их ишемию и,
как следствие, уменьшение притока 1§ и замедление миграции лейкоци­
тов в ткань почки. В целом это понижает эффективность реакций ИБН
и способствует инфицированию почек;
—пузырно-мочеточниковый рефлюкс (способствует инфицированию сли­
зистой оболочки лоханок и чашечек, а также интерстициальной ткани
почки в результате восходящего распространения микроорганизмов
из мочевого пузыря);
—иммунодефицитные состояния (способствуют внедрению микроорганиз­
мов в организм, в почки и размножению в них).
Пути проникновения инфекции в почки:
—гематогенный;
—лимфогенный; два этих пути называют нисходящими (микроорганизмы
попадают в микрососуды почки, клубочков, канальцев и далее «нисхо­
дят» с кровью в ткань чашечек и лоханок);
—урогенный — «восходящий» путь (микроорганизмы «восходят» к почке
по мочеотводящим путям).
Рис. 27.6. Факторы, способствующие возникновению пиелонефрита
Звенья механизма развития пиелонефрита:
—инвазия микроорганизмов в почки с развитием воспаления слизистых обо­
лочек чашечек, лоханок и/или интерстиция;
—генерализация инфекции по почечной ткани (проникновение микроорга­
низмов в канальцы и клубочки с развитием гломерулонефрита);
—формирование (как правило) участков некроза слизистой оболочки и аб­
сцессов почек;
—деструкция эпителия канальцев и десквамация погибших клеток;
—обтурация просвета канальцев клеточным детритом.
Указанные изменения сопровождаются нарушением процессов фильтрации,
реабсорбнии и секреции.
Острое течение процесса чревато развитием ОПН, длительное и/или реци­
дивирующее — ХПН, нефросклероза, артериальной гипертензии.
Нефротический синдром
Нефротический синдром — состояние, развивающееся при поражениях почек
различного генеза, приводящих к дефектам клубочковых капилляров.
Для нефротического синдрома характерен комплекс нефрогенных симпто­
мов — протеинурия (в основном альбуминурия), гипопротеинемия (гипоальбуминемия), гиперлипопротеинемия, липидурия, отеки.
Причины неф ротического синдром а
Как правило, нефротический синдром — это заключительный этап забо­
леваний и патологических процессов, приводящих к нарушениям клубочко­
вой фильтрации и канальцевой реабсорбции альбуминов, ЛП, ионов, других
органических и неорганических веществ.
Причины (как почечные, так и внепочечные) развития нефротического
синдрома показаны на рис. 27.7. Наиболее клинически значимые:
• патология почек (как причина первичного нефротического синдрома):
— острый и хронический гломерулонефрит (выявляется у 2/3 пациентов
с нефротическим синдромом);
— гломерулосклероз;
—липоидный нефроз;
—мембранозная гломерулопатия;
• внепочечная патология (как причина и/или фактор риска вторичного
нефротического синдрома):
—хронические инфекции (например, остеомиелит, туберкулез, сифилис,
малярия, вирусные гепатиты);
—патология системы крови (например, лимфомы, лейкозы, лимфограну­
лематоз сочетаются с нарушениями иммунитета и снижением противоинфекционной резистентности);
—злокачественные новообразования (бронхов, легких, желудка, толстой
кишки и других органов) нередко вызывают иммунопатологические
состояния с развитием нефротического синдрома;
—СД (вызывает существенные изменения почечной ткани, снижение
активности системы ИБН и противоинфекционной резистентности);
— болезни иммунной аутоагрессии и аллергические состояния (СКВ, ревмато­
идный артрит, склеродермия, васкулиты, лекарственная болезнь и др.).
Первичный нефротический синдром <
Гломерулонефрит
(острый и хронический)
1
I
Гломерулосклероз
Липоидный
нефроз
Мембранозная
гломерулопатия
'
П зтп ппгм я п п ч е к
Этиологические факторы
Рис. 27.7. Основные причины нефротического синдрома
Патогенез неф ротического синдром а
Основные звенья патогенеза и проявления нефротического синдрома пред­
ставлены на рис. 27.8.
На начальном этапе нефротического синдрома активируются механизмы,
вызывающие:
— повреждение клеточных мембран клубочкового аппарата (под влиянием
причинного фактора);
— формирование иммунопатологических реакций (о чем свидетельствуют
повышенные уровни в крови и ткани почек 1§, компонентов системы
комплемента, иммунных комплексов);
—развитие воспалительного процесса (в ткани почки нарушается микроцир­
куляция, повышается проницаемость стенок микрососудов, происходит
инфильтрация лейкоцитами, развиваются пролиферативные процессы).
„[Этиологические факторы_1_
Рис. 27.8. Основные звенья патогенеза и проявления нефротического синдрома
Позднее формируются важные патогенетические звенья нефротического син­
дрома:
— повышение проницаемости фильтрационного барьера клубочков для мелко-
и крупномолекулярных веществ;
—изменения процессов фильтрации и реабсорбции белков: избыточная филь­
трация белков в клубочках сочетается с их повышенной реабсорбцией
в канальцах почек; при хроническом течении это приводит к поврежде­
нию эпителия канальцев, развитию дистрофических изменений в них,
нарушению процессов реабсорбции и секреции ряда веществ;
— активация синтеза ЛП гепатоцитами (как реакция на гипопротеинемию);
— повышение проницаемости стенок клубочковых капилляров для органиче­
ских и неорганических веществ.
Указанные и другие изменения фильтрации и реабсорбции приводят к протеинурии. Характер протеинурии (потери с мочой белков) и последствия
потерь разных белков представлены на рис. 27.9.
Рис. 27.9. Белки, теряемые организмом при нефротическом синдроме (а), и послед­
ствия протеинурии (б)
Проявления неф ротического синдром а
Описанные выше звенья патогенеза нефротического синдрома обусловли­
вают соответствующие его проявления:
—протеинурию (в основном альбуминурию);
—гипопротеинемию (главным образом, в связи с потерей организмом аль­
буминов);
—дислипопротеинемию (вследствие нарушения синтеза ЛП в печени
и их экскреции почками);
—липидурию;
— микрогематурию (обычно при мембранозно-пролиферативной патоло­
гии нефрона);
— отеки (как следствие гипопротеинемии);
— полигиповитаминоз;
— гиперкоагуляцию белков крови и тромбоз (в связи с диспротеинемией,
в том числе белков системы гемостаза);
—снижение противоинфекционной резистентности организма;
— анемию (железорезистентная микроцитарная, гипохромная);
— ацидоз (выделительный почечный).
ПОЧЕЧНАЯ НЕДОСТАТОЧНОСТЬ
Почечная недостаточность — синдром, развивающийся в результате зна­
чительного снижения или прекращения выделительной функции, а также
нарушения других процессов в почках.
Для почечной недостаточности характерны прогрессирующее увеличение
содержания в крови продуктов азотистого обмена (азотемия) и нарастающие
расстройства жизнедеятельности организма.
В зависимости от скорости возникновения и дальнейшего развития раз­
личают ОПН и ХПН.
Острая почечная недостаточность
ОПН возникает «внезапно» и быстро прогрессирует. Это состояние потен­
циально обратимо. Однако нередко ОПН приводит к смерти пациентов.
Причины острой почечной недостаточности
Различают преренальные, ренальные и постренальные причины ОПН
(рис. 27.10).
Преренальные причины острой почечной недостаточности
Наиболее частые причины преренальной ОПН:
— массивная кровопотеря;
—коллапс;
—шок;
— острая сердечная недостаточность;
—тромбоз почечных артерий.
Все эти формы патологии обусловливают значительное снижение кровотока
в почках. Функции самих почек при действии указанных причин на начальных
этапах острой почечной недостаточности сохранены. Однако они не могут
реализоваться, главным образом, в связи со значительным уменьшением тока
крови в почках. В условиях их гипоперфузии снижается эффективное фильтра­
ционное давление в клубочках, и в крови накапливаются продукты (в том числе
токсичные), в норме удаляющиеся из организма при участии почек.
Рис. 27.10. Основные группы причин острой почечной недостаточности
Ренальные причины острой почечной недостаточности
Факторы этой категории вызывают прямое повреждение почек.
К таким факторам относятся:
—некронефроз (наблюдается примерно у 2/3 пациентов с ОПН; часто раз­
вивается после хирургических операций на почках);
— острая значительная локальная или тотальная ишемия почек;
—нефротоксические агенты (например, четыреххлористый углерод, неко­
торые антибиотики, сульфаниламиды, органические растворители,
НПВС, цитостатики);
—остро текущие патологические процессы, поражающие ткань почек (острые
гломерулонефриты, васкулиты, пиелонефриты). Указанные состояния
перерастают в ОПН примерно у 20% пациентов.
Постренальные причины острой почечной недостаточности
Они обусловливают нарушение (вплоть до прекращения) оттока мочи
по мочевыводящим путям. К таким факторам относят:
— обтурацию мочевыводящих путей (почечными камнями, опухолью, ино­
родными телами, например длительно находящимися в мочеточниках
катетерами; сгустком крови, воспалительным отеком);
— сдавление мочевыводящих путей (например, опухолями органов брюш­
ной полости, увеличенной маткой, тканью аденомы простаты, асцити­
ческой жидкостью);
—перегибы мочеточников (например, при мигрирующих почках, избыточ­
ной их длине).
Патогенез острой почечной недостаточности
Основные звенья патогенеза ОПН представлены на рис. 27.11 и перечис­
лены ниже.
Рис. 27.11. Основные звенья патогенеза острой почечной недостаточности
• Значительное и быстро нарастающее снижение объема клубочковой фильтра­
ции. Причины нарушения клубочковой фильтрации:
— гипоперфузия клубочков в результате ишемии обеих почек преренального
генеза (критическим считают уровень давления крови в афферентных
артериолах в 40-60 мм рт.ст.);
—констрикция почечных артериол, развивающаяся в связи с гипотензией
и гипоперфузией почек;
—микротромбоз и/или агрегация клеток крови в микрососудах почек (в наи­
большей мере последнее наблюдается при различных видах шока, кото­
рый сопровождается образующимся избытком факторов коагуляции
крови).
• Сужение или обтурация многих канальцев почек. Причины уменьшения про­
света почечных канальцев:
—накопление в поврежденных клетках гидрофильных ионов Са2+ (это вызыва­
ет отек и набухание эпителия канальцев, уменьшает просвет канальцев);
—закрытие просвета канальцев клеточным детритом (в связи с повреждени­
ем и гибелью эпителия), цилиндрами, состоящими из белка (например,
при воспалении или повышении проницаемости клубочкового филь­
тра), миоглобина (у пациентов с травмами мышц), гемоглобина (у боль­
ных с гемолизом эритроцитов).
• Подавление процессов экскреции и секреции эпителием канальцев под дей­
ствием нефротоксических факторов (препараты фосфора, соли тяжелых
металлов, фенолы, соединения мышьяка и др.). Тяжесть течения ОПН
в значительной мере обусловлена степенью альтерации канальцев и сни­
жения скорости клубочковой фильтрации;
•Дополнительное (к действию названных выше факторов) повреждение
клубочков, канальцев, интерстициальной ткани в связи с развитием воспа­
лительной и иммуноаллергических реакций в ответ на прямое повреждение
указанных структур. Этот механизм нередко способствует переходу острой
почечной недостаточности в хроническую форму.
ХРОНИЧЕСКАЯ ПОЧЕЧНАЯ НЕДОСТАТОЧНОСТЬ
ХПН — состояние (синдром), развивающееся в результате нарастающей
гибели и значительного уменьшения числа функционирующих нефронов
и характеризующееся существенным прогрессирующим (часто необрати­
мым) снижением функций почек.
Как правило, ХПН завершается гибелью пациентов.
Клиническая манифестация ХПН начинается, когда число нефронов сни­
жается до 30% от нормального. Уменьшение их количества до 15-10% сопро­
вождается развитием уремии.
Причины хронической почечной недостаточности
Как и при ОПН, в развитии ХПН выделяют преренальные, ренальные
и постренальные причины (рис. 27.12).
Рис. 27.12. Основные причины хронической почечной недостаточности
Преренальные причины ХПН:
—хронические артериальные гипертензии;
—медленно прогрессирующий стеноз почечных артерий;
—двусторонняя эмболия артерий почек.
Ренальные причины ХПН:
—хронические патологические процессы в почках (например, гломеруло­
нефриты, пиелонефриты, тубулоинтерстициальные нефриты, поликистоз, тубулопатии);
—хроническая патология других органов, обусловливающая вторичные
поражения почек (например, СД, СКВ, диспротеинозы).
Постренальные причины ХПН:
—факторы, вызывающие длительное нарушение оттока мочи (закрыва­
ющие изнутри или сдавливающие снаружи мочевыводящие пути).
Патогенез хронической почечной недостаточности
Патогенез ХПН заключается в прогрессирующем снижении (вплоть до пре­
кращения) клубочковой фильтрации, канальцевой секреции и реабсорбции.
Основа этих процессов — прогрессирующая гибель нефронов, замещение
их соединительной тканью (т.е. развитие нефросклероза). Это и приводит
к нарастающей недостаточности функций почек. Заключительным этапом
ХПН является уремия.
УРЕМИЯ
Уремия — синдром аутоинтоксикации организма продуктами метаболиз­
ма (нормального и нарушенного), а также экзогенными соединениями,
в норме выводящимися почками.
Причины уремии
Непосредственной причиной уремии является почечная недостаточность
(острая или хроническая).
Патогенез уремии
К ключевым звеньям патогенеза уремии относят повреждения тканей и орга­
нов при уремии и почечной коме:
—интоксикацию организма избытком аммонийных соединений (аммиак, про­
изводные аммония), образующихся в процессе трансформации мочеви­
ны в кишечнике;
—токсическое действие продуктов метаболизма ароматических аминокислот
(фенолов, индолов, скатолов);
— повреждение мембран и ферментов клеток указанными выше и другими
«уремическими токсинами»;
—нарушение энергетического обеспечения клеток;
—дисбаланс ионов и жидкости в клетках, а также дисиония в интерстиции
и крови; при ХПН нередко наблюдается избыток ПТГ в крови, что при­
водит к накоплению ионов Са2+ в клетках, а это, в свою очередь, ведет
к разобщению процессов окисления и фосфорилирования, дефициту
АТФ и нарушениям энергозависимых процессов;
—нарастающий ацидоз (вследствие потенцирования процесса накопления
кислых валентностей, обусловленного торможением ацидо- и аммониогенеза, экскреции «кислых» соединений почками, расстройств гемодинамики
(метаболический ацидоз) и газообмена в легких (респираторный ацидоз);
— расстройства электрогенеза в возбудимых клетках, в том числе мозга
и сердца; этим обусловлена потеря сознания при коме, усугубление рас­
стройств функций сердечно-сосудистой, дыхательной и других физио­
логических систем.
Уремия нередко завершается почечной комой. Как и любая другая, почеч­
ная кома характеризуется угнетением функции нервной системы и проявляет­
ся потерей сознания, гипо- или арефлексией, значительными расстройствами
функций органов и физиологических систем организма.
НЕФРОЛИТИАЗ
Нефролитиаз — состояние, характеризующееся наличием в ткани почек
плотных конкрементов (камни) из неорганических и органических комнонентов мочи.
Образование конкрементов в лоханках, чашечках и мочеточниках обознача­
ется как уролитиаз.
Причины нефро- и уролитиаза подразделяются на эндогенные и экзогенные:
— экзогенные — например, «жесткая» питьевая вода, однообразная гиповита-
минизированная пища (большое значение имеет дефицит витамина А);
—эндогенные — например, микрофлора мочевыводящих путей, ЖКТ,
половой системы; избыток продуктов нарушенного обмена веществ
при подагре, миеломной болезни, эндокринопатии, преимущественно
гиперпаратиреоз.
Факторы риска нефролитиаза и уролитиаза. К числу наиболее клинически
значимым относят:
—уменьшение содержания в моче солюбилизаторов (агенты, поддержи­
вающие соли мочи в растворенном состоянии: мочевина, креатинин,
ксантин, цитраты и др.), ингибиторов кристаллизации солей (неоргани­
ческий пирофосфат), комплексообразователей (М§2+, цитраты);
—повышение в моче уровня агентов, инициирующих процесс кристаллизации
солей в моче (мукопротеинов, солей пировиноградной кислоты, колла­
гена, эластина, сульфаниламидов);
—изменение рН мочи (при рН, равном 5, осаждаются соли мочевой кисло­
ты, при рН выше 7 — кальция фосфат, фосфорнокислый аммиак);
—повышение в моче содержания камнеобразующих солей (в основном каль­
ция);
— нарушения оттока мочи.
Механизмы формирования конкрементов
В конкрементах всегда (или почти всегда) обнаруживаются два компонен­
та — органический и минеральный. В связи с этим имеется две точки зрения
на механизм камнеобразования. Они сформулированы в виде кристаллизаци­
онной и коллоидной теорий.
Согласно кристаллизационной теории, начало образованию камня дает про­
цесс кристаллизации солей. При этом в состав конкремента включаются
(случайно) и органические компоненты (фибрин, коллаген, клеточный детрит
и др.).
Авторы коллоидной теории считают, что вначале формируется органическая
матрица, на которой затем кристаллизуются соли.
Наиболее значимые последствия нефро- и уролитиаза:
—гидронефроз с атрофией почки (почек);
—пиелонефрит;
— нефросклероз;
— абсцессы почек;
—почечная колика.
Проявления почечной патологии
Расстройства функций почек проявляются отклонением характеристик
мочи, крови, а также развитием общих нефрогенных синдромов.
ИЗМЕНЕНИЯ ПАРАМЕТРОВ МОЧИ
К изменениям параметров мочи (рис. 27.13) относят:
• изменения диуреза (количество выделяемой мочи):
—полиурия (увеличение объема мочи: выделение за сутки более 2000—
2500 мл мочи; причины полиурии — увеличение клубочковой фильтра­
ции и/или уменьшение канальцевой реабсорбции);
—олигурия (выделение в течение суток менее 500—300 мл мочи; обычно
бывает следствием уменьшения фильтрации и/или увеличения реаб­
сорбции);
—анурия (прекращение поступления мочи в мочевой пузырь; как правило,
это результат значительного снижения фильтрации, что может сочетать­
ся с увеличением реабсорбции);
• изменения относительной плотности мочи:
—гиперстенурия (увеличение плотности мочи выше нормы: более 1,029-1,030;
как правило, является следствием увеличения реабсорбции веществ);
—гипостенурия (уменьшение плотности мочи ниже нормы: менее 1,009;
наблюдается при нарушении концентрационной функции почек);
—изостенурия (мало меняющаяся в течение суток относительная плотность
мочи; свидетельствует об уменьшении эффективности канальцевой
реабсорбции и о снижении концентрационной способности почек);
• изменения состава мочи:
— колебания (за пределы нормы) содержания нормальных ее компонентов
(глюкозы, ионов, воды, азотистых соединений, свидетельствует о менее
эффективной канальцевой реабсорбции и о сниженной концентрацион­
ной способности почек);
—появление в моче отсутствующих в норме компонентов: эритроцитов (гема­
турия), лейкоцитов (пиурия), белка (протеинурия), аминокислот (аминоацидурия), осадка солей, цилиндров (канальцевых слепков, состо­
ящих из белка, клеток крови, эпителия канальцев, клеточного детрита).
• изменения ритма мочеиспускания:
—поллакиурия (частое мочеиспускание); причины полаккиурии — поли-
урия (увеличение объема мочи); раздражение мочевыводящих путей,
например, при их воспалении или при прохождении через мочевыводя­
щие пути мелких конкрементов — «песка» и др.);
— никтурия (преимущественное мочеиспускание ночью); причины — нару­
шение кровоснабжения почек, аденома простаты, патология почек
(амилоидоз) и/или мочевыводящих путей (уретрит, цистит).
-полиурия;
-олигурия;
-анурия
-
гиперстенурия;
гипостенурия;
изостенурия;
гипоизостенурия
- значительные
(за пределы нормы)
изменения содержания
нормальных компонентов
мочи;
- появление патологических
компонентов мочи
Рис. 27.13. Проявления расстройств мочеобразования и мочевыведения
ИЗМЕНЕНИЯ ОБЪЕМА И СОСТАВА КРОВИ
Эти изменения показаны на рис. 27.14. К ним относятся:
—гиперволемия (почечного генеза). Причины «почечной» гиперволемии —
снижение клубочковой фильтрации и/или увеличение канальцевой
реабсорбции;
—гиповолемия (почечного происхождения). Причины «почечной» гипово­
лемии — как правило, увеличение фильтрации и/или уменьшение реаб­
сорбции;
—азотемия (повышение уровня небелкового азота в крови). Причина азо­
темии — нарушение экскреторной функции почек (например, при гломерулонефрите, пиелонефрите, амилоидозе). Более половины небелко­
вого азота крови составляет азот мочевины, около 25% — аминокислоты,
остальное — азот мочевой кислоты («4%), креатин («5%), креатинин
(=2,5%) и другие небелковые соединения;
—гипопротеинемия (снижение уровня белка крови). Причина — наруше­
ние канальцевой реабсорбции альбуминов;
—диспротеинемия (нарушение оптимального соотношения отдельных
фракций белка в крови: глобулинов, альбуминов). Причина диспротеинемии — повышенное выделение альбуминов с мочой;
— гиперлипопротеинемия. Одна из наиболее частых причин — нефротиче­
ский синдром;
— ацидоз. Причины ацидоза — снижение эффективности ацидогенеза,
аммониогенеза, ионообменного механизма Ма+/К +, а также экскреция
почками соединений с «кислыми» свойствами;
—при различных заболеваниях почек могут развиваться также гипер(гипо)
фосфатемия, пшер(гипо)калиемия, гипер(гипо)натриемия, гипер(гипо)кальциемия, гипер(гипо)магниемия и другие изменения в содержании компо­
нентов крови.
Характер отклонений определяется конкретным заболеванием почек
и нарушением в них процессов фильтрации, реабсорбции и секреции.
----------------- Изменение показателей объема и состава крови--------------------I [_______________________ V __________________________ У _________________
Гипер-,
гиповолемия
Дисионии
Гипо-,
диспротеинемия
ЛГ
1Г
Гипер-,
Ацидоз
липопротеинемия
Азотемия
Рис. 27.14. Проявления расстройств экскреторной функции почек (изменение пока­
зателей объема и состава крови)
ОБЩИЕ НЕФРОГЕННЫЕ СИНДРОМЫ
Общие для большинства заболеваний почек синдромы представлены
на рис. 27.15.
Рис. 27.15. Проявления расстройств экскреторной функции почек (общие нефрогенные синдромы)
Принципы лечения почечной
патологии
Лечение больных с расстройствами функций почек базируется на этиотропном, патогенетическом и симптоматическом принципе.
Этиотропный принцип лечения патологии почек. Цель — устранение (сниже­
ние степени патогенного действия) причинного фактора.
Для этого используют, например, антибиотики, сульфаниламиды, а также
проводят лечение других болезней, вызвавших почечные заболевания.
Патогенетический принцип терапии почечной патологии. Он имеет цель разо­
рвать звенья патогенеза болезней почек.
С этой целью применяют иммунодепрессанты, иммуномодуляторы, антиаллергические препараты и проводят мероприятия по «разгрузке» почек
(гемодиализ, перитонеальный, гастроинтестинальный диализ).
Наиболее эффективный способ ликвидации токсичных веществ, накапли­
вающихся при почечной недостаточности, — гемодиализ с использованием
специального прибора — искусственной почки (гемодиализатор). Первый
такой прибор, использовавшийся в эксперименте на животном, был разрабо­
тан в 1913 г. В 1960 г. гемодиализ впервые использован для лечения пациентов
с ХПН.
Работа аппарата «искусственная почка» основана на принципе диффузии
из крови в специальный диализирующий раствор через полупроницаемую
мембрану небелковых соединений. Применение искусственной почки позво­
ляет нормализовать на некоторое время ряд параметров организма и облег­
чить состояние пациента. Однако гемодиализ не восполняет всех почечных
функций. С целью радикального устранения патологии почки (почек) исполь­
зуют пересадку донорского органа (трансплантация почки).
Симптоматический принцип терапии нефропатий. Цель — устранение
(или облегчение) вторичных страданий и последствий, вызванных патоло­
гией почек (анемия, отеки, гастриты, энтероколиты, тромбогеморрагические
расстройства, артериальная гипертензия и др.). С этой целью переливают
кровь и/или ее компоненты, проводят противоотечную терапию, устраняют
заболевания желудка и кишечника, системы кровообращения.
Глава 28
ТИПОВЫЕ ФОРМЫ ПАТОЛОГИИ
ЭНДОКРИННОЙ СИСТЕМЫ
Эндокринная система — совокупность анатомически, гистологически и цито­
логически дифференцированных структур, вырабатывающих гормоны.
Эндокринные клетки синтезируют и выделяют в жидкие среды организма
(кровь, лимфу, межклеточную жидкость, ликвор и др.) молекулы гормона.
ЭНДОКРИННЫЕ ЖЕЛЕЗЫ
В большинстве случаев гормоны синтезируются в анатомически автоном­
ных структурах — эндокринных железах, или железах внутренней секреции.
К ним относятся гипофиз, эпифиз (шишковидная железа), щитовидная желе­
за, околощитовидные железы, надпочечники (рис. 28.1).
Рис. 28.1. Органы и клетки, синтезирующие гормоны
ЭНДОКРИННЫЕ КЛЕТКИ ОРГАНОВ И ТКАНЕЙ
Ряд гормонов продуцируется совокупностью клеток или отдельными клет­
ками, не организованными анатомически в виде железы. Эти клетки находятся
в самых различных тканях и органах (см. рис. 28.1). К ним относят нейросекре­
торные клетки гипоталамуса, эндокринные клетки островков Лангерганса подже­
лудочной железы (а-, (3-, 8-клетки), эндокринные клетки ЖКТ (продуцирующие
гастрин, глюкагон, мотилин, секретин, соматостатин, холецистокинин, гастринрилизинг-гормон), интерстициальные клетки почек (вырабатывающие ПГЕ2
и эритропоэтин), интерстициальные клетки Лейдига яичка (продуцирующие
андрогены), фолликулярные клетки яичника (образующие эстрадиол, эстрон,
эстриол, ПГ) и его желтое тело (продуцирующее прогестерон и эстрогены),
кардиомиоциты правого предсердия (синтезируют атриопептин — натрийуретический фактор), эндокринные клетки легких (продуцирующие кальцитонин,
бомбезин, ПГ, лейцин-энкефалин), эпителиальные клетки вилочковой железы
(тимуса), вырабатывающие пептидные гормоны тимопоэтин и тимозины.
ГОРМОН
Термином «гормон» обозначают секретируемое эндокринными клетками
во внутреннюю среду организма БАВ, взаимодействующее с рецепторами клетокмишеней и изменяющее их функционирование.
Химическая структура гормонов различна. Основные их классы:
—олигопептиды (например, нейропептиды);
—полипептиды (например, инсулин);
—гликопротеины (например, ТТГ);
—стероиды (например, альдостерон и кортизол);
—производные тирозина (например, йодсодержащие гормоны щитовидной
железы — трийодтиронин Т3 и тироксин Т4);
—эйкозаноиды (например, ПГ и простациклины).
Рецепторы гормонов и вторые посредники
Рецептор гормона — белковая молекула, расположенная на поверхности цито­
леммы, в цитоплазме или в ядре, которая специфически взаимодействует с опре­
деленным гормоном и передает сигнал вторым посредникам.
(Подробнее о рецепторах и гормонах см. в приложении «Справочник тер­
минов».)
Варианты воздействия гормонов на клетки-миш ени
По расстоянию от клетки-продуцента гормона до клетки-мишени различа­
ют эндокринный, паракринный и аутокринный варианты регуляции.
Эндокринная или дистантная регуляция — секреция гормона происходит
во внутреннюю среду организма. Клетки-мишени могут отстоять от эндокрин­
ной клетки сколь угодно далеко. Пример — секреторные клетки эндокринных
желез, гормоны из которых поступают в систему общего кровотока.
Паракринная регуляция — клетка-продуцент биологически активного вещества
и клетка-мишень расположены радом. Молекулы гормона достигают мишени
путем диффузии в межклеточном веществе. Например, в париетальных клет­
ках желез желудка секрецию Н + стимулируют гастрин и гистамин, а подавля­
ют соматостатин и ПГ, секретируемые рядом расположенными клетками.
Аутокринная регуляция — клетка-продуцент гормона имеет рецепторы к нему
(т.е. клетка-продуцент гормона является его мишенью). Например, эндотелины,
вырабатываемые клетками эндотелия, воздействуют на эти же эндотелиаль­
ные клетки; Т-лимфоциты, секретирующие интерлейкины (ИЛ), имеющие
мишенями разные клетки, в том числе и Т-лимфоциты.
МЕХАНИЗМЫ НЕЙРОЭНДОКРИННОЙ РЕГУЛЯЦИИ
Функция эндокринной системы, как правило, тесно связана с нервной
деятельностью. В связи с этим сложилось представление о нейроэндокринной
регуляции. В общем виде речь идет о сочетанной деятельности корковых
и подкорковых нервных структур, эндокринных клеток и их мишеней, осу­
ществляющей регуляцию конкретных функций организма. В результате фор­
мируются контуры саморегуляции.
Регуляторные контуры нейроэндокринной
регуляции
Типичный пример регуляторного контура — половая функция женского орга­
низма.
В этом иерархическом контуре можно выделить следующие звенья,
или уровни: нейроны коры большого мозга, подкорковые структуры, нейро­
эндокринные клетки гипоталамуса, эндокриноциты передней доли гипофиза,
эндокринные клетки половых органов и клетки-мишени половых гормонов.
Сигналы каждого звена (уровня) оказывают регуляторное воздействие
на следующее звено контура в направлении сверху вниз.
Каждое звено контура направляет гуморальные или нервные сигналы
к выше расположенным уровням (чаще по принципу отрицательной обратной
связи — подавляя активность этих уровней).
В структурах мозга формируются относительно автономные генераторы
ритма.
Примером функционирования такого саморегулирующегося нейроэндо­
кринного контура является регуляция овариально-менструального цикла.
Общая этиология и общий
патогенез эндокринных
расстройств
Общие звенья патогенеза эндокринных расстройств
Выделяют следующие варианты инициальных звеньев патогенеза эндокрин­
ных расстройств:
— центрогенный;
— первично-железистый;
—постжелезистый.
Центрогенное инициальное звено патогенеза эндокринных форм
патологии
В основе его лежит нарушение механизмов нейрогуморальной регуляции желез
внутренней секреции со стороны нейронов коры большого мозга и/или гипоталамогипофизарной системы.
Как правило, центрогенное звено патогенеза эндокринопатий — следствие
расстройств функций коры головного мозга, гипоталамуса, аденогипофиза,
нейрогипофиза.
Причины инициации центрогенного звена патогенеза эндокринных расстройств:
• на уровне коры большого мозга:
—дефекты развития и органические повреждения головного мозга (чаще в резуль­
тате кровоизлияния, роста опухолей, образования гранулем, травм);
—действие токсинов и инфекционных агентов (например, этанола, наркоти­
ков, микробных эндо- и экзотоксинов);
— нарушения ВНД (как правило, невротические состояния, затянувшиеся
стресс-реакции, психозы);
• на уровне гипоталамуса и гипофиза:
—генные дефекты клеток гипоталамуса и/или гипофиза (мутации генов
либеринов, статинов, адено- и нейрогипофизарных гормонов, а также
ферментов синтеза этих БАВ);
—прямое повреждение гипоталамуса и/или гипофиза (например, при росте
и/или распаде опухоли, кровоизлияниях, сотрясении, сдавлении);
— воздействие на гипофиз и гипоталамус токсичных веществ экзо- и/или эндо­
генного происхождения инфекционной либо неинфекционной природы
(например, этанола, столбнячного токсина, нейротропных ЛС).
Расстройства ф ункций коры головного мозга и гипоталамогипофизарной системы приводят к нарушениям в образовании нейрогор­
монов гипоталамуса (либеринов, статинов, АДГ), а также тропных гормо­
нов аденогипофиза. Последнее, в свою очередь, вызывает расстройства
функций желез и клеток внутренней секреции, регулируемых тропными
гормонами аденогипофиза.
Первично-железистые эндокринны е расстройства
Они вызваны расстройствами синтеза, и/или инкреции в кровь, и/или интерстиций гормонов эндокринными железами и отдельными эндокринными
клетками (рис. 28.2).
Рис. 28.2. Типовые механизмы эндокринопатий: первично-железистые расстройства
П остж елезисты е эндокринопатии
Обусловлены различными нарушениями транспорта гормона, его рецепции
и пострецепторными сбоями в клетке-мишени.
Наиболее клинически значимые варианты постжелезистых эндокринопа­
тий показаны на рис. 28.3.
Разновидности механизмов
постжелезистых эндокринопатий
Транспортный
Метаболический!
!«Контргормональный»| I Рецепторный
Рис. 28.3. Типовые механизмы эндокринопатий: постжелезистые расстройства
• Транспортный вариант патогенеза постжелезистых эндокринопатий. Заключается
в чрезмерно сниженном или повышенном связывании гормонов с их транс­
портными белками (наиболее часто с альбуминами). В результате уменьшается
или возрастает уровень свободного, активного гормона (например, инсулина,
кортизола, йодсодержащих гормонов щитовидной железы).
• «Контргормональный» вариант патогенеза постжелезистых эндокринопатий.
Этот вариант патогенеза происходит несколькими путями, снижающими
или устраняющими эффективность гормонов:
—чрезмерная связь гормонов с транспортными белками; не связанные с ними
гормоны быстро инактивируются в крови, что проявляется недостаточ­
ностью их эффективности;
— высокий уровень 1§ к гормонам; выделены АТ, например, к инсулину,
АКТГ, СТГ;
—повышенное содержание ферментов, разрушающих гормоны: например,
повышение уровня инсулиназы, глутатионредуктазы или глутатионтрансферазы приводит к разрушению инсулина; а моноаминооксидазы
и/или катехол-о-метилтрансферазы — адреналина;
—изменение конформации молекул гормонов; наблюдается, например,
в условиях выраженного ацидоза в клетках и интерстициальной жидко­
сти или взаимодействия с гормонами токсинов, солей тяжелых метал­
лов, свободных радикалов;
—действие гормонов-антагонистов; например, избыток в крови катехола­
минов, кортизола, глюкагона, СТГ, тиреоидных гормонов препятствует
эффективному воздействию инсулина.
• Рецепторный (реактивный) вариант патогенеза постжелезистых эндокрино­
патий. Он заключается в нарушении взаимодействия гормона с его рецеп­
тором. Выделяют несколько разновидностей этого механизма:
—изменение числа рецепторов к гормону (увеличение или уменьшение);
— нарушение нормального соотношения высоко- и низкоаффинных рецепто­
ров к гормону;
— образование противорецепторных АТ (например, к рецепторам инсулина
или ТТГ);
— блокада рецепторов к гормонам негормональными лигандами, которые
имеют структуры, сходные с фрагментом молекулы гормона; описана
в отношении рецепторов инсулина, инсулиноподобных факторов роста,
тиреоидных гормонов;
—перекрестный эффект гормона (например, СТГ может активировать
рецепторы пролактина, в результате чего развивается галакторея).
• Метаболический вариант патогенеза постжелезистых эндокринопатий. Этот
механизм заключается в разнообразных нарушениях метаболизма гормонов.
Наиболее часто наблюдаются:
—расстройства катаболизма гормона — например, инсулина и стероидных
гормонов в гепатоцитах (при этом торможение разрушения глюкокортикоидов приводит к подавлению синтеза АКТГ);
—чрезмерное дейодирование Т4. Например, дейодирование наружного
кольца Т4, частично происходящее в щитовидной железе, начинает осу­
ществляться преимущественно в печени, что приводит к образованию
более активного гормона Т3 и гипертиреоидному состоянию; дейо­
дирование внутреннего кольца Т4 происходит в щитовидной железе,
печени и частично в почке, в результате чего образуется реверсивный
гТ3 (от англ. геуегзе), имеющий низкую физиологическую активность,
что обусловливает развитие гипотиреоидного состояния.
В основе большинства эндокринопатий лежит дефицит конкретного гормона
и/или его эффекта. Это определяет один из основных принципов лечения
при таких заболеваниях — заместительную терапию.
Нарушения функций
гипоталамо-гипофизарной
системы
Гкпоталамо-гипофизарная система включает:
—переднюю долю гипофиза (она имеет эпителиальное происхождение,
вместе с туберальной и промежуточной долями образует аденогипофиз,
осуществляет синтез тропных гормонов и экспрессию гена проопиомеланокортина);
—перикарионы секреторных нейронов гипоталамуса, синтезирующие рилизинг-гормоны, или либерины, антидиуретический гормон (АДГ), окситоцин, нейрофизины, орексины;
—гипоталамо-гипофизарный тракт (обеспечивающий транспорт гормонов
по аксонам нейросекреторных нейронов);
— аксовазальные синапсы (участвующие в секреции АДГ и окситоцина
в капилляры задней доли гипофиза, секреции либеринов в капилляры
срединного возвышения);
—портальную систему кровотока между срединным возвышением и перед­
ней долей гипофиза.
ТИПОВЫЕ ФОРМЫ ПАТОЛОГИИ ГИПОФИЗА
Типовые формы патологии аденогипофиза
Критерии дифференцировки типовых расстройств аденогипофиза пред­
ставлены на рис. 28.4.
Уровень продукции и/или
эффектов гормонов
Масштаб поражения
аденогипофиза
Время возникновения
в онтогенезе
1
4
___________
.
-= -------- 1 ___________
--------- . . п?-----т ------г?:-------- 5
Парциальные Тотальные
Парциальные
>
«Ранние»
«Поздние»)
«Ранние»
Первичные
(гипофизарные)
Происхождение
Проявления
Гиперфункции
(гиперпитуитаризм)
Г ипофункции
> (гипопитуитаризм)
■
Болезни
Синдромы
^ ------ - ------Тотальные
НПоздние»!
Вторичные
(гипоталамические)
Патологические состояния
Рис. 28.4. Типовые формы гипофизарных эндокринопатий
• Уровень продукции гормона (определяют по его содержанию в жидкостях орга­
низма) и/или по выраженности его эффектов. По этому критерию различают:
—гипофункциональные состояния (гипопитуитаризм);
—гиперфункциональные состояния (гиперпитуитаризм).
• Масштаб поражения аденогипофиза и характер расстройств в организме.
По этому признаку выделяют:
—тотальный гипо- и гиперпитуитаризм (нарушение продукции и/или эффек­
тов действия всех гормонов аденогипофиза);
—парциальный гипо- и гиперпитуитаризм (расстройство синтеза и /
или эффектов одного гормона аденогипофиза);
—субтотальный (расстройство синтеза и/или эффектов нескольких гормо­
нов аденогипофиза) гипо- и гиперпитуитаризм.
• Бремя возникновения эндокринопатии в онтогенезе. В связи с этим диффе­
ренцируют:
—«ранние» формы эндокринопатий (выявляются до завершения периода
полового созревания);
—«поздние» формы (развиваются после завершения периода полового
созревания).
• Происхождение эндокринопатий. По этому признаку выделяют:
—первичные формы (гипофизарные, т.е. вызванные прямым повреждени­
ем самого аденогипофиза);
—вторичные (гипоталамические: нейрогенные, центрогенные, т.е. обу­
словленные нарушением церебропитуитарного звена в механизме ней­
роэндокринной регуляции).
• Проявления эндокринных форм патологии (клинические, биохимические
и др.). По этому критерию выделяют конкретные болезни, синдро­
мы, патологические состояния (например, болезнь Иценко—Кушинга,
синдром гипофизарного ожирения, гиперпролактинемия, гипофизарная
кахексия — болезнь Симмондса).
Отдельные формы патологии аденогипофиза
Нозологические формы аденогипофиза характеризуются либо гипер-, либо
гипопитуитаризмом.
Гипопитуитаризм
Гипопитуитаризм — типовая форма патологии аденогипофиза, характе­
ризующаяся недостаточностью содержания и/или эффектов одного либо
нескольких его гормонов.
Причины гипопитуитаризма
Рис. 28.5. Наиболее частые причины недостаточности аденогипофиза (гипопитуитаризм)
Наиболее частые причины гипопитуитаризма показаны на рис. 28.5. К ним
относят:
— разрушение аденогипофиза (полное или частичное), например, новооб­
разованиями (злокачественными, доброкачественными, метастазами
других опухолей) или множественными кистами, при хирургических
вмешательствах (например, при удалении аденомы или кисты гипофи­
за), вследствие облучения аденогипофиза (например, при радиотерапии
рядом расположенных опухолей или новообразований самого аденоги­
пофиза), в результате реакций иммунной аутоагрессии (например, лим­
фоцитарный аутоагрессивный гипофизит);
—кровоизлияние в ткань гипофиза (например, у пациентов с артериальной
гипертензией или в результате травмы);
— ишемию и некроз гипофиза (например, после большой кровопотери);
—врожденные пороки развития аденогипофиза (аплазия гипофиза, энцефа­
лоцеле основания мозга и др.);
— генетические дефекты специализации отдельных клонов в клетах аденоги­
пофиза, вследствие чего нарушается образование СТГ, гонадотропных
гормонов и ТТГ;
—воспалительные процессы в гипофизе (например, при туберкулезе
или сифилисе);
—гипотрофию и/или гипоплазию аденогипофиза (синдром «пустого турец­
кого седла»).
Виды гипопитуитаризма
Основные виды недостаточности аденогипофиза представлены на рис. 28.6.
Парциальный гипопитуитаризм
«Чистых» парциальных форм недостаточности аденогипофиза обычно
не встречается. У пациента доминируют признаки недостаточности одного
из гормонов аденогипофиза, сочетающиеся с дефицитом эффектов других.
Наиболее часто в клинической практике выявляются:
—гипофизарная карликовость (гипофизарный нанизм, микросомия, наносомия), которая развивается при дефиците СТГ и/или соматолиберина (см. статьи «Гормоны роста» и «Соматолиберин» в приложении
«Справочник терминов»);
—гипофизарный гипогонадизм (гипофизарный евнухоидизм), наблюдаю­
щийся при дефиците и/или дефектах ФСГ, лютропина (Л Г) и их рецеп­
торов (см. статьи «Гормоны гонадотропные», «Гонадолиберин»
и «Гипогонадизм» в приложении «Справочник терминов»);
—гипофизарное (нейроэндокринное) ожирение (см. раздел «Ожирение»
в гл. 10 «Нарушения липидного обмена» и статью «Лептин» в приложе­
нии «Справочник терминов»);
—нарушения половой дифференцировки (см. статьи «Нарушения половой
дифференцировки», «Гермафродитизм», «Недостаточность ферментов»
в приложении «Справочник терминов»);
—пангипопитуитаризм. Понятие «тотальный гипопитуитаризм» (пангипопитуитаризм) применяют при повреждении 75—90% паренхимы адено­
гипофиза. Примеры этой патологии — послеродовой гипопитуитаризм
(синдром Шиена) и гипофизарная кахексия (болезнь Симмондса).
Рис. 28.6. Наиболее частые виды недостаточности аденогипофиза (гипопитуитаризм)
Проявления и механизмы гипопитуитаризма
Гипопитуитарные синдромы клинически весьма вариабельны, зависят
от масштаба и степени поражения гипофиза, основной патологии и многих
других факторов.
Вместе с тем, как правило, имеются три группы признаков гипопитуита­
ризма — признаки:
—полигормональной недостаточности;
нейросоматических расстройств;
психических нарушений (рис. 28.7).
Тотальный гипопитуитаризм
Полигормональная недостаточность
в результате дефицита гормонов
гипофиза
ТТГ
- гипофизарный
гипотиреоз;
I Гонадотропинов
-
евнухоидизм;
женский инфантилизм;
гипофизарное ожирение;
адипозогенитальная
дистрофия
1соматотропина!
- снижение массы тела;
- изменения кожи и ее
дериватов;
- костные дистрофии
Нейросоматические
расстройства
- гипотермия;
- нейровегетативные
расстройства;
-признаки повышения
внутричерепного
давления
кортикотропина
- гипофизарный
гипокортицизм
Психические
нарушения
- апатия;
- депрессия;
- расстройства
психики
Рис. 28.7. Основные проявления тотального гипопитуитаризма
Признаки полигормональной недостаточности — результат дефицита и/или
дефекта структуры конкретных гормонов аденогипофиза.
• СТГ-недостаточность проявляется:
—прогрессирующей потерей массы тела (в среднем 2—6 кг в месяц, в тяже­
лых случаях — до 25—30 кг);
—изменениями кожи и ее производных (сухость, морщинистость кожи,
ломкость волос и ногтей);
—дистрофическими и дегенеративными изменениями костной ткани (декаль­
цификация, остеопороз, повышенная ломкость костей, выпадение зубов).
• ТГГ-дефицит характеризуется появлением признаков гипотиреоза:
—вялости, апатии, гиподинамии;
—снижения интеллекта;
—низкой физической активности;
—дистрофических изменений в органах.
• Недостаточность гонадотропинов проявляется симптомами евнухоидизма
и инфантилизма:
—атрофией внутренних и наружных половых органов;
—инволюцией характерных половых признаков (у женщин — молочных
желез; у мужчин — яичек, предстательной железы, пениса; у тех и дру­
гих — исчезновение характерного оволосения; утрата полового чувства,
снижение половой потенции; у женщин отсутствие лактации и восста­
новления менструации при развитии синдрома после родов).
• Дефицит АКТГ приводит к развитию гипофизарного гипокортицизма, прояв­
ляющегося дефицитом эффектов глюко- и минералокортикоидов, а также
андрогенных стероидов. Для гипокортицизма характерны:
—общая слабость;
—мышечная гипотония, гиподинамия;
—снижение резистентности организма к возбудителям инфекций;
—артериальная гипотензия;
—гипогликемия на фоне относительного гиперинсулинизма;
—диспепсические расстройства (отсутствие аппетита, тошнота и рвота,
боли в животе в связи со спазмом ГМК кишечной трубки).
Нейросоматические расстройства проявляются признаками поражения ядер
гипоталамуса:
—гипотермией;
—вегетативными нарушениями (преходящая гипогликемия, полиурия,
гипотензивные реакции, коллапсы, тетанические судороги и др.);
— ограничением полей зрения, снижением остроты зрения;
— головными болями.
Три последних проявления вызваны повышением внутричерепного давления
(например, при внутричерепном росте новообразований или кровоизлияниях).
Психические нарушения наблюдаются при всех указанных выше разновид­
ностях гипоталамо-гипофизарной недостаточности. Чаще всего они характе­
ризуются:
— апатией и безучастным отношением к происходящему вокруг;
—депрессией;
—снижением эмоционального уровня оценки событий;
—психическими расстройствами (например, галлюцинациями, параноид­
ным психозом).
Гиперпитуитаризм
Гиперпитуитаризм — типовая форма патологии аденогипофиза, характери­
зующаяся избытком содержания и/или эффектов одного либо нескольких
его гормонов.
Причины гиперпитуитаризма
• Аденома передней доли гипофиза (в большинстве случаев гиперпитуитариз­
ма).
• Злокачественные опухоли аденогипофиза.
• Патология гипоталамуса (сопровождающаяся гиперпродукцией либеринов
и/или гипопродукцией статинов).
Виды гиперпитуитаризма
Гиперпитуитаризм характеризуется, как правило, парциальной патологией
(рис. 28.8). В практике врача наиболее часто встречаются следующие виды.
• Гипофизарный гигантизм или макросомия (чрезмерное увеличение роста,
размеров тела и внутренних органов; по времени возникновения в онто­
генезе является ранней формой эндокринопатии). Инициальные звенья
патогенеза гипофизарного гигантизма:
----------------
Виды гиперпитуитаризма
—
,,
11арциальныи
'г
^т
Гипофизарный Акромегалия
гигантизм
.г
------------
1 отальныи
-
1г
Гиперпролактинемия
I Гипермеланотропинем ия
..
(?)
Гипофизарный
гипертиреоидизм
Синдром
Гипофизарный
гипофизарного
гиперкортицизм
(«истинного»)
(болезнь Иценко-Кушинга)
преждевременного
полового развития
Рис. 28.8. Наиболее частые виды гиперпитуитаризма
—центрогенные (результат поражения нейронов коры и/или гипотала­
муса, влекущего за собой гиперпродукцию соматолиберина и СТГ и/
или меньшую выработку соматостатина);
—первично-железистые (гипофизарное; следствие повышенного синтеза
СТГ ацидофильными клетками аденогипофиза);
—постжелезистые (среди них наиболее часто встречается рецепторный, обу­
словленный повышенной чувствительностью тканей и органов к СТГ).
Проявления гигантизма и их механизмы (рис. 28.9):
Рис. 28.9. Основные проявления гипофизарного гигантизма
—увеличение роста, превышающее норму (обычно выше 200 см у мужчин
и 190 см у женщин). Описаны случаи роста 190 см в 10 лет и 250 см
в 18 лет. Причина — интенсивное эпифизарное и периостальное увеличе­
ние размера костей (главным образом линейного) под действием СТГ;
— несоответствие величины и массы внутренних органов размерам тела. Чаще
органы также увеличены (спланхномегалия); реже — относительно
уменьшены в сравнении со значительно ускоренным ростом. В связи
с этим возможно развитие функциональной недостаточности отдельных
органов (например, сердца и печени). Причина — разная чувствитель­
ность клеток, органов и тканей к СТГ В органах с высокой чувствитель­
ностью интенсивно гипертрофируются паренхима и фиброзная ткань;
— непропорциональное развитие мышц (при возникновении заболевания сте­
пень развития мышц обычно соответствует размерам тела; затем начинает
отставать — развивается слабость мышц, их гипотония, нередко гипотро­
фия; при физической нагрузке наступает быстрое утомление). Причины:
—дегенеративные изменения миофибрилл;
—разрастание соединительной ткани;
— гипергликемия, нередко СД. Причина — прямое гипергликемизирующее
действие СТГ и развитие относительного или абсолютного гипоинсулинизма на фоне повышенного уровня СТГ;
— гипогенитализм. Характеризуется недоразвитием внутренних и внешних
половых органов, нередко бесплодием. Причина — недостаточность син­
теза и/или эффектов гонадотропинов;
—психические расстройства (эмоциональная неустойчивость, раздражи­
тельность, нарушение сна, снижение умственной работоспособности,
психастения). Причины:
о поражение нейронов коры и подкорковых центров, определяющих
эмоциональное состояние индивида;
о длительная негативная стресс-реакция, вызванная у пациента фактом
заболевания;
о гипертиреоз, который нередко сочетается с гигантизмом.
• Акромегалия (от греч. акгоз — крайний, отдаленный, те§аз — огромный) —
диспропорциональное увеличение размера отдельных частей тела (чаще
кистей рук, стоп, внутренних органов), сочетающееся с существенными
нарушениями жизнедеятельности организма. По времени возникновения
в онтогенезе — поздняя форма эндокринопатии. Она развивается после
того, как завершается окостенение эпифизарных хрящей. Причина — повы­
шение уровня и/или эффектов СТГ. Проявления акромегалии, их механиз­
мы представлены на рис. 28.10:
[Повышение концентрации в крови и/или эффектов соматотропного гормона
1Г
Увеличение разме­
ров кистей и стоп
1Г
4
4
Половые
расстройства
Расстройства
психики
Парестезии
»
Огрубление
черт лица
1Г
Нарушения об­
мена веществ
»Г
Г
1
Спланхномегалия, Утолщение
Уплотнение
макроглоссия
кожи
мягких тканей
Рис. 28.10. Проявления акромегалии
—увеличение размеров кистей и стоп (за счет периостального роста костей,
стимулируемого СТГ);
— огрубение черт лица (увеличение нижней челюсти, носа, надбровных дуг,
скул; формирование толстых кожных складок);
—увеличение размеров внутренних органов (сердца, легких, печени, почек,
селезенки); на раннем этапе болезни функция их адекватна, но посте­
пенно развиваются признаки полиорганной недостаточности, сочета­
ющиеся с гиперплазией элементов соединительной ткани;
—утолщение кожи, уплотнение мягких тканей (в связи с разрастанием
их соединительнотканных элементов);
—увеличение языка (макроглоссия) с отпечатками зубов на нем;
—расстройства обмена веществ:
о углеводного — характеризуются стойкой гипергликемией (более
чем у 50% пациентов) и нередко — СД (примерно у 10% пациентов);
о жирового — проявляются повышением в крови уровня холестерина,
лецитина, ВЖК, КТ тел, ЛП. Причина — липолитическое, анаболиче­
ское, контринсулярное действие избытка СТГ;
—половые расстройства (снижение полового влечения, импотенция; у жен­
щин — дисменорея и нередко галакгорея). Причины — недостаточность
синтеза и/или эффектов гонадотропинов, при галакгорее — гиперсекреция
пролакгина;
—парестезии, особенно в области кистей и стоп (акропарестезии).
Механизмы: сдавление нервных стволов, проходящих в костных каналах
или углублениях, в связи с периостальным утолщением костей, гипер­
трофией и уплотнением мягких тканей.
• Гиперпролактинемия (см. статьи «Пролактин» и «Гиперпролактинемия»
в приложении «Справочник терминов»),
• Синдром гипофизарного преждевременного полового развития. Характе­
ризуется появлением отдельных или всех вторичных половых признаков,
в некоторых случаях наступлением половой зрелости (у девочек до 8-,
у мальчиков до 9-летнего возраста). Причина — преждевременная секреция
гонадолиберинов или гиперсекреция гонадотропинов.
Типовые формы патологии нейрогипофиза
Патология нейрогипофиза приводит к нарушениям водного баланса (см.
главу 12 «Типовые нарушения водного обмена», т. 1) в результате АДГ-эндокринопатий (недостаточность или избыточность эффектов АДГ).
К ним относятся центральные формы НД (недостаточность эффектов АДГ)
и синдром неадекватной секреции АДГ (избыточность эффектов АДГ).
Несахарный диабет
НД (несахарное мочеизнурение) развивается в результате недостаточной
эффективности АДГ. Разные формы заболевания (этиология, клинические
проявления, лечение) охарактеризованы в статье «Диабет несахарный» (см.
приложение «Справочник терминов»).
Патогенез несахарного диабета
Инициальные и ключевые звенья патогенеза представлены на рис. 28.11.
Основные проявления несахарного диабета и их механизмы
Они показаны на рис. 28.12:
—полиурия. Суточный диурез составляет обычно 3—15 л, иногда до 20—30 л.
При этом моча имеет очень низкую осмоляльность. Механизмы:
о недостаточная эффективность АДГ, что обусловливает снижение реаб­
сорбции жидкости в дистальных отделах канальцев почек. Вследствие
этого почки в большом количестве выводят неконцентрированную мочу
(относительная плотность всех порций равна обычно 1,000—1,005);
—гиперосмоляльность плазмы крови (более 290 мосм/кг Н20 ) внутриклеточ­
ной и других биологических жидкостей. Это обусловливает повышенную
фильтрацию жидкости в клубочках почек при нормальной реабсорбции
ионов, неорганических и органических соединений, гипогидратацию
клеток и тканей;
—гемоконцентрация, обусловленная полиурией;
—гипернатриемия. Механизм:
о активация выработки, высвобождения и эффектов альдостерона
в условиях нарастающей значительной гипогидратации организма
и развития гиповолемии. Это ведет к увеличению реабсорбции № +
в почках и увеличению его содержания в плазме крови;
—полидипсия (повышенное потребление жидкости, обусловленное пато­
логически усиленной жаждой). Количество выпиваемой пациентами
жидкости колеблется от 3 до 15, иногда до 30 л/сут. При отсутствии воз­
можности восполнить утрату жидкости развивается угрожающая жизни
гиперосмоляльная гипогидратация организма. Механизм:
о активация нейронов центра жажды гипоталамуса вследствие гиперосмоляльности плазмы крови и гипогидратации клеток организма.
Известно, что дефицит уже 1—2% воды в организме вызывает ощуще­
ние жажды, особенно при гиперосмоляльности крови.
Дефицит АДГ и/или его эффектов
4
„
1
IПолиурия1 Г иперосмоляльность |Гипернатриемия |1 Полидипсия
биологических
жидкостей организма
Рис. 28.12. Основные проявления несахарного диабета
Синдром неадекватной секреции АД Г
Синдром неадекватной секреции АДГ (СНСАДГ) развивается вследствие
избыточного действия АДГ и характеризуется олигурией и отеками.
Характеристика синдрома приведена в статье «Синдром неадекватной
секреции АДГ» (см. приложение «Справочник терминов»).
Патогенез СНСАДГ. Ведущую роль в развитии СНСАДГ играют два взаи­
мосвязанных инициальных звена — центрогенное и первично-железистое.
Центрогенное звено патогенеза СНСАДГ характеризуется нейрогенной
корково-подкорковой стимуляцией образования АДГ в гипоталамусе и его
транспорта в нейрогипофиз.
Первично-железистое. В этом случае речь идет о двух вариантах патоге­
неза — об избыточной продукции и о нейросекреции АДГ нейронами гипо­
таламуса и эктопической секреции гормона (например, мелкоклеточными
карциномами легких).
Проявления СНСАДГ и их механизмы показаны на рис. 28.13.
___________
М
Неадекватная секреция АДГ
Н
1Г
Увеличение Гипонатриемия
массы тела
________________
и
Повышение
содержания
№ * в моче
*Г
Психоневрологические
расстройства
Рис. 28.13. Основные проявления синдрома неадекватной секреции АДГ
• Олигурия. Механизм развития олигурии:
—значительная активация реабсорбции жидкости в дистальных отделах
почечных канальцев под влиянием АДГ;
—АДГ взаимодействует с У2-рецепторами эпителия в дистальных отделах
канальцев и собирательных трубочках почек. Это обусловливает образо­
вание цАМФ, встраивание водных каналов в цитолемму и торможение
экскреции жидкости, что приводит к увеличению реабсорбции воды
и снижению диуреза.
• Нарастание массы тела. Механизм:
—задержка жидкости в организме (гипергидратация). Вода накапливает­
ся в тканях и сосудистом русле, что и увеличивает массу тела. Важно,
что отеки при этом не развиваются в связи с уменьшением [Ыа+1
в интерстиции.
• Гипонатриемия. Считается кардинальным признаком синдрома. [Иа+]
снижается до 130—135 мэкв/л, а осмоляльность плазмы до 270 носмоль/кг.
Механизм:
—гипергидратация организма, вызванная олигурией, в сочетании с гиперволемией.
• Повышение содержания натрия в моче (обычно более 20 мэкв/л).
Механизм:
—гипергидратация организма, стимулирующая экскрецию Ыа+ почками.
• Психоневрологические расстройства. Характеризуются апатией, вялостью,
нарушением сознания, нередко судорогами. Механизм:
—набухание нейронов мозга, что характерно для состояния так называ­
емого водного отравления.
НАРУШЕНИЯ ФУНКЦИЙ НАДПОЧЕЧНИКОВ
Надпочечники — парные эндокринные железы, состоящие из коркового
вещества (мезодермального происхождения) и мозгового (нейроэктодермаль­
ного генеза). Фактически это две железы — кора, на долю которой приходится
около 80% массы железы, и мозговое вещество.
Кора надпочечников синтезирует кортикостероиды, хромаффинные клет­
ки мозговой части — катехоловые амины.
Каждый надпочечник в норме имеет массу около 4 г как у мужчин, так
и у женщин. При остром стрессе или гиполипидемии масса надпочечни­
ков может существенно уменьшаться. Напротив, при длительном стрессе
или некоторых хронических заболеваниях наблюдается гипертрофия и гипер­
плазия надпочечников с увеличением массы в 1,5—2 раза.
Кортикостероиды
В коре надпочечника синтезируются минералокортикоиды (основной
из них — альдостерон), глюкокортикоиды (основной глюкокортикоид — кор­
тизол: на его долю приходится 80% всех глюкокортикоидов; остальные глюко­
кортикоиды — 20% кортизон, кортикостерон, 11-дезоксикортизол и 11-дезок­
сикортикостерон) и дегидроэпиандростерон (рис. 28.14).
Рис. 28.14. Биосинтез стероидных гормонов в коре надпочечников
Катехоловые амины
Катехоламины (преимущественно адреналин) синтезируют хромаффинные
клетки.
Адреналин и норадреналин инкретируются в кровь из хромаффинных
клеток при активации симпатической нервной системы.
Катехоламины имеют широкий спектр эффектов (воздействие на гликогенолиз, липолиз, глюконеогенез, влияние на ССС). Вазоконстрикция, пара­
метры сокращения сердечной мышцы и другие эффекты катехоловых аминов
реализуются через а- и р-адренергические рецепторы на поверхности клетокмишеней (ГМК, секреторные клетки, кардиомиоциты). Недостаточность
катехоламинов в мозговой части надпочечников крайне редко приводит к раз­
витию серьезной патологии, но чрезмерная продукция адреналина (например,
при феохромоцитоме) «гарантирует» развитие артериальной гипертензии.
Типовые формы патологии надпочечников
Типовые формы патологии надпочечников подразделяются на две боль­
шие группы — гиперфункциональные и гипофункциональные состояния
(рис. 28.15).
------------- Этиологические ф акторы--------------->Г
17
Гипофункциональные состояния
Гиперфункциональные состояния
надпочечников
надпочечников
I
Л
Парциальные
Парциальные
т
|Гиперкортицизм| Г иперпродукция
катехоламинов
Т
мозговым
|Гиперальдостеронизм
веществом
*г
Синдром/болезнь Кортико-генитальный
Иценко-Кушинга
синдром
Тотальные I
Г ипоальдостеронизм
Надпочечниковая
недостаточность
(болезнь Аддисона)
Рис. 28.15. Типовые формы патологии надпочечников
Гиперфункциональные состояния
Кора надпочечников. К гиперфункциональным состояниям коры надпо­
чечников относятся синдромы:
—гиперальдостеронизм;
—гиперкортизолизм;
—адреногенитальный синдром.
Мозговая часть надпочечников. Гиперкатехоламинемия, как правило, наблю­
дается при опухоли из хромаффинных клеток — феохромоцитоме.
Гипофункциональные состояния
К гипофункциональным состояниям относится недостаточность коры над­
почечников (например, болезнь Аддисона и гипоальдостеронизм).
Гиперальдостеронизм
Гкперальдостеронизм — общее название синдромов, возникающих вследствие
гинерсекреиии и/или нарушений обмена альдостерона и характеризующихся
наличием отеков, асцита, гипокалиемии и реноваскулярной артериальной
гипертензии.
Синдром гиперальдостеронизма может быть первичным или вторичным.
В некоторых случаях развивается псевдогиперальдостеронизм.
Первичный гиперальдостеронизм
Причины:
— альдостеронпродуцирующая аденома клубочковой зоны коры одного из над­
почечников;
— первичная гиперплазия клубочковой зоны коры надпочечников. При этих
состояниях развивается синдром Конна (около 80% всех случаев пер­
вичного гиперальдостеронизма). Синдром Конна — расстройство, харак­
теризующееся чрезмерной секрецией альдостерона и проявляющееся
головными болями, полиурией, слабостью, артериальной гипертензией,
гипокалиемическим алкалозом, гиперволемией и пониженной активно­
стью ренина.
Проявления гиперальдостеронизма, их причины и механизмы (рис. 28.16).
Основные проявления гиперальдостеронизма:
—высокий уровень альдостерона в крови; причина — гиперпродукция гормо­
на в клубочковой зоне коры надпочечников;
— снижение содержания (активности) ренина и ангиотензина II в плаз­
ме крови. Причина — подавление активности ренин-ангиотензиновой
системы в условиях гиперальдостеронизма и гиперволемии, потенци­
рующих торможение синтеза и секреции ренина;
—гипернатриемия и гипокалиемия. Причина — активация реабсорбции № +
и стимуляции экскреции К+ в канальцах почек в результате непосред­
ственного влияния на них избытка альдостерона;
—артериальная гипертензия. Причина и механизм —увеличение [Ыа1] в плазме
крови (гиперосмия), что обусловливает цепь следующих явлений: актива­
ция осморецепторов и стимуляция секреции АДГ в задней доле гипофиза
-> повышение реабсорбции жидкости в дистальных отделах канальцев
почек, пропорциональное гиперосмии -> увеличение ОЦК в суженном
сосудистом русле -> повышение сердечного выброса и увеличение АД;
—снижение остроты зрения (иногда слепота). Причина — нарушение крово­
снабжения сетчатки глаза в связи с изменениями в ее микрососудах (утол­
щение стенки, микроаневризмы, повышенная извитость) и расстройства­
ми микрогемоциркуляции (замедление тока крови, ишемия, стаз);
—изменение показателей мочи — гипостенурия (из-за низкого содержания
№ + в моче), олигурия на начальном этапе болезни (в связи с повышенной
реабсорбцией № +), полиурия и никтурия на последующих этапах заболе­
вания, протеинурия. Причины — дистрофия эпителия почечных каналь­
цев и гипосенситизация рецепторов эпителия канальцев почек к АДГ
вследствие снижения уровня К+ в клетках;
—расстройства нервно-мышечной возбудимости: парестезии, мышечная сла­
бость и гипотония, судороги, вялые (нейрогенные) параличи. Причины —
гипернатриемия, увеличение уровня На1 в миоцитах и нервных клетках,
гипокалиемия, дефицит К+ в клетках, алкалоз. Указанные отклонения
приводят к нарушениям электрогенеза и дистрофическим изменениям.
Гиперальдо стеронизм ——
■
г
Г иперальдостеронемия
г
Гипернатриемия| Гипокалиемия1 |Расстройства зрения
*
Активность ренина в крови:
-н и з к а я при первичном;
-вы сокая при вторичном
гиперальдостеронизме
Г
г
1Г
Расстройства
Артериальная Нефропатии |
нервно-мышечной гипертензия
возбудимости
Вторичный гиперальдостеронизм
Причины вторичного гиперальдостеронизма — состояния, вызывающие сни­
жение ОЦК и/или АД. Это обусловливает активацию ренин-ангиотензиновой
системы и вторично — гиперпродукцию альдостерона обоими надпочечника­
ми. Наиболее часто к этому приводят:
—сердечная недостаточность;
—нефроз (с гипоальбуминемией), оба указанных выше состояния сопро­
вождаются ишемией почек;
—гломерулонефрит;
—гидронефроз;
—нефросклероз;
—цирроз печени;
—полиурия.
Последствия вторичного гиперальдостеронизма
Названные и другие состояния приводят к стимуляции синтеза ренина
и избыточному образованию ангиотензина (в отличие от первичного гипе­
ральдостеронизма! ).
Проявления вторичного гиперальдостеронизма:
—высокий уровень альдостерона в крови;
—повышенная активность ренина плазмы крови;
—другие проявления, аналогичные тем, которые наблюдаются при первич­
ном гиперальдостеронизме.
Гиперкортизолизм
Синдромы гиперкортизолизма (гиперкортицизма) возникают в результате
существенною увеличения уровня кортикостероидов (в первую очередь
кортизола) в крови.
Виды и причины гиперкортицизма
Синдром Иценко—Кушинга. Характеризуется высоким уровнем кортизола
в крови при низком содержании в ней АКТГ. Причина синдрома — гиперпро­
дукция глюкокортикоидов клетками пучковой зоны коры надпочечников.
Характеристика синдрома Иценко-Кушинга приводится в статье «Синдром
Иценко—Кушинга» (см. приложение «Справочник терминов»).
Болезнь Иценко-Кушинга. Характеризуется высоким содержанием в крови
как АКТГ, так и глюкокортикоидов (см. статью «Болезнь Иценко-Кушинга»
в приложении «Справочник терминов»).
Синдромы эктопической (гетеротопной) гиперсекреции АКТГ (см. статьи
«Синдром паранеопластический эндокринный» и «Аденоматоз семейный
полиэндокринный» в приложении «Справочник терминов»),
Ятрогенный синдром Иценко-Кушинга. Развивается при длительном введении
в организм препаратов глюкокортикоидов с лечебной целью. При этом, как пра­
вило, наблюдается гипотрофия коркового вещества обоих надпочечников.
Основные проявления гиперкортицизма и их причины показаны на рис. 28.17.
Рис. 28.17. Основные проявления гиперкортизолизма. * Частота проявления (сред­
няя арифметическая), %
• Артериальная гипертензия. Выявляется в среднем у 75% пациентов с гиперкортизолизмом. Причины:
—сосудистые и другие эффекты кортизола (включая задержку натрия);
—увеличение выработки альдостерона в клубочковой зоне коры надпо­
чечника и его уровня в крови (наблюдается при опухолях, гипертрофии
и гиперплазии коры надпочечников с поражением клубочковой и пуч­
ковой его зон).
• Кушингоидная внешность. Наблюдается не менее чем у 85—90% пациентов
как результат липосинтетического эффекта глюкокортикоидов. При избы­
точном образования жира происходит его перераспределение с накопле­
нием в области шеи («бизоний горб»), живота и груди при уменьшении
количества жира на конечностях. Лицо при этом приобретает округлую —
«лунообразную» — форму.
• Мышечная слабость и гиподинамия. Наблюдаются более чем у 80% пациен­
тов. Причины:
—гипокалиемия;
—уменьшение внутриклеточного [К+] и увеличение внутриклеточного [№ +];
—уменьшенное содержание глюкозы в мышечных волокнах (обусловлено
контринсулярным эффектом избытка кортизола);
—дистрофические изменения скелетных мышц. Это обусловливает нару­
шения электрогенеза, электромеханического сопряжения, что ведет
к нарушению контрактильного процесса.
• Остеопороз. Выявляется почти у 75% больных. Причина:
—ингибирующий эффект кортизола на синтез коллагена и всасывание
кальция. Этим обусловлено увеличение катаболизма белков костной
ткани, торможение протеосинтеза в костях, нарушения фиксации Са2+
белковой матрицей кости.
• Гипергликемия и нередко СД. Выявляются, соответственно, примерно у 75
и 20% пациентов с гиперкортизолизмом. Причина — контринсулярные
эффекты избытка кортизола.
• Красно-багровые или фиолетовые «полосы растяжения» — стрии — на коже
(чаще на животе, плечах, бедрах, молочных железах). Наблюдаются более
чем у половины пациентов. Причины:
—активация катаболизма белков и угнетение протеосинтеза в ткани кожи,
что ведет к дефициту в ней коллагена, эластина и других белков, форми­
рующих структуры кожи;
—просвечивание в области стрий микрососудов подкожной клетчатки.
Багровый или фиолетовый цвет стрий обусловлен застоем венозной
крови в микрососудах клетчатки.
• Снижение противоинфекционной резистентности организма. У пациентов
с гиперкортизолизмом часто развиваются инфекции: пиелонефриты,
циститы, гнойничковые поражения кожи, трахеобронхиты и др. Причина —
иммунодепрессия, вызванная избытком глюкокортикоидов.
Кортико-генитальный (адреногенитальный) синдром
Адреногенитальный синдром — типовая форма патологии, характеризую­
щаяся дисфункцией коры надпочечников (чрезмерной секрецией андроге­
нов) и проявляющаяся признаками вирилизации.
Практически все случаи адреногенитального синдрома — врожденные (см.
статью «Синдром адреногенитальный» в приложении «Справочник терминов»).
Причина — недостаточность одного из ферментов, необходимых для син­
теза кортизола. Дефицит кортизола стимулирует выработку АКТГ, что при­
водит к гиперплазии коры надпочечников и избыточной продукции АКТГзависимых стероидов, синтез которых при данной недостаточности фермента
не нарушен (в основном надпочечниковых андрогенов: дегидроэпиандростерона, андростендиона и тестостерона).
Виды
Разновидности адреногенитального синдрома представлены на рис. 28.18.
Рис. 28.18. Виды кортико-генитального синдрома
Врожденный адреногенитальный синдром. Встречается в 95% случаев гипер­
плазии надпочечников. Клинические варианты синдрома:
—вирильная форма (простая: неосложненная вирилизирующая форма);
— сольтеряющая форма (вирилизм с гипотензивным синдромом);
— гипертензивная форма (вирилизм с гипертензивным синдромом).
(Причины и механизмы см. статьи «Синдром адреногенитальный»
и «Недостаточность ферментов» в приложении «Справочник терминов».)
Приобретенный адреногенитальный синдром
Причина синдрома — андростерома (доброкачественная или злокачествен­
ная опухоль, развившаяся из аденоцитов сетчатой зоны коры надпочечника).
Такие опухоли синтезируют избыточное количество андрогенов. Андростерома
может развиться в любом возрасте.
Проявления приобретенного кортико-генитального синдрома могут отли­
чаться от проявлений врожденной формы нормальным или незначительным
повышением содержания в крови АКТГ.
Характерные проявления кортико-генитального синдрома представлены
на рис. 28.19.
Рис. 28.19. Основные проявления кортико-генитального синдрома
Общие проявления
• Врожденная вирилизация наружных половых органов у девочек (пенисо­
образный клитор, мошонкообразные большие половые губы). Внутренние
половые органы под влиянием андрогенов не меняются: матка и яичники
развиваются, как правило, в соответствии с возрастной нормой. Этот при­
знак обозначают так же, как женский псевдогермафродитизм, или вирилизм
по гетеросексуальному типу. Причина — избыток в организме андрогенов,
вызывающих маскулинизацию наружных половых органов.
• Макросомия (увеличенные масса тела и рост новорожденных). Наблюдается
как у девочек, так и у мальчиков. В первые годы жизни больные дети растут
быстрее, чем их сверстники. Однако в 12—14 лет происходит прекращение
эпифизарного роста трубчатых костей, и такие дети остаются низкорос­
лыми, непропорционального телосложения, с сильно развитой мускулату­
рой. Причина — анаболическое действие избытка андрогенов.
• Гирсутизм — рост волос на теле по мужскому типу — ранний признак вири­
лизма (он может появиться в возрасте 2—5 лет) в виде избыточного оволо­
сения: на лице (усы, борода), лобке, в подмышечных впадинах, на груди,
спине, конечностях. Причина — гиперпродукция андрогенов и осущест­
вление их действия.
• Маскулинизация — развитие мужских вторичных половых признаков у инди­
видов генетически женского пола. Это проявляется атрофией (гипотрофи­
ей) молочных желез и матки, различными нарушениями менструального
цикла или отсутствием менструаций, телосложением по мужскому типу,
низким голосом, изменением поведения (по «мужскому типу»: появление
властолюбия, стремления к лидерству, увлечение техникой, мужскими
видами развлечений и т.п.). Причина — высокий уровень андрогенов
в крови и их действие на ткани и клетки-мишени.
• Раннее ложное половое созревание мальчиков по изосексуальному типу.
Проявляется преждевременным формированием вторичных половых при­
знаков и наружных половых органов, сохранением темпа развития поло­
вых желез, свойственного данному возрасту (отсутствие сперматогенеза),
и изменением телосложения (низкий рост, сильно развитая мускулатура,
короткие мускулистые ноги — феномен «ребенок-Геркулес»),
Проявления, свойственные сольтеряющей форме кортико-генитального син­
дрома (помимо указанных выше).
• Артериальная гипотензия: стойкое уменьшение АД ниже нормы; встречает­
ся практически у всех пациентов.
• Коллапсы. Выявляются у многих пациентов.
Причины гипотензии и коллапсов:
—гипонатриемия;
—гиперкалиемия;
—гиповолемия;
—гипогидратация организма вследствие дефицита альдостерона и его
меньшего участия в регуляции водно-солевого обмена.
Проявления, характерные для гипертензивной формы кортико-генитального
синдрома (помимо указанных выше общих проявлений).
Артериальная гипертензия — стойкое увеличение АД сверх нормы.
Причина — избыток в крови минералокортикоида 11-дезоксикортикостерона
при недостаточности 11-гидроксилазы.
Гиперкатехоламинемия
Гиперкатехоламинемия наблюдается при опухолях из хромаффинных клеток —
феохромоцитомах, развивающихся как изолированно, так и при некоторых фор­
мах семейного полиэндокринного аденоматоза (см. статьи «Феохромоцитома»
и «Аденоматоз семейный полиэндокринный» в приложении «Справочник
терминов»).
Проявления гиперкатехоламинемии рассмотрены в разделе «Артериальная
гипертензия при эндокринопатиях надпочечников» в главе 23 и представлены
на рис. 28.20.
Надпочечниковая недостаточность
Гипофункциональные состояния надпочечников называют надпочечниковой
недостаточностью.
Виды
Виды надпочечниковой недостаточности представлены на рис. 28.21. Среди
множества состояний, сопровождающихся надпочечниковой недостаточно­
стью, наибольшее клиническое значение имеют:
— болезнь Аддисона;
—надпочечниковый криз;
— синдром Уотерхауса—Фридериксена;
— адренолейкодистрофия (сочетание лейкодистрофии и болезни Аддисона);
— аутоиммунный полигландулярный синдром;
—гипоальдостеронизм.
Рис. 28.21. Виды надпочечниковой недостаточности
Болезнь А д ди со н а
Болезнь Аддисона — хроническая первичная недостаточность коры надпо­
чечников, которая возникает при двустороннем поражении надпочечников,
приводящем к их недостаточности, т.е. к уменьшению (или прекращению)
секреции глюкокортикоидов и минералокортикоидов.
Причины болезни Аддисона:
— аутоагрессивный процесс (примерно в 80% случаев);
—туберкулез.
Как синдром, хроническая недостаточность коры надпочечников присутству­
ет при множестве наследуемых заболеваний. Характеристика заболевания приве­
дена в статье «Болезнь Аддисона» (см. приложение «Справочник терминов»).
Различают первичную, вторичную и ятрогенную формы болезни Аддисона.
Первичная форма болезни Аддисона (железистая, надпочечниковая) обусловлена
прямым поражением надпочечников, сопровождающимся гибелью его клеток (пре­
имущественно коркового вещества) и дефицитом кортикостероидов. Причины
первичной формы болезни Аддисона изложены в статье «Болезнь Аддисона» (см.
приложение «Справочник терминов») и показаны на рис. 28.22.
Причинные факторы
надпочечниковой недостаточности
-----------------------------------------
Первичной
Туберкулезное
поражение
1/ нтоксикации
Амилоидоз
Иммунная
аутоагрессия
♦
Врожденная
гипоплазия
-
Вторичной
*
Опухоли или
их метастазы
*
Опухоли
гипоталамуса
----------------------- 1
1
Врожденная недостаточность Кровоизлияние
ферментов синтеза стероидов
в гипофиз
*
Ишемия
гипофиза
------------Опухоли
гипофиза
Врожденная гипосенситизация Облучение в обгтс ти гипо­
к АКТГ
физа и/или гипсталамуса
Рис. 28.22. Основные причины хронической тотальной недостаточности надпочеч­
ников
Вторичная форма тотальной надпочечниковой недостаточности (центрогенная,
гипоталамо-гипофизарная). Причина — центрогенные расстройства в систе­
ме нейроэндокринной регуляции (поражение гипоталамуса и/или гипофиза,
сопровождающееся дефицитом кортиколиберина и/или АКТГ).
Ятрогенная форма болезни Аддисона. Причина — прекращение введения
в организм глюкокортикоидов после длительного их применения с лечебной
целью.
Развивающееся при этом состояние обозначают термином «синдром
отмены глюкокортикоидов» или ятрогенная надпочечниковая недостаточность.
Обусловлена эта форма патологии продолжительным угнетением функции
гипоталамо-гипофизарно-надпочечниковой системы и атрофией коры надпо­
чечников. Главным провоцирующим фактором ятрогенной надпочечниковой
недостаточности обычно бывает стресс, особенно длительный.
Общие проявления болезни Аддисона представлены на рис. 28.23.
I Дефицит гормонов надпочечников и/или чувствительности клеток к ним
4
Мышечная сла­
бость, утомляемость
1Г
4
Гемоконцентрация
*г
Г иперпигментация
кожи и слизистых
оболочек
Уменьшение
оволосения тела
Г
Артериальная
гипотензия
Полиурия |
Г ипогидратация
организма
1Г
Нарушения
полостного и
мембранного
пищеварения
>Мышечная слабость и быстрая физическая утомляемость. Причины:
—дисбаланс ионов в биологических жидкостях и миоцитах (уменьшение
[№ +], избыток К+; нарушение транслокации Са2+ через плазматическую
мембрану, мембраны саркоплазматической сети и митохондрий в мышцах).
Причина ионного дисбаланса: недостаточность эффектов альдостерона;
— гипогликемия, дефицит глюкозы в миоцитах;
— недостаточность энергообеспечения миоцитов. Причина гипогликемии
и энергодефицита миоцитов — недостаточность глюкокортикоидов:
—уменьшение массы миоцитов и дистрофические изменения в них. Причина —
недостаточность анаболического эффекта надпочечниковых андрогенов.
•Артериальная гипотензия. Механизмы развития артериальной гипотензии
при надпочечниковой недостаточности рассмотрены вразделе «Эндокринные
артериальные гипотензии» (см. гл. 23) и представлены на рис. 28.24.
Н а д п о че ч н и ко в а я недостаточность
,
Снижение
тонуса ГМК
артер>иол
1
Уменьшение чувствительности
рецепторов ГМК артериол
..
Снижение
сократительнои
функции миокарда
Гиповолемия
> Артериальная гипотензия
Рис. 28.24. Основные механизмы артериальной гипотензии при надпочечниковой
недостаточности
• Полиурия. Причина — снижение реабсорбции жидкости в канальцах почек
вследствие гипоальдостеронизма.
• Гипогидратация организма и гемоконцентрация. Причина — снижение объе­
ма жидкости в сосудистом русле, приводящее к гиповолемии.
• Нарушение полостного и мембранного пищеварения, нередко приводящие
к развитию синдрома мальабсорбции. Причины мальабсорбции:
—недостаточность секреции желудочного и кишечного сока (обусловлена
нарушениями в кровоснабжении стенок желудка и кишечника и дефи­
цитом кортикостероидов);
—профузный понос.
• Механизмы мальабсорбции: экскреция избыточного количества Ыач в про­
свет кишечника (в связи с гипоальдостеронизмом), приводящая к повы­
шению осмоляльности кишечного содержимого; это вызывает транспорт
жидкости в кишечник и так называемый осмотический понос, при кото­
ром теряется не только жидкость, но и вещества, не всосавшиеся через
стенку кишечника.
• Гипогликемия. Причина — дефицит глюкокортикоидов, приводящий к тор­
можению глюконеогенеза.
• Гиперпигментация кожи и слизистых оболочек. Они характерны для первич­
ной надпочечниковой недостаточности, при которой гипофиз не поражен.
Механизм — повышение (в условиях дефицита кортизола) секреции аде­
ногипофизом как АКТГ, так и меланоцитстимулирующего гормона.
• Уменьшение оволосения тела, особенно в подмышечной области
и на лобке. Причина — недостаточность надпочечниковых андрогенов.
Надпочечниковый криз
К острой недостаточности коры надпочечников относятся надпочечни­
ковый (гипоадреналовый) криз и аддисонов криз — осложнение болезни
Аддисона.
Причины надпочечникового криза:
— разрушение обоих надпочечников при травме (например, при автомобиль­
ной катастрофе, падении с большой высоты, попадании под завалы);
—двустороннее кровоизлияние в мозговое вещество и ткань коры надпочеч­
ников (например, в родах, при передозировке гепарина натрия, остро
или молниеносно протекающем сепсисе). В последнем случае говорят
о синдроме Уотерхауса—Фридериксена;
—удаление надпочечника, пораженного гормонпродуцирующей опухолью.
Недостаточность развивается в результате гипо- или атрофии коркового
вещества второго надпочечника.
Проявления острой недостаточности коры надпочечников представлены
на рис. 28.25.
Рис. 28.25. Основные проявления надпочечниковой недостаточности
• Острая артериальная гипотензия. Причины:
—острая недостаточность катехоламинов;
—дефицит минералокортикоидов;
—гиповолемия.
Указанные факторы вызывают снижение сердечного выброса, тонуса сосу­
дов и ОЦК.
• Гипогидратация организма. Причины:
—недостаток минералокортикоидов (обусловливает потерю организмом
натрия и воды);
—рвота (особенно выражена при тяжелых инфекциях и интоксикациях).
• Нарастающая недостаточность кровообращения (центрального, органотка­
невого, микрогемоциркуляции). Причины:
-О С Н ;
—снижение тонуса ГМК стенки артериальных сосудов;
—уменьшение ОЦК.
Каждое из названных изменений само по себе и особенно в совокупности
нередко приводит к коллапсу и обморокам. Острая тяжелая недостаточность
кровообращения — главная причина смерти большинства пациентов с гипоадреналовым кризом.
Гипоальдостеронизм
Характеристика парциальной надпочечниковой недостаточности — гипоальдостеронизма — приведена в статье «Гипоальдостеронизм» (см. приложе­
ние «Справочник терминов»).
Проявления гипоальдостеронизма показаны на рис. 28.26.
Рис. 28.26. Основные проявления гипоальдостеронизма
НАРУШЕНИЯ ФУНКЦИЙ ЩИТОВИДНОЙ ЖЕЛЕЗЫ
Щитовидная железа секретирует йодсодержащие гормоны (Т3 и Т4), а также
пептидный гормон кальцитонин.
Кальцитонин
Кальцитонин (тиреокальцитонин) вырабатывают светлые клетки щито­
видной железы. Функциональный антагонист кальцитонина — паратиреокрин (паратиреоидный гормон, ПТГ), синтезирующийся в главных клетках
паращитовидных желез. Эндокринные регуляторы обмена Са2+ и фосфатов
рассмотрены в разделе «Нарушения функций паращитовидных желез» (см.
гл. 28), а также в статьях «Кальцитонин» и «Паратиреокрин» (см. приложение
«Справочник терминов»).
Й одсодерж ащ ие гормоны
Йодсодержащие гормоны вырабатываются эпителиальными клетками стенки
фолликулов. Фолликулярные клетки щитовидной железы и секретируемые ими
гормоны относятся к системе гипоталамус — гипофиз — щитовидная железа.
Регуляторные взаимоотношения в этой системе представлены на рис. 28.27.
--------Г ипоталамус
Дофамин Т,гТиролиберин Т
.
— ► Аденогипофиз №
Глюкокортикоиды 4- —
Эстрогены Т ~
— * --------
ТиреотропныйТ
гормон
Соматотропный
гормон 4-
Т..Т. 1
Щитовидная железа
Рис. 28.27. Регуляция синтеза гормонов в гипоталамо-гипофизарно-тиреоидной
системе: Т — стимулирующий эффект;
— тормозящий эффект
Тиреоидны й статус организма
Выделяют три варианта тиреоидного статуса:
—эутиреоидию — отсутствие отклонений;
—гипотиреоз — характеризуется признаками недостаточности эндокрин­
ной функции;
—гипертиреоз — проявляется симптомами избыточного действия тиреоид­
ных гормонов.
Типовые формы патологии щитовидной железы
Многочисленные заболевания щитовидной железы, характеризующиеся
изменением уровня и/или эффектов йодсодержащих гормонов, относятся
к двум группам — гипертиреоидные состояния (гипертиреозы) и гипотиреоидные состояния (гипотиреозы).
Гипертиреозы
Гипертиреоидные состояния (гипертиреозы) характеризуются избыточ­
ными эффектами йодсодержащих гормонов в организме. Нередко эти
состояния называются также тиреотоксикозами.
Термин «тиреотоксикоз» обычно применяют для обозначения сходных,
но все же различающихся состояний:
—чрезмерно выраженного гипертиреоза;
— гипертиреоза, вызванного избытком экзогенных тиреоидных гормонов
(например, в результате неправильно рассчитанной дозы лечебных пре­
паратов или по ошибке).
Причины гипертиреозов
Различные факторы вызывают повреждения на разных уровнях нейро­
эндокринной регуляции (гипоталамо-гипофизарно-тиреоидная система),
синтеза, транспорта и реализации действия тиреоидных гормонов. В связи
с этим выделяют причины первичного, вторичного и третичного гиперти­
реоза.
Причины первичного гипертиреоза
Причинные факторы первичного гипертиреоза поражают саму щитовидную
железу или эктопическую тиреоидную ткань. Наиболее частые причины первич­
ного гипертиреоза представлены на рис. 28.28. К ним относят:
—зоб (гипертиреоз вызван увеличением массы и размеров щитовидной
железы);
—диффузный токсический зоб (болезнь Грейвса) — наиболее частая причина
гипертиреоза;
—узловой токсический зоб (болезнь Пламмера) — гипертиреоз вследствие
автономно функционирующей аденомы щитовидной железы (в том
числе множественных аденом);
—тиреоидит подострый (болезнь де Кервена). Тиреотоксикоз развивается
вследствие разрушения фолликулов железы и поступления избытка
тиреоидных гормонов в кровь;
—Т3- и Т4-секретирующие эктопические опухоли (представляют собой тера­
томы яичника или метастазы фолликулярного рака щитовидной железы
в различных органах);
—тиреотоксикоз, вызванный йодом (феномен «йод-Базедов»);
—передозировку тиреоидных гормонов (наиболее часто это бывает либо врачеб­
ной ошибкой, либо результатом приема гормонов с целью похудания).
Рис. 28.28. Наиболее частые причины первичного гипертиреоза
Причины вторичного гипертиреоза
Наиболее значимые причины вторичного гипертиреоза — ТТГ-секретирующая аденома гипофиза и селективная резистентность аденогипофиза
к гормонам щитовидной железы (в крови существенно повышены уровни Т3
и Т4, но в силу низкой чувствительности и/или уменьшенного числа рецеп­
торов к Т3 и Т4 в тиреотрофах аденогипофиза не происходит адаптивного
уменьшения синтеза ТТГ).
Причины третичного гипертиреоза
Причинные факторы третичного гипертиреоза воздействуют на нейроны
гипоталамуса. Как правило, это наблюдается при:
—невротических состояниях, сопровождающихся избыточным образова­
нием тиролиберина;
—состояниях, вызывающих длительную активацию норадренергических ней­
ронов гипоталамуса. При этом происходит стимуляция синтеза Т3 и Т4
через нисходящие пути симпатической нервной системы.
Основные клинические формы гипертиреоза
Основные клинические формы гипертиреоза в виде диффузного и узлового
токсического зоба рассмотрены в статьях «Гипертиреоз», «Подострый тиреои­
дит» и «Зоб» (см. приложение «Справочник терминов»).
Проявления гипертиреозов и их механизмы
Признаки гипертиреоидных состояний при разных клинических формах
сходны, хотя каждое конкретное заболевание, сопровождающееся гиперпро­
дукцией тиреоидных гормонов, имеет специфику.
Особенности проявлений в определенной мере зависят от тяжести и про­
должительности заболевания, а также от преимущественно пораженной
физиологической системы, органа или ткани.
Наиболее характерные проявления гипертиреозов приведены в статье
«Гипертиреоз» (см. приложение «Справочник терминов») и рассмотрены ниже.
• Проявления поражения нервной системы и ВНД при гипертиреозе представ­
лены на рис. 28.29.
Рис. 28.29. Наиболее характерные признаки нарушения функции нервной системы
и психической деятельности при гипертиреозе
Рис. 28.30. Наиболее характерные признаки сердечно-сосудистых расстройств
при гипертиреозе
• Проявления расстройств системы кровообращения при гипертиреозе пока­
заны на рис. 28.30. К их числу относятся:
—синдром «тиреотоксическое сердце». Он характеризуется триадой рас­
стройств:
о нарушениями ритма сердца (синусовая тахикардия более 130-150
импульсов в минуту, мерцательная аритмия, трепетание предсердий);
о гиперфункцией и гипертрофией миокарда (при длительном течении тирео­
токсикоза), нередко сочетающимися с мелкоочаговым кардиосклерозом;
о сердечной недостаточностью (особенность сердечной недостаточно­
сти при гипертиреозе — сравнительно высокий сердечный выброс
как результат тахикардии);
—увеличение линейной и объемной скорости кровотока;
—повышение систолического АД (обусловлено высоким сердечным выбро­
сом); часто развивается систолическая артериальная гипертензия;
—снижение диастолического АД (вызвано компенсаторным расширением
резистивных сосудов в ответ на повышение сердечного выброса).
• Проявления нарушений функции системы пищеварения при гипертиреозе:
—изменения аппетита — у многих молодых пациентов аппетит повышен
в связи с активацией окислительных процессов, возрастанием основно­
го обмена и теплопродукции; у отдельных пациентов, чаще у пожилых,
аппетит снижен вплоть до анорексии;
— нарушение пищеварения в желудке и кишечнике (характеризуется усиле­
нием перистальтики желудка и кишечника, частым стулом, нарушением
желчеобразования и желчевыделения). Указанные изменения (наряду
с другими физиологическими и метаболическими эффектами повышен­
ного уровня тиреоидных гормонов) приводят к похуданию.
• Офтальмопатическая симптоматика при гипертиреозе (рис. 28.31). «Глаз­
ные симптомы» относятся к числу патогномоничных для гипертиреоза.
Однако они могут встречаться и у некоторых пациентов с гипотиреозом
(например, при хроническом аутоиммунном тиреоидите Хашимото):
—офтальмопатия Грейвса. Выявляется более чем у 45% пациентов. В осно­
ве патогенеза офтальмопатии лежат реакции иммунной аутоагрессии.
- «Гневный взгляд» при фиксации взора
Рис. 28.31. Наиболее характерные «глазные симптомы» гипертиреоза
Проявления офтальмопатии:
—экзофтальм — смещение глазного яблока вперед. Причины: отек ретроорби-
тальной клетчатки, лимфоцитарная инфильтрация тканей глазницы (среди
лимфоцитов обнаруживается множество С0 8 клеток — Т-киллеров
и СБ4+-клеток — Т-хелперов), мукоидный отек ретробульбарной клет­
чатки, фиброз и дегенерация глазодвигательных мышц;
—диплопия — удвоение предметов при взгляде на них, а также ограничение
подвижности глазного яблока. Причины — фиброз глазодвигательных
мышц, склероз их, дегенерация миоцитов глазодвигательных мышц;
—сухость, эрозии и изъязвления роговицы глаза. Причины — нарушение
смыкания век в связи с их отеком и экзофтальмом; расстройства образо­
вания и оттока слезной жидкости;
—резь в глазах, слезотечение, светобоязнь. Причины — сухость роговицы
глаза, кератит;
—слепота. Причины — сдавление зрительного нерва отечной тканью, дис­
трофические изменения в зрительном нерве, кератит;
—симптом Дальримпля (широкое раскрытие глазных щелей и появление
полоски склеры между верхним веком и радужной оболочкой глаза);
—симптом Кохера—Грефе (отставание верхнего века от движения глазного
яблока при взгляде вниз);
— симптом Штельвага (редкое мигание).
Описанные симптомы придают лицу характерное выражение испуга,
а фиксация взора имитирует «гневный взгляд».
• Расстройства метаболизма при гипертиреозе (рис. 28.32). Они проявляются:
—повышением основного обмена и теплопродукции. Это сопровождается
потливостью и плохой переносимостью тепла;
— отрицательным азотистым балансом, повышением уровня остаточного
азота в крови и моче;
—усилением катаболизма белков (в связи с активацией протеаз), сочета­
ющимся с усилением глюконеогенеза при участии аминокислот;
—повышенной мобилизацией жира из депо, активацией липолиза и после­
дующим окислением его продуктов;
— активацией обмена холестерина (синтез, утилизация тканями, транспорт
в гепатоциты и выведение печенью);
—усилением гликогенолиза и торможением глюкогенеза, что сочетается
с повышенной адсорбцией углеводов в кишечнике. В целом, это при­
водит к гипергликемии. Последняя потенцируется в связи с активацией
симпатико-адреналовой системы.
Рис. 28.32. Наиболее характерные признаки изменения обмена веществ при гипертиреозе
• Проявления поражения опорно-двигательного аппарата при гипертиреозе.
Для них характерны:
—тиреотоксическая миопатия. Она проявляется слабостью, гипотрофией
мышц, дистрофическими изменениями в них. Это обусловливает труд­
ности при длительной ходьбе, переносе тяжестей, подъеме по лестнице.
Иногда развивается преходящий (через несколько часов или суток)
«тиреотоксический мышечный паралич»;
- остеопороз — разрежение костной ткани. У пациентов с тиреотоксико­
зом резорбция костной ткани доминирует над ее образованием. Это,
как правило, сопровождается гиперкальциемией и кальциурией.
• Проявления поражений кожи и подкожной клетчатки у пациентов с гипертиреозом:
— кожа теплая, влажная, особенно на ладонях. В значительной мере это свя­
зано с повышением основного обмена, теплопродукции и потливостью;
—претибиальная микседема: одно- или двусторонний локальный слизистый
отек мягких тканей на передней поверхности голеней. Характеризуется
утолщением (слизистый отек — микседема), гиперпигментацией кожи
с контурированными волосяными фолликулами (вид апельсиновой
кожуры).
• Дисбаланс тиреоидных гормонов и ТТГ при гипертиреозе. Он характеризуется:
—повышенным уровнем общей и свободной фракций Т3 и Т4 (за редким
исключением, когда имеется патологически высокая чувствительность
тканей к Т3 и Т4);
—значительным снижением содержания ТТГ при первичном гипертиреозе;
— повышенным содержанием ТГГ (в сочетании с высокой концентрацией
Т3 и Т4) при вторичном (гипофизарном) и третичном (гипоталамическом)
тиреотоксикозе.
• Поглощение радиоактивного йода щитовидной железой существенно меняется:
— повышено при усиленном образовании тиреоидных гормонов (т.е. при пер­
вичном, вторичном и третичном гипертиреозе);
— снижено при введении избытка тиреоидных гормонов в организм
и при их поступлении в кровь из распадающейся ткани железы (например,
при тиреоидитах или опухоли).
• Инициация синтеза тиреоспецифических 1§ при гипертиреозе. В крови паци­
ентов с болезнью Грейвса выявляются специфические иммуноглобулины
к различным Аг щитовидной железы:
—тиреостимулирующие 1§ — маркёры диффузного токсического зоба;
— \% к Аг тиреоцитов (к белкам микросом, йодидпероксидазе).
Т и р ео т о к си ч еск и й кр и з
Тиреотоксический криз — острое, тяжелое, чреватое смертью проявление
(осложнение) тиреотоксикоза. Характеризуется прогрессирующим («взры­
вообразным») усугублением течения гипертиреоза.
Наиболее частые причины тиреотоксического криза:
—травмы и хирургические вмешательства (нередко даже удаление зуба);
— стрессовые ситуации;
—инфекционные болезни и/или интоксикации;
—физическое перенапряжение, роды.
Главные звенья патогенеза тиреотоксического криза:
— острое и значительное увеличение содержания в крови тиреоидных гормонов;
—нарастающая острая надпочечниковая недостаточность (как результат
стресс-реакции, сопровождающей тиреотоксический криз);
— избыточная активация симпатико-адреналовой системы (приводит к гипер­
катехоламинемии и реализации ее цитотоксических эффектов).
Проявления тиреотоксического криза перечислены на рис. 28.33.
Исход тиреотоксического криза зависит от своевременности его диагности­
ки и эффективности лечения.
Рис. 28.33. Главные проявления тиреотоксического криза
Летальность при тиреотоксическом кризе достигает 60%.
Гипотиреозы
Гипотиреоз — состояние, обусловленное недостаточностью эффектов
тиреоидных гормонов щитовидной железы.
Частота зарегистрированных случаев гипотиреоза достигает 10 на 1000 насе­
ления в общей популяции (у новорожденных — 0,025%, у людей старше 65 лет —
3%). Преобладающий возраст пациентов — старше 40 лет. Преобладающий
пол — женский (соотношение заболевших среди женщин и мужчин — 7,5:1).
Причины гипотиреоза
Физические, химические и/или биологические тиреотропные патогенные
факторы обусловливают развитие разных форм гипотиреоза, действуя на:
— щитовидную железу (вызывая первичный, или железистый, гипотиреоз);
—гипофиз (приводя к вторичному, или гипофизарному, гипотиреозу);
—гипоталамические центры (обусловливая третичный, или гипоталамический, центрогенный гипотиреоз);
—механизмы транспорта, метаболизм, рецепцию тиреоидных гормонов
(вызывая постжелезистый гипотиреоз).
На практике различают первичный и вторичный (гипоталамо-гипофизарный) гипотиреоз.
Первичный гипотиреоз
Первичный гипотиреоз (90% случаев гипотиреоза) развивается при поражении
ткани самой щитовидной железы и сопровождается повышением уровня ТТГ.
Причины первичного гипотиреоза:
— врожденное нарушение развития щитовидной железы (раннее выявление
этой патологии может предотвратить развитие существенных невроло­
гических осложнений; в России эту патологию выявляют по уровню ТТГ
на пятый день жизни новорожденного);
—повреждение щитовидной железы аутоагрессивными 1§ (приводит к раз­
витию иммунного тиреоидита — болезни Хашимото, наиболее частой
причины первичного тиреоидита);
— идиопатическая атрофия щитовидной железы (при ней часто выявляют
антитиреоидные АТ, повреждающие тиреоидные клетки);
—воздействие радиоактивного йода на щитовидную железу (например,
при лечении препаратами йода пациентов с диффузным токсическим
зобом; гипотиреоз при этом развивается почти у 50% больных);
— субтотальная тиреоидэктомия;
—длительное применения антитиреоидных средств;
—дефицит йода в организме;
— феномен Вольфа—Чайкова — гипотиреоз, вызванный введением в организм
препаратов йода (обычно в большой дозе). Развивается у пациентов с гипер-
тиреозом (например, при тиреоидите Хашимото, диффузном токсическом
зобе), а также у детей, матери которых во время беременности принимали
препараты йода. Патогенез феномена Вольфа-Чайкова включает несколь­
ко взаимосвязанных звеньев (рис. 28.34). К числу основных относят:
о подавление окисления йодидов в их более реакционноспособную форму
(вследствие угнетения активности йодидпероксидазы);
о торможение связывания йодида с тирозильными остатками в молекуле
тиреоглобулина и, как следствие, образование моно- и дийодтирозина;
о ингибирование окислительной конденсации моно- и дийодтирозина в трии тетрайодтиронин вследствие ингибирования активности йодидпе­
роксидазы;
о уменьшение интенсивности гидролиза тиреоглобулина в тироцитах
в результате подавления кинетических свойств протеаз и пептидаз.
Рис. 28.34. Основные механизмы развития феномена Вольфа—Чайкова (гипотиреоз
вследствие острой передозировки йода)
Вторичный и постж елезисты й гипотиреоз
Вторичный гипотиреоз развивается при поражении гипоталамо-гипофизарной
системы. Это обусловливает недостаточное выделение соответственно тиролиберина и ТТГ с последующим снижением функций щитовидной железы.
Причины гипофизарного, гипоталамического и постжелезистого гипоти­
реоза представлены на рис. 28.35. К ним относятся:
— гипопитуитаризм различного происхождения (для развития гипотиреоза
существенное уменьшение секреции ТТГ);
—дефекты синтеза, транспорта или взаимодействия тиролиберина с его
рецепторами (что приводит к гипоталамическому гипотиреозу);
—инактивация циркулирующих в крови Т3 и Т4, ТТГ (1§, протеазами, чрез­
мерной связью с транспортными белками и др.);
—низкая чувствительность тканей к тиреоидным гормонам (например,
при уменьшении числа рецепторов к ним и/или их чувствительности
к Т 3 и Т 4);
— образование гормонально-неактивного гТ3 (реверсивного Т3).
Последние три группы вызывают постжелезистый гипотиреоз.
Рис. 28.35. Основные причины вторичного и постжелезистого гипотиреоза
Основные формы гипотиреоза
Основные формы гипотиреоза (хронический аутоиммунный тиреоидит —
болезнь Хашимото, микседематозная кома) рассмотрены в статьях «Гипотиреоз»,
«Кома» и «Тиреоидит» (см. приложение «Справочник терминов»).
Кретинизм
Выделяют:
—спорадический (врожденный);
—эндемический (эндемический зоб).
Врожденная (спорадическая) форма кретинизма. Наиболее частые причи­
ны спорадического кретинизма изложены в разделе «Генетические аспекты»
(см. статью «Гипотиреоз») и представлены на рис. 28.36.
Эндемическая форма кретинизма
Причины эндемического зоба:
—дефицит йода в воде и пище. Наблюдается на определенных территори­
ях, наиболее часто — в горных (Альпы, Гималаи, Кавказ, Кордильеры,
Карпаты, Тянь-Шань), а также в некоторых равнинных районах
(Центральной Африки, Южной Америки, Восточной Европы);
—избыток в среде обитания (воде, продуктах питания) веществ, тормозящих
или блокирующих синтез тиреоидных гормонов. Эти вещества называются
тиреостатическими. К ним относятся производные тиоурацила, тиомочевины, тиоцианаты, роданиты. Важно, что при попадании в организм
тиреостатических веществ даже добавка йода в пищу не предотвращает
развития гипотиреоза;
— недостаток в организме ряда микроэлементов, необходимых для синтеза
и реализации эффектов йодсодержащих тиреоидных гормонов. К наиболее
важным из них относят кобальт, молибден, цинк и медь.
Инициальным и основным патогенетическим звеном обеих разновидностей
кретинизма является дефицит Т3 и Т4, а последующие звенья патогенеза —
результат недостаточно эффективного действия йодсодержащих тиреоидных
гормонов.
Рис. 28.36. Наиболее частые причины спорадического кретинизма
Проявления кретинизма и их механизмы
Наличие зоба — важный признак эндемического кретинизма. Причина раз­
вития зоба — избыточная продукция ТТГ (как результат дефицита Т3 и Т4).
При этом воздействие ТТГ на щитовидную железу в условиях дефицита йода
проявляется стимуляцией ее роста (гиперплазия): железа увеличивается в раз­
мере, но уровень Т3 и Т4 по-прежнему низок.
Другие общие проявления кретинизма и их выраженность зависят от воз­
раста ребенка, в котором диагностирован гипотиреоз, от того, насколько свое­
временно начато его лечение. Однако имеются и существенные отличия.
Для эндемического кретинизма характерны:
—зоб;
—значительно колеблющаяся степень проявлений расстройств жизнедеятель­
ности (включая и интеллект — от несколько пониженного 10 до кретинизма);
— глухота, сочетающаяся с немотой (оба расстройства имеют центрогенное
происхождение, обусловленное нарушением развития нервной системы
в условиях гипотиреоза).
Для всех форм кретинизма, как правило, характерны:
— отставание в физическом развитии, что проявляется малым ростом
(нередко — карликовым);
—грубые черты лица (обусловлены отечностью мягких тканей);
— большой язык (из-за своего отека он часто не вмещается во рту);
— широкий плоский («квадратный») нос с западанием его спинки;
—далеко расставленные глаза (глазной гипертелоризм);
—большой живот (нередко с наличием пупочной грыжи);
—задержка роста и смены зубов;
—длительное незаращение родничков черепа;
—нарушения психического развития (более или менее выраженное сниже­
ние интеллекта, вплоть до идиотии, а у детей старшего возраста — пло­
хая успеваемость в школе). Указанные выше признаки являются резуль­
татом множественных генетических дефектов.
М икседем а
Микседема — тяжелая форма гипотиреоза, развивающаяся, как правило,
у взрослых и подростков.
Характерным признаком микседемы является слизистый отек кожи и подкож­
ной клетчатки, при котором отсутствует ямка при надавливании.
Инициальное звено патогенеза микседемы — недостаточная деятельность
тиреоидных гормонов, чаще в результате первичного гипотиреоза (около 95%
случаев).
Проявления микседемы и их механизмы
Указанные ниже признаки характерны для всех разновидностей гипотиреоза.
Однако их комбинация и выраженность у конкретных пациентов различна.
Нарушения функций нервной системы и ВНД при микседеме
Основные проявления расстройств нервной деятельности показаны
на рис. 28.37. Наиболее значимые среди них:
— энцефалопатия гипотиреоидная (характеризуется снижением интеллекта
и психической активности; замедлением мышления и речи, затормо­
женностью; сонливостью и медлительностью; нарушением памяти;
частыми депрессивными состояниями; гипорефлексией;
— парестезии;
— мозжечковая атаксия;
— снижение тонуса симпатико-адреналовой системы (обусловливает, поми­
мо прочего, нарушение моторики ЖКТ, запор и уменьшение потоотде­
ления).
Рис. 28.37. Наиболее характерные признаки нарушения функций нервной системы
и психической деятельности при гипотиреозе
Основной причиной энцефалопатии и других названных выше нейропатий
является недостаточная эффективность тиреоидных гормонов, что тормозит
дифференцировку нервных структур и их функций, а также развитие ВНД,
особенно у детей.
Расстройства функции ССС при гипотиреозе
Наиболее значимые из них представлены на рис. 28.38. К ним относятся:
— брадикардия;
— снижение сердечного выброса с развитием сердечной недостаточности;
—кардиомегалия (развивается в связи с дилатацией полостей сердца вслед­
ствие дистрофических изменений в миокарде, обусловленных недоста­
точностью метаболического действия тиреоидных гормонов; скопления
жидкости в полости перикарда);
— кардиалгии — боли в сердце (обусловлены недостаточностью коронарно­
го кровотока и нарушениями обмена веществ в миокарде);
— снижение линейной и объемной скорости кровотока;
— нарушения микрогемоциркуляции в тканях, вызванных в основном сер­
дечной недостаточностью.
Рис. 28.38. Наиболее характерные признаки сердечно-сосудистых расстройств
при гипотиреозе
Расстройства пищеварения при гипотиреозе:
—снижение аппетита, нередко тошнота;
—недостаточность переваривающей функции желудка и кишечника (вслед­
ствие гипоацидного гастрита, гипотонии и гипокинезии кишечника
и желчных путей; они приводят к расстройствам полостного и мембран­
ного пищеварения, частым проявлениям запора, кишечной непроходи­
мости).
Нарушения функций почек при гипотиреозе:
— снижение экскреторной функции почек (вызвано уменьшением объема
перфузии почек кровью и, как следствие, фильтрационного давления,
а также гипонатриемией);
—гипотония мышц и гипокинезия (приводят к замедлению оттока мочи
и частому инфицированию мочевыводящих путей. В этих условиях раз­
виваются инфекционные уретриты, цистит, воспаления мочеточников
и лоханок почек).
Нарушения обмена веществ при гипотиреозе
Метаболические расстройства патогномоничны для гипотиреоидных
состояний. К ним относятся (рис. 28.39):
—уменьшение интенсивности окислительных процессов и основного обмена
(сопровождается падением теплопродукции и гипотермией, что проявля­
ется у пациентов зябкостью даже при нормальной температуре воздуха);
—торможение процессов протеосинтеза, сочетающееся с активацией протеолиза;
— развитие гиперлипопротеинемии с повышением содержания в крови холе­
стерина и триглицеридов (это способствует прогрессированию атеро­
склероза; указанные изменения обусловлены в значительной мере сни­
жением активности ЛПЛазы);
—гипогликемия вследствие уменьшенной интенсивности всасывания глю­
козы в кишечнике и нарушений мобилизации гликогена в гепатоцитах,
что вызвано подавлением активности фосфорилазы;
—накопление избытка кислых гликозаминогликанов в коже, подкожной клет­
чатке, сердце, легких, почках, а также в жидкости серозных полостей;
—повышенное содержание ионов натрия в клетках и интерстициальной жид­
кости;
—повышенное содержание жидкости в тканях.
Три последних изменения лежат в основе развития особого отека, харак­
терного для гипотиреоза, — микседемы.
Рис. 28.39. Наиболее характерные расстройства обмена веществ при гипотиреозе
Поражения кожи и ее производных у пациентов с гипотиреозом
Они являются обязательным компонентом клинической картины гипоти­
реоза и включают:
— развитие микседемы. Причины формирования микседемы:
о значительное повышение гидрофильности соединительной ткани
вследствие большего содержания в ней глюкуроновой и хондроитинсерной кислоты, а также накопления в клетках и межклеточной
жидкости избытка № + (этому способствует снижение выработки пред­
сердного натрийуретического фактора);
о задержка жидкости в тканях в связи с повышением эффектов АДГ
в условиях пониженного уровня Т3 и Т4;
о связывание большого количества жидкости тканевым коллоидом (содер­
жащим избыток гликозаминогликанов и № + с образованием муцина:
слизеподобного соединения); накопление муцина приводит к утол­
щению кожи и подкожной клетчатки. В связи с этим кожа не соби­
рается в складки, поверхность ее сухая, шелушащаяся, холодная,
бледная с желтоватым оттенком (вследствие накопления каротина,
превращение которого в витамин А в печени замедлено); образование
избытка муцина в органах обусловливает увеличение их размеров,
нарушения микроциркуляции крови в них и развитие дистрофиче­
ских процессов;
—одутловатость лица, огрубение его черт, гипомимичность (маскообразность), отек периорбитальной клетчатки (в основном как результат мик­
седемы);
—ломкость волос, легкое их выпадение, хрупкость ногтей (обусловлены дис­
трофическими изменениями в коже, подкожной клетчатке, нарушением
их кровоснабжения);
—отечность голосовых связок (в результате голос становится низким и гру­
бым);
—увеличение языка (на боковых поверхностях его видны отпечатки зубов;
нечеткая, затрудненная речь);
—асептический полисерозит (проявляется накоплением избытка серозной
жидкости в полостях перикарда, брюшной, грудной и др. Причина: гене­
рализованная реакция иммунной аутоагрессии по отношению к изме­
ненным белковым Аг серозных оболочек).
Патология опорно-двигательного аппарата при гипотиреозе
Она характеризуется:
—миопатиями (проявляются миалгиями, снижением мышечной силы,
повышенной утомляемостью; причина указанных изменений — выпаде­
ние эффектов Т3 и Т4 по отношению к мышечной ткани);
— артропатиями — поражением суставов (характеризуются болями в суста­
вах, дегенеративно-деструктивными изменениями суставных поверхно­
стей).
Расстройства роста и развития организма при гипотиреозе
У детей это выражается задержкой роста (в результате дефицита Т3 и Т4,
а также недостаточным содержанием и/или деятельностью СТГ).
Нарушенное содержание гормонов в крови при гипотиреозе
Гипотиреоз вызывает дисбаланс гормонов щитовидной железы и гипофиза
в организме, для которого характерны:
—снижение содержания общих и свободных фракций Т3 и Т4; исключе­
ние составляет постжелезистый (рецепторный) вариант гипотиреоза.
При нем уровень тиреоидных гормонов находится в нормальных преде­
лах, но чувствительность тканей к ним значительно понижена;
—уровень ТГГ меняется по-разному в зависимости от происхождения гипоти­
реоза:
о повышен при первичном гипотиреозе;
о понижен при вторичном (гипофизарном и гипоталамическом) гипо­
тиреозе;
о может быть нормальным или даже повышенным при снижении чув­
ствительности тироцитов к ТТГ;
— различные результаты пробы с введением в организм экзогенного тиролиберина:
о при первичном гипотиреозе (т.е. при поражении щитовидной железы)
она положительная. Это означает, что секреторная реакция аденогипо­
физа на тиролиберин не нарушена);
о при гипофизарном гипотиреозе проба отрицательная (так как отсутствует
или существенно снижена продукция ТТГ);
о при гипоталамическом гипотиреозе прирост концентрации ТТГ значи­
тельно растет во времени и достигает максимума более чем через 60—80
мин (в норме до 30 мин).
Гипотиреоидная кома
Гипотиреоидная (микседематозная) кома — крайне тяжелое, нередко смер­
тельное проявление гипотиреоза (летальность при ней достигает 75%). Является
конечным этапом любой разновидности гипотиреоза при его неправильном
лечении или в его отсутствие.
Факторы риска гипотиреоидной комы:
—переохлаждение (особенно при малоподвижности пациента);
— недостаточность кровообращения любого генеза;
— острые инфекции (например, грипп, пневмония, менингит);
— интоксикации (в том числе транквилизаторами, опиатами, барбитурата­
ми, анестетиками);
— стрессовые ситуации;
—кровотечения (например, желудочно-кишечные, маточные, носовые);
— состояния, приводящие к гипогликемии и/или гипоксии (например, длитель­
ное голодание, ДН, хронические анемии, сердечная недостаточность).
Проявления гипотиреоидной комы и их механизмы
Основные признаки гипотиреоидной комы представлены на рис. 28.40.
Рис. 28.40. Основные проявления гипотиреоидной комы
К ни м относят:
— значительную брадикардию (обусловлена недостаточностью кардиости­
мулирующего действия Т3 и Т4 в условиях их низкой концентрации
и снижением кардиотропных эффектов катехоламинов, что характерно
для гипотиреоза вообще);
— выраженную артериальную гипотензию, вплоть до коллапса;
- Д Н , которая вызвана:
о снижением альвеолярной вентиляции (в результате уменьшения возбу­
димости дыхательного центра, нарушения проходимости дыхательных
путей из-за отека их стенок);
о уменьшением легочного кровотока (в связи с недостаточностью крово­
обращения);
о затруднением диффузии газов через аэрогематическую мембрану вслед­
ствие ее отека;
—нарастающие гипоксия и ацидоз, обусловленные:
о ДН;
о недостаточностью кровообращения;
о анемией (часто развивающейся при гипотиреозе в результате нару­
шения всасывания в ЖКТ витамина В12, фолиевой кислоты, железа;
развития аутоагрессивных иммунных реакций; расстройств кроветво­
рения);
о нарушением аэробного обмена веществ;
о снижением функции почек по компенсации сдвигов КЩР;
— почечная недостаточность (результат нарушенного кровообращения);
—прогрессирующая гипотермия (вызвана нарастающим снижением эффек­
тивности экзотермических реакций организма; в связи с этим гипотиреоидную кому нередко называют гипотермической);
—угнетение сознания вплоть до полной его потери.
Клиническая характеристика гипотиреоидной комы приведена в статье
«Кома микседематозная» см. приложение «Справочник терминов».
НАРУШЕНИЯ ФУНКЦИЙ ПАРАЩИТОВИДНЫХ ЖЕЛЕЗ
Четыре небольшие по размеру паращитовидные железы расположены
на задней поверхности и под капсулой щитовидной железы. Функция желез —
синтез и секреция Са2+-регулирующего пептидного гормона паратиреокрина
(ПТГ). ПТГ вместе с кальцитонином и катакальцином, а также с витами­
ном О регулирует обмен кальция и фосфатов.
Типовые формы патологии паращ итовидной железы
Различные заболевания, обусловленные изменением уровня и/или
эффектов ПТГ, могут рассматриваться как относящиеся к состояниям
гиперпаратиреоидным (гиперпаратиреозы) или гипопаратиреоидным (гипопаратиреозы).
Гиперпаратиреоидные состояния
Гиперпаратиреозы характеризуются повышенными эффектами ПТГ.
Различают первичные (железистые), вторичные (гиперкальциемические) и третичные гиперпаратиреозы, а также псевдогиперпаратиреоз
(рис. 28.41).
Виды гиперпаратиреоза \----------------Первичные
(первично-железистые)
4Вторичные
(гиперкальциемические)
Третичные|
Наиболее частые причины
Патология паращитовидных
желез (ведущая к их
автономной функции):
- аденомы;
- гиперплазия;
- новообразования
- нефропатии;
- патология кишечника;
- остеопатии
- хронический вторичный
гиперпаратиреоз,
ведущий к развитию
аденомы с автономной
функцией
Рис. 28.41. Гиперпаратиреоидные состояния. Виды и причины
Первичный гиперпаратиреоз
Первичные гиперпаратиреозы — патология самих паращитовидных желез.
Причины первичного гиперпаратиреоза:
—автономно функционирующая аденома (или несколько аденом, наблюда­
ется в 75—80% случаев первичного гиперпаратиреоза);
—первичная гиперплазия желез (10—15% пациентов с гиперпаратиреоццизмом);
—карцинома паращитовидной железы (менее 5% случаев).
Вторичный гиперпаратиреоз
Вторичный гиперпаратиреоз — результат длительной гипокальциемии, как пра­
вило, в сочетании с гиперфосфатемией и вторичным развитием гиперфункции
и гиперплазии паращитовидных желез.
Наиболее частые причины вторичного гиперпаратиреоза:
—патология почек, приводящая к гипокальциемии (наиболее частая причи­
на; при этом выявляется снижение активности 1а-гидроксилазы):
о ХПН (она сопровождается снижением экскреции фосфатов и развити­
ем гиперфосфатемии; это приводит к снижению уровня Са2+ в крови
и стимуляции функции паращитовидных желез);
о тубулопатии (сочетающиеся с гипокальциемией);
—патология кишечника:
о синдром мальабсорбции, сопровождающийся нарушением всасывания
кальция в кишечнике;
о стеаторея (повышенное выведение с калом жира, жирных кислот,
их соединений, а также связанных с ними солей кальция);
—патология костной ткани:
о остеомаляция (размягчение костей и деформация их в связи с дефици­
том в них солей кальция и фосфорной кислоты);
о деформирующая остеодистрофия (болезнь Педжета, характеризующая­
ся резорбцией костной ткани, дефицитом в ней кальция, деформацией
костей;
—гиповитаминоз Б (см. статью «Рахит» в приложении «Справочник тер­
минов»).
Третичный гиперпаратиреоз
Причина третичного гиперпаратиреоза — длительно протекающий вторич­
ный гиперпаратиреоз; последний приводит к развитию аденом в паращито­
видных железах, приобретающих способность автономного функциониро­
вания и гиперпродукции ПТГ. В этих условиях разрушается обратная связь
между уровнем Са2+ в крови и секрецией ПТГ.
Псевдогиперпаратиреоз
Псевдогиперпаратиреоз — гиперпродукция ПТГ эктопическими опухолями.
Наблюдается при семейном полиэндокринном аденоматозе и паранеопла­
стических синдромах.
Проявления гиперпаратиреоза
Проявления гиперпаратиреоза рассмотрены в статье «Гиперкальциемия»
и представлены на рис. 28.42. К основным проявлениям гиперпаратиреоза
относят:
Рис. 28.42. Основные проявления гиперпаратиреоза
—изменения костной ткани:
о остеопороз (генерализованное уменьшение объема и плотности кост­
ной ткани, в том числе в результате потери солей кальция);
о деформации костей (результат остеопороза; кости изогнуты и уплоще­
ны; в наибольшей мере деформируются бедренные и тазовые кости,
грудина, ребра, грудные и поясничные позвонки);
о множественные переломы костей (обычно переломы происходят в труб­
чатых костях, ребрах, позвонках);
о расшатывание и выпадение зубов (следствие остеопороза челюстей
и образования в них кист);
—патология почек (изменения в почках обусловлены гиперкальциемией
и повышенным выведением кальция с мочой, что сопровождается
повреждением клеток эпителия почечных канальцев, нарушени­
ем их экскреторной функции). В целом, совокупность изменений
в почках нередко приводит к развитию прогрессирующей почечной
недостаточности и уремии. Патология почек при гиперпаратиреозе
проявляется:
о полиурией — значительным увеличением суточного диуреза. Причины:
• повышенная экскреция кальция с мочой. Известно, что, помимо
стимуляции осмотического диуреза, избыток кальция повреждает
эпителий почечных канальцев, потенцируя полиурию уже незави­
симо от экскреции Са2+;
• нарушение реабсорбции жидкости в почках, обусловленное дистрофи­
ческими и структурными изменениями в клетках канальцев почек);
• снижение чувствительности канальцевого эпителия к АДГ, являющее­
ся следствием повышенной концентрации в крови ПТГ;
о вторичной полидипсией (обусловлена потерей значительного количе­
ства жидкости с мочой и гиперосмией в результате гиперкальциемии);
о нефро- и уролитиазом (образование множественных камней в ткани
почки и/или мочевыводящих путях соответственно); встречается
у 20—25% пациентов; причиной нефро- и уролитиаза являются гиперкальциемия и гиперкальциурия;
—нервно-мышечная патология:
о миастения (мышечная слабость) и в связи с этим — быстрая физиче­
ская утомляемость;
о миалгии (боли в отдельных группах мышц, чаще в нижних конечностях).
У пациентов развивается «утиная походка» («переваливание» с одной ноги
на другую) и плоскостопие. И то, и другое изменение — результат гипер­
кальциемии и повышенного содержания Са2+ во внеклеточной среде;
—желудочно-кишечные расстройства (их причины — нарушение кро­
воснабжения и трофики ЖКТ в результате кальцификации стенок его
сосудов):
о язвенная болезнь (преимущественно двенадцатиперстной кишки);
о гастриты;
о энтероколиты (нередко с множественными эрозиями и язвами);
о нарушения аппетита;
о тошнота и рвота;
—сердечно-сосудистые расстройства:
о развитие стеноза, и/или недостаточности аортального, и/или митраль­
ного клапана в результате кальцификации створок клапанов и дефор­
мации их;
о артериальная гипертензия, обычно почечного генеза; ее причины:
• активация системы ренин — ангиотензин — альдостерон;
• включение ренопривного механизма гипертензии в связи с нефрокальцинозом и нефроскперозом (приводят к уменьшению массы почечной
ткани и образованию в ней вазодепрессорных ПГ и кининов);
• сердечная недостаточность (как результат указанных выше нарушений,
а также расстройств транслокации кальция в кардиомиоцитах);
—нарушения ВНД:
• психастения (быстрая психическая истощаемость);
• повышенная раздражительность;
• плаксивость, депрессивные состояния, сменяющиеся психическим
возбуждением; нарушения сна, сонливость днем;
— гиперпаратиреоидный гиперкальциемический криз (наиболее тяжелое,
чреватое смертью пациента осложнение гиперпаратиреоза; летальность
при гиперкальциемическом кризе достигает 50%). Причинами криза могут
быть:
о состояния, прямо или опосредованно приводящие к повышению уровня
Са2+ в крови: переломы костей, обогащенная кальцием пища, гипогидра­
тация, прием ощелачивающих, антацидных и содержащих кальций ЛС;
о инфекции;
о интоксикации.
Инициальные патогенетические факторы криза:
— острое значительное повышение Са2+ крови до 3,5—5 ммоль/л (14—
20 мг%) и выше;
— снижение содержания фосфатов, К+, М§2+ в сыворотке крови.
Проявления криза:
— острые желудочно-кишечные расстройства (тошнота, рвота, диарея, силь­
ная жажда, острые боли в животе);
—почечная недостаточность (а при ее наличии — нарастание степени недо­
статочности) с развитием уремии и комы;
—прогрессирующие нарушения психики (с развитием заторможенности
и ступора либо с возбуждением, галлюцинациями и бредом);
—недостаточность кровообращения, вплоть до коллапса.
Гипопаратиреоидные состояния
Гипопаратиреоидные состояния (гипопаратиренш, гипопаратиреоидизм,
недостаточность паращитовидных желез) характеризуются снижением
содержания в крови и/или выраженности эффектов ПТГ в организме.
Выделяют две разновидности гипопаратиреоза:
—железистый;
—внежелезистый (псевдогипопаратиреоз).
Гипопаратиреоз
Первичный (железистый) гипопаратиреоз развивается в результате отсутствия,
повреждения или удаления паращитовидных желез (рис. 28.43).
Рис. 28.43. Наиболее частые причины первичного железистого гипопаратиреоза
П севдогипопаратиреоз
Внежелезистый (периферический) гипопаратиреоз называют также псевдогипопаратиреозом.
Псевдогипопаратиреоз (например, болезнь Олбрайта) — наследуемое забо­
левание, характеризующееся избыточно повышенной резистентностью органовмишеней к ПТГ.
Проявления псевдогипопаратиреоза
Проявления псевдогипопаратиреоза рассмотрены в статье «Гипокальциемия» и представлены на рис. 28.44.
--------- -----..
|Катаракта|
11рпяпгтния гипопаратиреоза
|------4,
..
Повышение
Нервно-психические
нервно-мышечной
расстройства
возбудимости
Г
Г
Гипокальциемия,
сочетающаяся с
гиперфосфатемией
1
Расстройства
дыхания
Тетанус|
Судороги
различных
групп мышц
Нарушения
мочеиспускания
1
Желудочнокишечные
расстройства
Рис. 28.44. Основные проявления гипопаратиреоза
• Гипокалыдаемия (уменьшение содержания Са2+ в межклеточной жидкости
и крови), которая, как правило, сочетается с гиперфосфатемией. Причины
гипокальциемии:
—нарушение абсорбции Са2+ в кишечнике;
—торможение мобилизации Са2+ из костей;
—уменьшение реабсорбции Са2+ в канальцах почек;
—дефицит активной формы витамина Б 3 — холекальциферола.
Последствия гипокальциемии:
—расстройство трансмембранного распределения и соотношения ионов:
кальций/фосфор, натрий/калий между цитоплазмой и интерстицием;
—гипомагниемия (уменьшение содержания М§2+ в межклеточной жидко­
сти и крови), что потенцирует транспорт Ыа' в клетки и выход из них К+;
—нарушения электрогенеза возбудимых структур;
—повышение возбудимости нервных и мышечных клеток, формирование
состояния судорожной готовности, развитие тетануса и судорог.
• Повышение нервно-мышечной возбудимости с развитием тетануса и судорог:
—тетанус — состояние длительного сокращения, максимального напря­
жения мышц, обычно симметричных групп (сгибателей конечностей),
в тяжелых случаях — мышц лица. Причина тетануса — чрезмерно частые
импульсы возбуждения, поступающие к мышце по нервным волок­
нам. В этих условиях расслабления мышечных волокон не наступает.
Наблюдается примерно у 90% больных;
—судороги — непроизвольное сокращение групп мышц, сменяющее­
ся их расслаблением (клонические судороги) либо продолжающееся
в течение длительного времени (тонические судороги). Судороги сопро­
вождаются сильной болью и наблюдаются более чем у 50% пациентов
с гипопаратиреозом. Судороги определенных групп мышц характеризу­
ются своеобразными проявлениями:
о тонические судороги лицевой мускулатуры напоминают сардониче­
скую гримасу;
о тоническая судорога мышц спины приводит к опистотонусу;
о судороги мышцы гортани и бронхов (ларингоспазм и бронхоспазм
соответственно) могут вызвать асфиксию;
о судороги мышц пищевода, желудка, кишечника, мочевого пузыря
сопровождаются болями, нарушением прохождения пищи по пищево­
ду, поносом или запором, расстройствами мочеиспускания.
• Расстройства функций органов, тканей и физиологических систем:
—нарушения нервной и психической деятельности; характеризующиеся:
о эпилептиформными приступами;
о нейрогенными расстройствами чувствительности и движений (вслед­
ствие кальцификации структур мозга, чаще диэнцефальной области,
базальных ганглиев и мозжечка);
о отеком мозга (наблюдается при затянувшихся приступах тетании
в связи с нарушением мозгового кровообращения, развитием гипок­
сии, расстройствами обмена ионов и жидкости);
о повышенной нервной возбудимостью, сочетающейся с сильной
мышечной возбудимостью;
о бессонницей;
о депрессией, приступами тоски;
о развитием невротических состояний;
о психическими расстройствами (наблюдаются при длительной и выра­
женной гипокальциемии);
—расстройства кровообращения, которые включают:
о нарушения центральной и органотканевой гемодинамики;
о микрогемоциркуляции (обусловлены изменениями сердечного выбро­
са, колебаниями тонуса артериол, изменениями ОЦК).
Все эти показатели меняются по-разному, в зависимости от доминиро­
вания симпатико-адреналовой или парасимпатической системы. У каждо­
го пациента они могут колебаться (вплоть до альтернативных изменений)
как при различных эпизодах тетании и судорог, так и в межприступный
период. Так, тахикардия и артериальная гипертензия могут смениться брадикардией и артериальной гипотензией и коллапсом;
—нарушения дыхания, проявляющиеся:
о альвеолярной гиповентиляцией;
о нередко признаками асфиксии (при ларингоспазме и/или бронхоспаз­
ме);
—расстройства функций пищеварения, характеризующиеся:
о нарушениями глотания;
о признаками пилороспазма;
о рвотой;
о болями в животе;
о запором, сменяющимся поносом.
Причины расстройств пищеварения:
—спазм мышц ЖКТ;
—нарушение сбалансированности симпатических и парасимпатических
влияний на органы пищеварения;
—нарушения мочеиспускания; результат спазма мышц мочевого пузыря;
—развитие катаракты; обусловлено кальцификацией ткани хрусталиков
при длительном течении гипопаратиреоза.
Другие расстройства при гипопаратиреозе обычно не носят обязательного
характера и могут встречаться с разной частотой у различных пациентов.
Как правило, они развиваются при длительном течении патологии. К ним
относятся:
—изменения в костях (остеосклероз, периостоз трубчатых костей, обыз­
вествление реберных хрящей);
—кальцификация стенок артерий, связок, сухожилий;
—нарушения роста зубов, дефекты эмали, кариес;
—изменения производных эктодермы (шелушение кожи, ранняя седина,
выпадение волос);
—другие расстройства.
НАРУШЕНИЯ ЭНДОКРИННОЙ ФУНКЦИИ
ПОЛОВЫХ ЖЕЛЕЗ
Развитие половой систем ы
В пренатальном развитии выделяют начальный, зародышевый и плодный
периоды.
Начальный период (концептус, проэмбрион, предэмбрион) — первая неделя
после оплодотворения.
Зародышевый период (эмбрион) — со второй по восьмую неделю развития
включительно. Эмбрион — общность клеток или существо, формирующееся
в стадии первичной полоски, но не ранее.
Плодный период (плод) — от девятой недели развития до конца беременности.
Половые признаки
Первичные половые признаки (детерминация пола, закладка гонад и их раз­
витие, некоторые этапы гаметогенезов) определяются при оплодотворении
и в эмбриональном периоде. Их развитие продолжается в плодном периоде
и после рождения.
Вторичные половые признаки. С начала пубертатного периода и вплоть до завер­
шения полового созревания формируются вторичные половые признаки.
Половая диф ференцировка
Генетическая детерминация пола происходит при оплодотворении.
Зигота генетически мужского пола содержит 22 пары аутосом и поло­
вые хромосомы ХУ, т.е. 46,ХУ. Кариотип зиготы генетически женского пола
46,XX.
Генетический мужской пол определяет У-хромосома, в том числе ген 5ЯУ,
относящийся к семейству ДНК-регуляторных генов 5ох. Ген 8 К У кодирует
регуляторный фактор ТОР {Те$1\$-Ое1егт1тщ РасЮг).
Хромосома 17 содержит бох-подобный ген 8ЯА1, мутации которого приводят
к реверсии пола (генетические мужчины имеют женский фенотип) и камптомелической дисплазии (2/3 больных генотипа XV имеют женский фенотип).
Гаметогенез
Первичные половые клетки образуются в стенке желточного мешка и на 5-й
неделе эмбриогенеза начинают мигрировать в гонадные валики: это зачатки
индифферентных гонад. Половые железы развиваются из гонадных валиков.
Критическим периодом развития индифферентных гонад является 8-я неделя
внутриутробного развития. До 45—50-х суток зачатки гонад не имеют поло­
вой дифференцировки. Под влиянием регуляторного фактора ТОР, а также
под влиянием генов 5ох гонадные валики развиваются как яички; при отсут­
ствии эффектов этих факторов развиваются яичники. Дифференцировку
других структур определяют мужские половые гормоны и мюллеров ингиби­
рующий фактор, которые продуцируются в яичках плода.
В плодном периоде первичные половые клетки дифференцируются в овогонии в развивающихся яичниках или в сперматогонии в яичках.
На этапе от ово- или сперматогоний до гамет различают несколько стадий,
в течение которых осуществляется и мейоз.
Сперматогенез начинается не ранее наступления половой зрелости. Мейоз
обусловливает образование сперматозоидов с разными половыми хромосома­
ми: сперматозоиды содержат либо Х-, либо У-хромосому. Известны случаи
происходящей при кроссинговере транслокации из хромосомы У в хромосо­
му X локуса 5КУ. кодирующего регуляторный фактор ТО Р.
Овогенез. В претерпевающих дифференцировку яичниках овогонии всту­
пают в стадию размножения, образуя овоциты первого порядка. К 7 мес
внутриутробного развития стадия размножения обрывается, овоциты первого
порядка в профазе I мейотического деления приобретают оболочку из фолли­
кулярных клеток (образуется примордиальный фолликул) и вступают в дли­
тельный период покоя, вплоть до наступления половой зрелости.
Овариально-менструальный цикл. На пике уровня лютеинизирующего гор­
мона (ЛГ) завершается первое мейотическое деление. Сигналом для заверше­
ния второго мейотического деления является оплодотворение: овоцит второго
порядка делится с образованием зрелой яйцеклетки (гаплоидный набор хро­
мосом) и второго полярного (направительного) тельца.
Половое созревание
Нормальное половое созревание (пубертат) происходит при переходе
от половой незрелости к взрослому состоянию половой зрелости. В этот
период вызванная ФСГ и ЛГ секреция половых стероидных гормонов ведет
к развитию вторичных половых признаков и репродуктивной способности.
Синтез и секрецию половых гормонов регулирует гормональная цепочка:
«гонадолиберин гипоталамуса — гонадотропины гипофиза».
Типовые формы патологии в результате
эндокринопатий половых желез
Типовые формы патологии, обусловленные нарушениями эндокринной
функции половых желез, подразделяются на три группы:
—нарушения половой дифференцировки;
—расстройства полового развития у девочек и половой функции у женщин;
—нарушения полового развития у мальчиков и половой функции у мужчин.
Нарушения половой дифференцировки
Вследствие расстройств половой кифференцировки рождается ребенок,
имеющий черты и г^жского, и женского пола, но не являющийся фено­
типически (!) ни мужчиной, ни женщиной.
Этиология, патогенез, проявления и терапия разных форм этой пато­
логии рассмотрены в статьях «Нарушения половой дифференцировки»,
«Синдром Клайнфелтера», «Синдром Шерешевского-Тернера» (см. прило­
жение «Справочник терминов»),
Эндокриногенные расстройства полового развития
и половой функции у лиц генетически женского пола
Половое созревание (пубертатный период) у девочек начинается в возрасте
8—13 лет и происходит в течение 3—4 лет.
К наиболее существенным признакам пубертата относятся определяемые
изменениями эндокринного статуса:
—рост и развитие молочных желез (телархе);
—лобковое и подмышечное оволосение;
— начало менструаций (менархе). Менструации появляются в среднем в воз­
расте 12,5 года; кровотечение обычно длится 4—5 сут; в течение первых
двух лет менструальный цикл может быть нерегулярным, а овуляций нет
до 17—18 лет примерно у 20% девушек;
— становление регулярного менструально-овариального цикла.
Половая функция женского организма (циклическое освобождение из яич­
ника овоцита, циклическая продукция женских половых гормонов, цикли­
ческие изменения слизистой оболочки маточных труб, матки, влагалища,
молочных желез) определяется функционированием эндокринного контура
между корой большого мозга, гипоталамусом, аденогипофизом, яичником
и органами-мишенями женских половых гормонов.
Типовыми формами патологии полового созревания и половой функции явля­
ются:
—преждевременное половое созревание;
—задержка полового созревания;
—эндокринная гипо- и гиперфункция яичников.
П реж девременное половое созревание у лиц
генетически женского пола
Половое созревание считается преждевременным, если какой-либо из вто­
ричных половых признаков появляется у девочек младше 7,5 года.
Различают три разновидности преждевременного полового созревания лиц
генетически женского пола:
—центральное (истинный пубертат);
— периферическое (ложный пубертат, псевдопубертат);
—частичное (неполное) преждевременное половое созревание.
Истинный преждевременный пубертат у девочек
90% всех случаев преждевременного полового созревания составляет истин­
ный (зависящий от гонадотропинов) пубертат. У девочек он встречается значи­
тельно чаще, чем у мальчиков. Истинным этот вариант пубертата называется
потому, что половое созревание организма происходит хотя и преждевременно,
но по обычной (как и в норме) схеме: активация гипоталамуса и синтез гонадолиберинов -> секреция гонадотропных гормонов -» синтез половых гормонов
формирование вторичных женских половых признаков.
Причины истинного преждевременного пубертата:
—преждевременная активация синтеза гонадолиберина; развивается при опу­
холях диэнцефальной области (например, при гамартомах, астроцитомах,
глиомах, пинеаломах, арахноидальных кистах), травмах головного мозга,
аномалиях развития головного мозга, гидроцефалии, энцефалитах;
—гиперпродукция гонадотропинов аденогипофизом; встречается обычно
при новообразованиях и аденомах аденогипофиза, энцефалитах, менин­
гитах, травмах головного мозга.
Проявления истинного преждевременного полового развития у девочек:
—изосексуальность развития организма (т.е. соответствие генетическому
и гонадному женскому полу);
—комплексность («гармоничность») развития (включающее ускорение роста
тела, телархе, лобковое и подмышечное оволосение, формирование дру­
гих характерных вторичных половых признаков);
—завершенность развития (характеризуется менархе и преждевременным
началом овуляций).
Преждевременный псевдопубертат (ложное преждевременное половое разви­
тие) у лиц генетически женского пола
Преждевременный псевдопубертат характеризуется ускорением роста тела
(как и при истинном преждевременном половом развитии). Однако псевдопу­
бертат никогда не завершается половым созреванием (нет овуляции и менархе).
Причина преждевременного псевдопубертата — автономный избыточный
синтез эстрогенов в яичниках или надпочечниках. Наблюдается, как прави­
ло, при гормонально-активных опухолях (например, при эстрогенпродуцирующей гранулезоклеточной бластоме яичника, кортикостероме, лютеоме
или кисте яичника).
Проявления ложного преждевременного полового развития у девочек показа­
ны на рис. 28.45. К ним относят:
— бивалентное (двоякое — как изосексуальное, так и гетеросексуальное)
направление полового развития организма:
Оизосексуалыюе половое развитие (совпадающее с генетическим и гонад­
ным женским полом) происходит при синтезе избытка эстрогенов;
о гетеросексуальное половое развитие (не совпадает с генетическим
и гонадным женским полом): у девочек формируются вторичные
мужские половые признаки. Причина гетеросексуального развития
организма — продукция избытка андрогенов опухолями надпочечни­
ков или яичников, а также гиперплазированной тканью коры надпо­
чечника (в том числе в связи с недостаточностью 21-гидроксилазы;
см. статьи «Гиперплазия коры надпочечника врожденная» и «Синдром
адреногенитальный» в приложении «Справочник терминов»);
—нарушение гармоничности развития организма — ускоренный рост тела,
мужской тип телосложения, лобковое оволосение, гирсутизм сочетается
с отставанием телархе;
— отсутствие завершенности полового развития (не наблюдается овуляции
и появления менархе).
Рис. 28.45. Проявления ложного преждевременного полового развития у девочек
Частичное преждевременное половое развитие
Неполное преждевременное половое развитие характеризуется ранним появле­
нием какого-либо одного или отдельных вторичных половых признаков при отсут­
ствии других.
Причины неполного преждевременного пубертата:
—преждевременное начало синтеза эстрогенов в яичниках, как правило,
в избыточном количестве (вызывает преждевременное телархе);
—избыточное образование андрогенов в коре надпочечников (приводит
к преждевременному лобковому и подмышечному оволосению);
— повышенная чувствительность клеток-мишеней к эстрогенам (например,
клеток молочной железы).
Проявления частичного преждевременного пубертата:
—телархе (обычно в возрасте до 2—4 лет, реже — после 6 лет);
— преждевременный рост лобковых и подмышечных волос. Нередко этот
признак имеет преходящий характер (обусловлен временным увеличе­
нием секреции андрогенов в коре надпочечников и/или чувствительно­
сти к ним тканей-мишеней).
Задержка полового созревания
Торможение полового созревания характеризуется отсутствием вторичных поло­
вых признаков к 14-летнему возрасту, а также отсутствием менструаций к 16-лет­
нему возрасту (первичная аменорея) при наличии вторичных половых признаков.
Виды и причины задержки полового развития у девочек
Различают первичные и вторичные формы гипогонадизма.
Первичный гипогонадизм (яичниковый, гипогонадотропный). Является
результатом наследуемой, врожденной или приобретенной яичниковой недо­
статочности (рис. 28.46).
Рис. 28.46. Основные причины первичного гипогонадизма у девочек
Причины первичного гипогонадизма у девочек:
—недостаточность яичников (наследуемая или врожденная);
—синдром Шерешевского-Тернера (при кариотипе 45,ХО яичниковая
недостаточность развивается у 60% пациентов, при мозаицизме, напри­
мер, 45,ХО/46,ХХ, — в 20% случаев);
—дисгенезия гонад у пациентов с кариотипом 46,XX или 46,ХУ;
—мутации генов, кодирующих синтез ферментов половых гормонов (напри­
мер, 17а-гидроксилазы, десмолазы, ароматазы, 17р-гидроксистероиддегидрогеназы);
—слабая чувствительность яичников к гонадотропным гормонам (синдром
резистентных яичников);
—поликистоз яичников;
—приобретенная недостаточность яичников — например, при иммунной аутоа­
грессии в «адрес» белков яичников, инфекционном поражении яичников,
воспалительных процессах — оофорите, облучении яичника, применении
химиотерапевтических средств (длительном и/или в высоких дозах).
Вторичный гипогонадизм (гипогонадотропный, внеяичниковый)
Внеяичниковый гипогонадизм обусловлен дефицитом гонадотропных гормо­
нов (ФСГ, ЛГ) транзиторного (преходящего) или постоянного (хронического)
характера.
Наиболее частые причины вторичного гипогонадизма:
— факторы, вызывающие дефицит гонадотропинов и транзиторный вторич­
ный гипогонадизм. К ним относятся:
о длительное состояние стресса;
о хронические истощающие заболевания (например, ХМЛ, остеомие­
лит, туберкулез);
о эндокринопатии (например, СД, синдром Иценко-Кушинга, гипотиреоидные состояния. Указанные формы патологии обусловливают
нарушение синтеза гонадотропинов в аденогипофизе и вторично­
яичниковую недостаточность);
—состояния, приводящие к хроническому вторичному гипогонадизму:
о патология гипоталамуса (обычно речь идет о ВПР переднего мозга, напри­
мер, проявляющихся торможением миграции нейронов, нейросекретирующих гонадолиберины, в область гипоталамуса. Введение таким паци­
ентам гонадолиберинов вызывает повышение уровней ФСГ и ЛГ в крови,
что свидетельствует о нормальном функционировании аденогипофиза);
о патология гипофиза (встречается сравнительно редко: например,
при тяжелых энцефалитах; травмах, кровоизлиянии или новооб­
разованиях в области турецкого седла; дистрофических процессах
в аденогипофизе. После введения гонадолиберинов этим пациентам
не повышается содержание гонадотропных гормонов в крови либо это
повышение незначительно).
Гипофункция яичников
Эндокринная недостаточность яичников подразделяется на первичную
и вторичную.
Первичная яичниковая недостаточность (первичный гипогонадизм). Это
состояние обусловлено патологией яичников. Оно заключается в недостаточ­
ной продукции ими половых гормонов, а также в нарушениях менструального
цикла. В связи с этим в крови, как правило, обнаруживают компенсаторно
увеличенный уровень ФСГ
Причины первичного гипогонадизма те же, что и при вторичном гипогонадизме, — вызывающие задержку полового развития у девочек (см. выше).
Вторичная яичниковая недостаточность (вторичный, или внеяичниковый гипо­
гонадизм). Является результатом дефицита либо гонадолиберинов гипоталаму­
са, либо гонадотропных гормонов аденогипофиза.
Причины внеяичниковой недостаточности те же, что и при вторичном гипогонадизме (см. выше).
Проявления первичной и вторичной эндокринной недостаточности яичников
весьма сходны. К основным среди них относят следующие:
—нарушения менструального цикла, проявляющиеся дисфункциональны­
ми маточными кровотечениями. Их характеристика приведена в ста­
тье «Кровотечение маточное дисфункциональное» (см. приложение
«Справочник терминов»);
—аменорея — отсутствие менструаций более 6 мес у женщин с ранее периоди­
ческим их наступлением (вторичная аменорея), а также отсутствие менархе
у девочек старше 16 лет (первичная аменорея). Характеристика аменореи
изложена в статье «Аменорея» (см. приложение «Справочник терминов»);
— бесплодие — отсутствие беременности в течение одного года регулярной
половой жизни без использования методов контрацепции. Бесплодие
регистрируется у 10-15% супружеских пар (из них около 30—40% случа­
ев — результат мужского бесплодия). Характеристика бесплодия изло­
жена в статье «Бесплодие» (см. приложение «Справочник терминов»).
Эндокринная гиперфункция яичников характеризуется гиперандрогенией
или гиперэстрогенией (рис. 28.47).
Виды гиперфункции яичников у женщин и ее основные причины|
Гиперандрогения
Основные причины:
- гиперпродукция люлиберина;
- гиперсекреция андрогенов
надпочечниками;
- гиперинсулинемия;
-о п у х о л и яичников;
- недостаточность
Зр-гидроксистероиддегидрогеназы
т
'Гиперэстрогения
Основная причина:
-трансф орм ация в коже и
жировой клетчатке избытка
андрогенов в эстрогены
Рис. 28.47. Виды гиперфункции яичников и ее основные причины
Гиперандрогенные состояния характеризуются повышенной продукцией и/или
эффектами действия андрогенов.
Гиперандрогении разной степени выраженности выявляются у 10-15%
женщин.
Наиболее частые причины гиперандрогении:
—гиперпродукция люлиберина нейронами гипоталамуса и/или ЛГ клетками
аденогипофиза (наблюдается, например, при опухолях, гипертрофии
или гиперплазии их). Избыток ЛГ обусловливает гиперплазию внеш­
ней оболочки и зернистого слоя фолликулов, а также гиперплазию
стромы яичников. Указанные изменения сопровождаются увеличени­
ем синтеза андрогенов в яичниках и появлением признаков вирилиза­
ции организма;
—избыток андрогенных стероидов надпочечникового генеза, особенно в
период полового созревания. Эти андрогены трансформируются в пери­
ферических тканях в эстрон, который активирует продукцию люлибери­
на. ЛГ же приводит к увеличению синтеза андрогенов яичниками. В тка­
нях они превращаются в эстрон, что замыкает порочный круг, нередко
самопотенцирующийся;
—избыток инсулина в организме. Это активирует синтез лютеотропного
гормона и далее андрогенов как в яичниках, так и в надпочечниках;
—опухоли яичника, продуцирующие избыток андрогенов (например, лейдигомы, синтезирующей тестостерон, из клеток Лейдига);
—недостаточность Зр-гидроксистероиддегидрогеназы. Недостаточность этого
фермента приводит к избыточному содержанию в организме дегидроэпиандростерона.
Проявления гиперандрогенных состояний:
—повышенное содержание в крови андростендиона и тестостерона;
—увеличение соотношения в крови гонадотропинов ЛГ/ФСГ; обычно более 3;
—гирсутизм;
—аменорея;
— бесплодие;
— ожирение.
Гиперэстрогенные состояния характеризуются избыточным образованием
и/или эффектами эстрогенов в организме.
Наиболее частая причина гиперэстрогении — повышенное содержание
в крови андрогенов яичникового и/или надпочечникового генеза. Андрогены
в коже и жировой ткани трансформируются в эстрогены, что ведет к увели­
чению их концентрации в крови.
Проявления гиперэстрогенных состояний:
—повышение содержания в крови и моче эстрогенов;
— снижение уровня гонадотропных гормонов (по механизму обратной связи);
— преждевременное половое созревание у девочек по изосексуальному типу;
—ускоренный рост организма (опережающий возрастную норму);
—нарушения менструального цикла, обычно в виде меноррагии. Последняя
обусловлена длительно повышенным уровнем эстрогенов и нарушением
периодических колебаний содержания прогестерона в крови.
Эндокриногенные нарушения полового развития
и половой функции у лиц генетически мужского пола
Пубертатный период у мальчиков начинается в возрасте 9,5—13,5 года
и продолжается около 3 лет. Увеличение яичек обычно является первым
признаком полового созревания. К другим признакам полового созревания
относят акне, пубертатную гинекомастию и ряд других.
Проявляются эндокриногенные нарушения полового развития и половой
функции у лиц мужского пола следующими типовыми формами патологии:
—преждевременным половым развитием;
—задержкой полового развития;
—гипофункцией яичек.
Преж девременное половое развитие
Преждевременный (первичный) пубертат — состояние, характеризующееся
появлением всех или отдельных вторичных половых признаков у мальчиков
до 9-летнего возраста.
Нередко преждевременный пубертат сочетается с эмоциональными и пове­
денческими расстройствами, а также нарушением социальной адаптации.
Различают истинное и ложное преждевременное развитие у мальчиков.
Истинное преждевременное половое развитие мальчиков — результат гипер­
функции гипоталамо-гипофизарной системы. Характеризуется полным пре­
ждевременным половым развитием, которое включает вирилизацию и акти­
вацию сперматогенеза в яичках.
Причина истинного преждевременного пубертата у мальчиков — пре­
ждевременная активация секреции гонадолиберина нейронами гипоталамуса
и, как следствие, гонадотропных гормонов аденогипофиза. Обычно разви­
вается при опухолях в области заднего гипоталамуса, серого бугра, эпифиза,
при травматических повреждениях диэнцефальной области мозга, участву­
ющей в синтезе гонадолиберинов; энцефалитах, состояниях и патологических
процессах, повышающих внутричерепное давление (абсцессы, новообразова­
ния, гидроцефалия, нарушение оттока ликвора и др.).
Ложный пубертат у мальчиков. В отличие от первичного, ложное (вторичное)
преждевременное половое развитие у мальчиков возникает в результате авто­
номной гиперпродукции андрогенов или хорионического гонадотропина.
Ложный пубертат характеризуется неполным преждевременным поло­
вым развитием у мальчиков — появлением у них признаков вирилизации,
но без активации сперматогенеза.
Преждевременное половое созревание подразделяют (в зависимости от соот­
ветствия или несоответствия полового развития генетическому полу):
—на изосексуальное (оно совпадает с генетическим полом и характеризует­
ся признаками вирилизации у мальчиков);
—гетеросексуальное (не совпадает с генетическим мужским полом и соче­
тается с феминизацией мальчиков).
Наиболее частые причины преждевременного ложного пубертата:
— андрогенпродуцирующие опухоли яичек (например, лейдигомы, синтезирую­
щие тестостерон; сертолиомы и арренобластомы, образующие андрогены);
—гиперплазия клеток Лейдига и синтез ими избытка тестостерона;
—увеличение содержания в крови андрогенов, секретируемых в коре надпо­
чечников при их гиперплазии или поражении их опухолью.
Проявления преждевременного пубертата
Проявления истинного и ложного преждевременного полового созревания —
результат избытка в организме мужских половых гормонов. Это вызывает:
—признаки вирилизации (появление лобкового и подмышечного оволосе­
ния, огрубение голоса, увеличение яичек и полового члена и др.; первые
симптомы вирилизации могут наблюдаться уже в двухлетнем возрасте);
—низкий рост (обусловлен преждевременным прекращением эпифизарно­
го роста костей).
Задержка полового созревания
Задержкой пубертата считается отсутствие у мальчиков признаков полового
созревания к 14-летнему возрасту.
Причины задержки полового созревания:
—дефицит гонадолиберинов гипоталамуса и/или гонадотропинов аденоги­
пофиза (при повреждении ядер гипоталамуса или гипофиза опухолями,
в результате кровоизлияния или травмы, воспалительных процессов
и др.);
—сниженная выработка тестостерона яичками (в результате их травмы,
роста новообразований, нарушения развития, воспаления яичек —
орхита и т.п.);
—пониженная чувствительность тканей-мишеней к действию тестостерона.
Проявления задержки пубертата:
—признаки евнухоидизма:
о недоразвитые яички и пенис;
о слабовыраженные вторичные половые признаки или их отсутствие;
о женоподобный (девичий) голос;
о ожирение;
о диспропорция скелета (размах рук более чем на 6-8 см превышает рост,
что обусловлено запаздывающим завершением эпифизарного роста
трубчатых костей);
о сниженное содержание тестостерона в крови (при нормальном уровне
глобулина, связывающего половые гормоны).
Мужской гипогонадизм
Причины тестикулярной недостаточности:
—те же, что и при задержке полового развития мальчиков (см. выше);
—дефекты яичек:
о первичные (наследуемые или врожденные); наблюдаются, например,
у пациентов с различными вариантами синдрома Клайнфелтера. При этом
снижается продукция андрогенов и нарушается сперматогенез;
о вторичные (приобретенные); развиваются при травме, вирусном
или бактериальном воспалении, нарушении кровообращения, воздей­
ствии радиации, ЛС (например, наркотиков, спиронолактона) и др.
Проявления мужского гипогонадизма:
—снижение полового влечения;
—импотенция;
—бесплодие — неспособность мужчины к зачатию ребенка на протяже­
нии одного года регулярной половой жизни без использования кон­
трацептивов и независимо от возможности совершения полового акта
(не менее 35% случаев бесплодия обусловлено нарушением половой
функции мужчин. Характеристика мужского бесплодия приведена в ста­
тье «Бесплодие» приложения «Справочник терминов»).
Наряду со сказанным выше, половое развитие у мужчин существенно
не нарушено. В связи с этим телосложение и тембр голоса у них, как правило,
в диапазоне нормы.
Глава 29
ТИПОВЫЕ ФОРМЫ ПАТОЛОГИИ НЕРВНОЙ
СИСТЕМЫ
ОБЩАЯ ЭТИОЛОГИЯ РАССТРОЙСТВ
НЕРВНОЙ ДЕЯТЕЛЬНОСТИ
Общая этиология нейропатий рассматривает:
—причины повреждения;
—условия, определяющие патогенный эффект причин ее повреждения
(факторы риска).
Причины повреждения нервной системы
Факторы, вызывающие повреждение элементов нервной системы, могут
быть экзогенными и эндогенными.
Экзогенные причины повреждения нервной систем ы
Экзогенные нейротропные повреждающие факторы могут быть различной
природы:
- физической (например, механическая травма, ионизирующая радиация,
снижение или значительное увеличение р 0 2 во вдыхаемом воздухе);
- химической (этиловый, метиловый и другие спирты; ядохимикаты:
фосфорорганические соединения, применяемые в сельском хозяй­
стве, например пестициды, или в быту, в частности хлорофос; нервно­
паралитические отравляющие вещества, например зарин; фармаколо­
гические препараты центрального действия, в том числе наркотики;
токсины растительного происхождения, например стрихнин, кураре);
- биологической (например, нейротропные вирусы: возбудители бешен­
ства, полиомиелита, герпеса; микроорганизмы: возбудители сифилиса,
лепры; микробные токсины: ботулинический, дифтерийный, столбняч­
ный).
—психогенной (например, психотравмирующие ситуации, устрашающие
образы, звуки, ощущения). Наиболее часто в клинической практике
патогенную роль играет слово как стрессорный раздражитель, а также
другие воздействия, реализующие свое влияние условнорефлекторно,
через вторую сигнальную систему.
Эндогенные причины повреждения нервной систем ы
Причины эндогенного характера, вызывающие повреждение нервной
системы, представлены на рис. 29.1.
Рис. 29.1. Эндогенные причины повреждения нервной системы
Факторы риска
К наиболее значимым условиям, определяющим патогенность факто­
ров, которые воздействуют на нервную систему (факторы риска), относят
интенсивность, длительность, частоту и периодичность воздействия, а также
состояние нервной системы в момент действия патогенного агента и состоя­
ние гематоэнцефалического барьера.
Интенсивность, длительность, частота и периодичность воздействия. Значи­
тельные и труднообратимые нарушения нервной деятельности могут воз­
никать под влиянием не только сильных, но и слабых патогенных факторов
при определенных режимах их воздействия. Например, небольшие дозы
алкоголя, наркотиков, ЛС при достаточной продолжительности и частоте
их потребления способны вызывать грубые нарушения ВНД, движений, чув­
ствительности и другие нейрогенные расстройства.
Состояние нервной системы в момент действия патогенного агента. Состояние
нервной системы в момент действия патогенного агента определяется ее
генетическими особенностями (например, типом ВНД) и предшествующими
структурно-функциональными повреждениями.
Клинические проявления последних к моменту действия данного пато­
генного фактора могут быть компенсированными (скрытыми) или носить
характер следовых реакций. На таком фоне патогенность повреждающих воз­
действий увеличивается.
Состояние гематоэнцефалического барьера. Патологическая проницаемость
гематоэнцефалического барьера для экзо- и эндогенных факторов может
возникать при действии ионизирующей радиации, интоксикации спиртами,
микробными токсинами, в условиях охлаждения организма, развития тяжелых
стрессовых ситуаций, различных шоковых состояний. Повышение проницае­
мости гематоэнцефалического барьера имеет особое значение для нарушения
иммунной автономии головного мозга и развития вследствие этого состояний
иммунной аутоагрессии с поражением нервной системы (например, рассеян­
ного склероза, энцефаломиелита).
ОБЩИЙ ПАТОГЕНЕЗ
РАССТРОЙСТВ НЕРВНОЙ ДЕЯТЕЛЬНОСТИ
В этом разделе рассмотрены механизмы повреждения нейронов и наруше­
ния межнейронных взаимодействий, а также механизмы расстройств интегра­
тивной деятельности нервной системы.
Повреждение нейронов
Механизмы повреждения нейронов могут носить специфический и неспе­
цифический характер.
Н еспециф ические механизмы повреждения нейронов
Общие механизмы повреждения клеток подробно рассмотрены в главе 5
«Повреждение клетки» (см. т. 1). Неспецифические механизмы повреждения
нейронов представлены на рис. 29.2.
Рис. 29.2. Основные механизмы повреждения нейронов (неспецифические)
Нарушения энергетического обеспечения нейронов
Энергетическое обеспечение нейронов нарушается в результате действия
нескольких механизмов.
• Уменьшенное поступление в нейроны глюкозы и кислорода. Причины:
—гипоксемия;
—гипогликемия;
—анемия;
—снижение мозгового кровообращения;
—увеличение диффузионного пути глюкозы и 0 2 от микрососуда до ней­
ронов (например, при отеке мозга).
Глюкоза — основной энергетический субстрат нервных клеток.
Аэробное расщепление субстратов обеспечивает образование почти 80%
энергии, потребляемой мозгом.
В норме головной мозг утилизирует около 20% глюкозы плазмы крови
(ГПК) и всего потребляемого организмом кислорода. Нейроны весьма чув­
ствительны к дефициту кислорода и нарушению их энергообеспечения.
Наличие в нервных клетках гликогена как резервного источника энергии
лишь отчасти обеспечивает функционирование жизненно важных отделов
нервной системы.
В конечном счете дефицит глюкозы и кислорода приводит к гибели нейро­
нов, в первую очередь — коры головного мозга (корковые нейроны погибают
при аноксии и при отсутствии глюкозы в течение нескольких минут).
• Снижение активности ферментов биологического окисления. Причины:
—ингибирование и/или денатурация ферментов (например, при отравлении
цианидами и солями тяжелых металлов или при эццогенной интоксика­
ции);
—недостаточная активность энзимов (например, при генетических дефек­
тах, авитаминозах В,, В2);
—нарушение компартментализации ферментов (например, при дезорганиза­
ции мембран митохондрий под воздействием ионизирующей радиации,
высокой температуры, продуктов нарушенного обмена).
• Разобщение процессов окисления и фосфорилирования в митохондриях ней­
ронов. Причины:
—избыток в нейронах Са2+;
—накопление в нервных клетках жирных кислот.
• Нарушение транспорта энергии от мест образования макроэргических соеди­
нений до мест расходования энергии. Основная причина:
—потеря нейронами ВВ-фракции КФК при повреждении их мембран.
• Расстройство процессов использования энергии в нейронах. Наиболее частая
причина — снижение активности АТФаз (в условиях ацидоза, интоксика­
ции, ионного дисбаланса).
Расстройства синтеза белка
Наиболее частые причины нарушения белкового синтеза в нейронах:
—дефицит аминокислот;
— нарушения энергообеспечения нейронов;
—снижение активности ферментов протеосинтеза;
—деструкция шероховатой эндоплазматической сети (субстанция Ниссля),
в которой реализуется протеосинтез.
Расстройства синтеза белка приводят к катастрофическим последствиям
для всех сторон жизнедеятельности нервных клеток и в итоге к их гибели.
Дисбаланс ионов и жидкости
Ионный гомеостаз нейронов обеспечивается работой энергозависимых
калиевых, натриевых, кальциевых и других ионных каналов.
Типичные проявления ионного дисбаланса (возникающие, например,
при энергодефиците):
—накопление № + и Са2+ в нейронах;
—избыток К+ во внеклеточном пространстве.
Последствия дисбаланса ионов в нейронах:
—стойкая деполяризация плазмолеммы нейрона (приводит к прекращению
нормальной функции нервных клеток);
— повышение осмотического давления в нейронах, их набухание и последу­
ющая гибель.
Повреждение мембран нейрона
Этот процесс является универсальным механизмом большинства наруше­
ний нервной деятельности.
Наиболее частые причины повреждения мембран:
—чрезмерное образование активных форм 0 2 и усиления свободнорадикаль­
ного перекисного окисления липидов;
—активации эндогенных фосфолипаз;
—механическое растяжение мембран;
—воздействие на мембраны амфифильных соединений.
Последствия повреждения мембран:
—прекращение функциональной активности нейронов (электрогенез);
—нарушение всех проявлений жизнедеятельности нервных клеток (напри­
мер, синаптическая передача, синтез белка, аксонный транспорт и др.);
—гибель нейронов путем аутолиза, апоптоза и некроптоза (см. раздел
«Гибель клетки» в главе 5 «Повреждение клетки», т. 1).
Апоптоз
Важная роль апоптоза — генетически контролируемого процесса гибели
нейронов — доказана для патогенеза многих форм невропатологии, особенно
для нейродегенеративных заболеваний (например, для болезней Паркинсона,
Альцхаймера (Альцгеймера); старческой деменции; бокового амиотрофиче­
ского склероза и др.).
Причины апоптоза нейронов:
—гипоксия нервной ткани любого типа, особенно действующая длитель­
ное время (например, при атеросклерозе церебральных сосудов; опухо­
лях головного или спинного мозга, сдавливающих сосуды);
—внутриклеточный ацидоз (например, при значительной ишемии ткани
мозга);
—избыточная генерация радикалов кислорода, липидов и других веществ
в ткани мозга (например, в условиях гипоксии и/или гипероксигенации
мозга, отравлении нейротоксическими агентами).
Механизмы апоптоза изложены в разделе «Апоптоз» в главе 5 «Повреждение
клетки» (см. т. 1).
Описанные выше механизмы повреждения нейрона тесно взаимосвяза­
ны. Они нередко потенцируют друг друга, образуя порочные круги (агси1и$
уШозиз). Формирование таких порочных кругов — неспецифическая, стандарт­
ная реакция нейрона на разные патогенные воздействия.
Специф ические механизмы повреждения нейронов
Патогенетическую основу большинства форм нарушений нервной дея­
тельности составляют раздельные или сочетанные расстройства специфи­
ческих для нейрона процессов метаболизма различных нейромедиаторов
(рис. 29.3).
• Расстройство синтеза нейромедиаторов. Наиболее частые причины:
—нарушение энергетического обеспечения процесса биосинтеза нейроме­
диаторов;
—дефицит субстратов их синтеза;
—генетические дефекты ферментов биосинтеза нейромедиаторов;
—ингибирование ферментов их синтеза.
• Замедление аксонного транспорта нейромедиаторов. Наиболее частые
причины нарушения аксонного транспорта рассмотрены далее (раздел
«Механизмы нарушений межнейронных взаимодействий»),
• Нарушение выделения нейромедиатора в синаптическое пространство (см.
статью «Нейромедиаторы» в приложении «Справочник терминов»),
• Изменение скорости удаления нейромедиатора (см. статью «Нейромеди­
аторы» в приложении «Справочник терминов»).
• Нарушение взаимодействия нейромедиатора с его рецепторами (см. статью
«Нейромедиаторы» в приложении «Справочник терминов»).
Рис. 29.3. Основные механизмы повреждения нейронов (специфические)
Запуск описанных выше неспецифических и специфических механизмов
повреждения нейрона обусловливает нарушение восприятия, анализа, генера­
ции и проведения возбуждения нейронами.
М еханизмы нарушений межнейронного взаимодействия
Механизмы нарушений межнейронных взаимодействий изображены
на рис. 29.4.
Рис. 29.4. Механизмы нарушений взаимодействия нейронов
В основе нарушений межнейронных взаимодействий находятся расстрой­
ства физико-химических процессов и форм функционального взаимодействия
нейронов.
Расстройства физико-химических процессов
Существует несколько вариантов расстройств физико-химических процес­
сов, обеспечивающих межклеточные взаимодействия в нервной системе:
—нарушения мембранного электрогенеза (включая формирование и прове­
дение ПД по аксонам);
— расстройства синаптической передачи (включая секрецию и рецепцию
нейромедиатора);
—нарушения аксонного транспорта нейромедиатора.
Расстройства форм функционального взаимодействия нейронов
Среди форм функциональных взаимодействий нейронов выделяют жестко
детерминированную и вероятностно-статистическую формы.
Жестко детерминированная (жестко программная) стандартная форма ответ­
ной реакции определяется генетически запрограммированными особенностя­
ми реагирования нейронов на воздействия и наличием анатомических связей
между нейронами, составляющими ту или иную функциональную совокуп­
ность (ядра, проводящие пути, нейронные сети и т.п.).
Стохастическая (вероятностно-статистическая) форма реагирования нейро­
нов означает, что один и тот же стимул может вовлекать в ответные реакции
разные нейроны или вызывать неодинаковые ответы. Зависимость между
воздействием и ответной реакцией носит вероятностный характер. В основе
этой формы взаимодействия лежит изменчивость текущего функционально­
го состояния нейронов и функциональная избыточность нейронов и связей
между ними.
Стохастичность межнейронного взаимодействия обеспечивает высо­
кую функциональную пластичность, а также значительные компенсаторно­
приспособительные возможности нервной системы, предотвращающие воз­
можность «случайных» патогенных последствий при различных воздействиях.
В норме обе формы межнейронного взаимодействия сбалансированы
и дополняют друг друга. Нарушение этого баланса приводит к расстройствам
нервной деятельности. Например, в агональной стадии процесса умирания
наблюдается переход нейронов дыхательного центра на жестко программный
режим деятельности. Обеспечивая необходимый минимум легочной венти­
ляции, дыхательный центр утрачивает возможность реагирования на допол­
нительные воздействия. В условиях дефицита энергии дыхательный центр
не реагирует на афферентные стимулы от синокаротидных рефлексогенных
зон, рецепторов растяжения легких и других регионов. Такое состояние дыха­
тельного центра называют функциональной деафферентацией.
Механизмы расстройств интегративной деятельности
нервной системы
Механизмы расстройств интегративной деятельности нервной системы
заключаются в нарушениях функционирования одного или нескольких зве­
ньев нервной системы: афферентного, центрального и эфферентного.
• Афферентные нарушения интегративной функции нервной системы свя­
заны с расстройствами восприятия различных воздействий и проведения
сигнала от афферентных структур к нервным центрам.
• Центрогенные расстройства характеризуются расстройствами процессов
анализа афферентных сигналов, синтеза и генерации эфферентного сиг­
нала нервными центрами.
• Эфферентные нарушения заключаются в расстройствах проведения сигна­
лов из центра и их восприятия исполнительными структурами.
Указанные расстройства могут приводить к нарушениям деятельности
функциональных и физиологических систем организма, а при тяжелых пора­
жениях — к их распаду.
Типовые формы нарушений
деятельности нервной системы
Типовые формы нарушений деятельности нервной системы представлены
на рис. 29.5.
Рис. 29.5. Типовые формы расстройств деятельности нервной системы
Все многообразные типовые формы расстройств деятельности нервной
системы подразделяются на три группы (по критериям зависимости от интен­
сивности нервных влияний на ткани и органы, адекватности ответа нервной
системы на них и нарушенного вида нервной деятельности).
По критерию интенсивности выделяют:
— патологическое усиление нервных влияний на ткани и органы;
— патологическое ослабление их.
По адекватности ответа нервной системы на воздействия говорят о фазовых
состояниях.
По критерию преимущественно нарушенного вида нервной деятельности выде­
ляют:
—нейрогенные расстройства движений;
—нарушения чувствительности;
—расстройства трофики тканей-мишеней;
— нарушения ВНД.
ПАТОЛОГИЧЕСКОЕ ОСЛАБЛЕНИЕ НЕРВНЫХ ВЛИЯНИЙ
Патологическое ослабление нервного влияния на эффекторные структуры
возникает при нарушениях центрального или эфферентного звена нервной
системы.
Причины ослабления нервных влияний на ткани-мишени
Причины патологического ослабления нервных влияний — органические
повреждения и/или функциональные изменения центрального аппарата нерв­
ной регуляции, а также нарушения в эфферентном звене системы нервного
контроля.
• Органические повреждения структур центральной нервной системы. Наиболее
часто они бывают результатом:
—механических травм головного и/или спинного мозга, а также органных
и тканевых нервных структур (например, нервных сплетений кишечни­
ка, брюшной и грудной полости, малого таза и др.);
—воспалительных процессов в нервной ткани (например, энцефалит, менин­
гит, неврит);
—нейрогенных опухолей (например, головного или спинного мозга
и их оболочек);
—дегенеративных процессов в нервной ткани (например, при боковом амио­
трофическом склерозе, гибели нейронов коры большого мозга при болез­
ни Альцгеймера, стрионигральной дегенерации при атаксии и др.), нару­
шения кровообращения (ишемия, венозная гиперемия, стаз).
Возникающие при органических повреждениях нервных образований рас­
стройства усугубляются развивающимся в нейронах вокруг очага повреждения
(эту зону обозначают как пенумбра) состоянием так называемого охранитель­
ного торможения. Оно предотвращает или уменьшает дальнейшее нарастание
патологических изменений в нейронах. Однако это увеличивает степень дефек­
та нервной функции. Например, при локальном нарушении кровообращения
в двигательном анализаторе выраженность паралича определяется, с одной
стороны, размером очага органического повреждения, а с другой — тормо­
жением деятельности близлежащих неповрежденных нейронов, что усиливает
дефект регуляции движений. Со временем (по мере снятия охранительного
торможения) двигательная функция может в какой-то степени восстановиться
даже при сохранении структурных повреждений мозговой ткани.
• Функциональные расстройства в нервных центрах. Причинами их обычно
являются:
—подавление возбудительного процесса (например, при наркозе);
—гиперактивация ядер ЦНС, оказывающих тормозное влияние на эффекторы
(например, в условиях чрезмерной активации нейронов ретикулярной
формации продолговатого мозга, оказывающей тормозное нисходящее
влияние на структуры спинного мозга, развиваются расстройства дви­
жений, вплоть до парезов мышц, а также снижается чувствительность
тканей. В основе этих феноменов лежит угнетение полисинаптических
рефлексов в спинном мозге под влиянием тормозных стимулов от ней­
ронов ретикулярной формации).
• Расстройства в эфферентном звене системы нервного контроля. Их вызывают:
—нарушение проведения нервных импульсов по нейронам (частичное или пол­
ное);
— расстройства транспорта веществ по аксонам;
—расстройства восприятия нервных воздействий клетками-мишенями
(например, в условиях гипоксии, дисбаланса ионов, изменения числа
и аффинности рецепторов к нейромедиаторам).
Возникающий при значительном снижении или выпадении нервно­
го контроля комплекс метаболических, нейромедиаторных и структурно­
функциональных изменений в постсинаптических нейронах, тканях и органах
получил название «денервационный синдром».
ПАТОЛОГИЧЕСКОЕ УСИЛЕНИЕ НЕРВНЫХ ВЛИЯНИЙ
НА ЭФФЕКТОРНЫЕ СТРУКТУРЫ
Патологическое усиление нервных влияний на структуры-мишени раз­
вивается вследствие первичного либо вторичного чрезмерного повышения
уровня и/или длительности процесса возбуждения в нейронах.
Первичное чрезмерное усиление возбуждения нейронов
Причинами избыточного первичного нейрогенного возбуждения нейронов
могут быть:
—увеличение притока возбуждающей афферентации к нейронам (например,
при психогенном стрессе, болевом раздражении, повреждениях чув­
ствительных нервов);
—пролонгирование действия возбуждающих нейромедиаторов на нейроны
(например, при повышенном выделении нейромедиатора в синаптиче­
скую щель, снижении скорости его разрушения и/или удаления);
—повышение чувствительности нейронов к возбуждающим сигналам (напри­
мер, в результате избыточной деполяризации нейронов, вызванной
повышенным содержанем К+ в межклеточном пространстве).
Вторичное чрезмерное повышение интенсивности и/или продолжительности
возбуждения нейронов, уже находившихся в состоянии повышенной активности
Причинами вторичной гиперактивации нейронов могут быть:
—деафферентация нейронов обусловливает блокаду поступления тормозных
сигналов к нейронам или нервным образованиям (например, перерезка
в эксперименте ствола мозга между передним и задним четверохолмием
по Шеррингтону вызывает характерный «феномен растормаживания» —
децеребрационную ригидность);
—уменьшение выделения тормозных нейромедиаторов снижает степень тор­
можения нейронов (например, блокирование столбнячным токсином
секреции глицина из вставочных нейронов спинного мозга приводит
к развитию судорог в виде опистотонуса, что характерно для столбняка);
— блокада постсинаптических рецепторов тормозных нейромедиаторов обуслов­
ливает чрезмерную активацию нейронов (например, блокирование стрих­
нином глициновых рецепторов вызывает развитие своеобразных судорог).
Тормозные механизмы нервной системы весьма чувствительны к различ­
ным патогенным воздействиям. В связи с этим «феномен растормаживания»
рассматривается как один из основных механизмов развития многих наруше­
ний нервной деятельности.
Гиперакгивация нервных структур может привести к формированию «застой­
ных очагов возбуждения». Их функционирование проявляется различными
нейропатологическими синдромами: приступами эпилептических судорог
или болей (например, таламического болевого синдрома, фантомных болей).
Образование таких очагов (например, в центрах гипоталамуса) может приво­
дить к выраженным вегетативным расстройствам: артериальной гипертензии,
полифагии, гиперсекреции желудка, аритмиям сердца и др.
ФАЗОВЫЕ СОСТОЯНИЯ В НЕРВНОЙ СИСТЕМЕ
По критерию, отражающему адекватность ответа нервной системы на раз­
личные воздействия, выделены такие характерные для нарушения ее деятель­
ности феномены, как фазовые состояния, развитие патологических рефлексов
и снижение общей резистентности и степени адаптируемости организма
к меняющимся условиям существования.
Фазовые состояния — типовое нарушение адекватных соотношений между
интенсивностью и/или характером («качество») ответной реакции нервной
системы и характеристиками раздражителя, вызывающего данную реакцию.
Ответы нервной системы в количественном или качественном отноше­
нии при фазовых состояниях не соответствуют ни параметрам раздражителя,
ни потребностям организма.
Причины развития фазовых состояний
Фазовые состояния формируются в результате:
— генетически детерминированных (первичных) патологических изменений в раз­
ных структурах нервной системы. Так, конкордантность по рассеянному
склерозу среди монозиготных близнецов превышает 50%, а у близких род­
ственников (родителей, братьев и сестер) риск возникновения этой болез­
ни в 8 раз выше, чем в общей популяции. Это (наряду с влиянием факторов
внешней среды) отражает взаимодействие нескольких генетических факто­
ров между собой (так называемая полигенная зависимость);
— приобретенных (вторичных) структурно-функциональных нарушений в
нервной системе (например, при ишемии, росте опухолей, энцефалитах,
интоксикации).
Последствия формирования и стабилизации фазовых состояний в нервной
системе:
—утрата сложившихся в процессе индивидуальной жизни межнейронных
отношений, функциональных совокупностей нейронов или их систем
(функциональный распад нервной системы);
— формирование патологических, не свойственных данному индивиду функ­
циональных связей между нейронами и нервными образованиями («пато­
логическая интеграция» их), новых функциональных совокупностей
нейронов и систем (образование «патологической системы»);
—снижение пластичности и адаптивных возможностей нервной системы
в целом или отдельных ее функциональных образований.
Развитие фазовых состояний наиболее характерно для патологии высшей
нервной деятельности и нейровегетативных реакций.
Основные виды фазовых состояний:
—уравнительное фазовое состояние (характеризуется одинаковыми ответа­
ми нервных структур на воздействия разной интенсивности);
—фазовое состояние средних раздражителей (проявляется максимальным
ответом только на раздражители средней интенсивности);
—парадоксальное фазовое состояние (характеризуется слабой реакцией
или ее отсутствием на сильный раздражитель, а также сохранением
или усилением реакции на слабые раздражители);
—наркотическое фазовое состояние (проявляется последовательным выпа­
дением реакций на слабые, а затем и на сильные раздражители);
—тормозное фазовое состояние (характеризуется отсутствием реакции
на любой раздражитель);
—ультрапарадоксальное фазовое состояние (проявляется качественным
изменением соотношения между характером раздражителя и вызываемой
им реакцией; при ультрапарадоксальном состоянии негативные реакции
развиваются в ответ на положительные раздражители, и наоборот).
Для фазовых состояний характерны:
—постоянная смена их во времени («временная мозаика»);
—перемещение их в пространстве: в разных участках нервной системы («про­
странственная мозаика»);
—формирование на их основе патологических рефлексов: условных и /
или (чаще) безусловных;
—снижение общей резистентности и степени адаптируемости организма
к меняющимся условиям существования.
НЕЙРОГЕННЫЕ РАССТРОЙСТВА ДВИЖЕНИЙ
Нейрогенные расстройства движений характеризуются патологическими
изменениями количества движений, их темпа и координации.
Виды нейрогенных расстройств движений
Выделяют следующие виды наиболее распространенных нейрогенных рас­
стройств движения:
—гипокинезии — ограничение объема и скорости произвольных движений;
—гиперкинезии — выполнение избыточных непроизвольных движений;
—гиподинамии — снижение двигательной активности и силы мышечных
сокращений при движении;
—атаксии — нарушение координации движений.
Системы регуляции движений
К системам, осуществляющим регуляцию движений, относятся пирамид­
ная и экстрапирамидная системы, а также структуры, отвечающие за регуля­
цию координации движений.
Аксоны всех нисходящих путей заканчиваются на мотонейронах спинного
мозга.
Пирамидная си стем а контроля движений
Нисходящие пути:
—корково-спинномозговой путь передний (передний пирамидный путь,
IгасШз согйсозртаШ уеШгаИз)] он образован аксонами нейронов, рас­
положенных в двигательной зоне коры (предцентральная извилина,
8уги$ ргесеШгаШ). Волокна пути проходят через внутреннюю капсулу
и в переднем канатике, заканчиваются в передних рогах, перекрещива­
ясь посегментно;
—корково-спинномозговой путь латеральный (латеральный пирамидный
путь, Тгас1и$ соШсозртаИз ШегаИз) начинается в коре предцентральной
извилины, проходит через внутреннюю капсулу и после перекреста
в продолговатом мозге проходит в боковом канатике, заканчиваясь
в передних рогах спинного мозга.
Уровни наиболее частого поражения нервных структур пирамидной системы
(регулирующей функцию поперечнополосатых мышц и произвольные дви­
жения):
—тела нейронов центрального двигательного анализатора, расположенные
преимущественно в предцентральной извилине двигательной зоны
коры — пирамидные нейроны;
—корково-ядерные и корково-спинномозговые пути;
—вставочные нейроны спинного мозга (через которые пирамидные нейроны
влияют на мотонейроны передних рогов спинного мозга и черепных
нервов);
—тела мотонейронов передних рогов спинного мозга и двигательных ядер
черепных нервов;
— аксоны мотонейронов;
—нервно-мышечные синапсы.
Экстрапирамидная си сте м а регуляции движений
Она включает следующие нисходящие пути:
—красноядерно-спинномозговой путь (пучок фон
Монакова, 1гас1ш
гиЬгозртаНз); нисходящий путь экстрапирамидной системы, начинается
от красного ядра, проходит в стволе мозга и боковом канатике; заканчи­
вается в передних рогах спинного мозга;
—ретикулоспинальный путь (1гас1из геЯси1озртаПз) — эфферентный путь
экстрапирамидной системы; он начинается в ретикулярной формации
продолговатого мозга, заканчивается в передних рогах спинного мозга;
контролирует тонус скелетной мускулатуры и висцеральные двигатель­
ные функции, например автоматизм дыхания;
Уровни частого поражения структур экстрапирамидной системы, регулиру­
ющих мускулатуру, которая обеспечивает непроизвольные («автоматические»)
движения:
—нейроны коры (премоторная зона, поясная извилина и др.);
—подкорковые ядра стриопаллидарной системы, мозжечка, спинного мозга;
—проводящие пути стриопаллидарной системы (например, красноядерно­
спинномозговой или ретикулоспинальный).
С и стем а координации движений
К уровням часто поражаемых нервных структур, обеспечивающих коорди­
нацию движений, относят:
—нейроны лобной и височной области коры головного мозга;
—нейроны ядер мозжечка;
—проводящие пути (от нейронов коры мозга и мозжечка к нейронам гипо­
таламуса, красного ядра среднего мозга, вестибулярных ядер, ретикуляр­
ной формации ствола мозга и др.);
—синаптические структуры.
Характеристика типовых форм расстройств движения
В этом разделе рассмотрены основные виды наиболее распространенных
нейрогенных расстройств движения: гипокинезии, гиперкинезии и атаксии.
Гипокинезии
Гипокинезии — нейрогенные ограничения объема, количества и скорости
движений.
Гипокинезии, как правило, сочетаются со снижением двигательной актив­
ности и силы мышечных сокращений — с гиподинамией.
С учетом различных критериев выделяют несколько типов гипокинезий
(рис. 29.6).
В зависимости от выраженности нарушения движений выделяют парезы
и параличи:
—парез — уменьшение амплитуды, скорости, силы и количества произволь­
ных движений;
—паралич — полное отсутствие произвольных движений.
В зависимости от распространенности (масштаба) расстройств движения
выделяют различные плегии, от моно- до тетраплегии:
—моноплегия — паралич одной конечности (руки или ноги);
—параплегия — паралич обеих рук или обеих ног;
—гемиплегия — паралич левой или правой половины тела;
—триплегия — паралич трех конечностей;
—тетраплегия — паралич рук и ног.
В зависимости от изменения тонуса мышц различают:
—спастические гипокинезии — при них повышен тонус мышц, как правило,
одной группы, например сгибателей рук или разгибателей ног; наблю-
даются при поражении центральных мотонейронов на любом участке
кортико-спинального (пирамидного) пути;
ригидные гипокинезии — характеризуются длительно повышенным тону­
сом одной или нескольких групп мышц-антагонистов, например отво­
дящих и приводящих, сгибательных и разгибательных. В последнем слу­
чае (при одновременном повышении тонуса сгибателей и разгибателей)
конечность или туловище длительно сохраняет приданную ей (ему) позу
(так называемая восковидная ригидность, следствие поражения экстра­
пирамидной системы);
вялые гипокинезии — при них понижен тонус мышц в области иннерва­
ции поврежденного нервного ствола или центра (например, при пора­
жении мотонейронов или передних корешков спинного мозга).
Гипокинезииг
По выраженности!
По преимущественно пораженным
нервным структурам
1
Парезы
Центральные
Паралич*
Экстрапирамидные
По распространению I
Периферические
Нервно-мышечные
(миастенические)
По изменению
тонуса мышц
Моноплегии
Гемиплегии
. . IСпастические!
Параплегии
Триплегии
Вялые
Ригидные
Тетраплегии
Рис. 29.6. Виды гипокинезий
В зависимости от преимущественно пораженных нервных структур выделяют
центральные, периферические, экстрапирамидные и миастенические (нервно­
мышечные) формы гипокинезий.
Центральные параличи и парезы
Причины центрального (пирамидного, спастического) паралича или пареза:
— поражения центральных пирамидных нейронов двигательного анализатора
(например, при ишемии или геморрагиях мозга);
— повреждение проводящих (кортико-спинальных) путей пирамидной системы
(например, в результате травм, опухолей, ишемии аксонов нейронов).
Проявления центральных параличей и парезов
• Гиперрефлексия — повышение сегментарных сухожильных и периостальных
рефлексов (увеличение амплитуды ответа и расширение зоны вызывания
рефлекса).
• Мышечная гипертония — повышение тонуса мышц по спастическому типу.
Обычно носит неравномерный характер (например, на руке тонус повы­
шается преимущественно в приводящих мышцах плеча, сгибателях пред­
плечья, а на ноге — в разгибателях бедра и голени, приводящих мышцах
бедра, сгибателях стопы). Со временем это может приводить к контракту­
рам — стойким ограничениям движений в суставах и необычным положе­
ниям конечностей.
• Патологические рефлексы (например, Бабинского, Россолимо, Бехтерева).
Эти рефлексы подразделяются на разгибательные и сгибательные. Первые
являются одним из наиболее ранних и постоянных проявлений поражения
пирамидного пути. Указанные признаки обусловлены повышением сег­
ментарных рефлексов спинного мозга вследствие ослабления тормозных
нисходящих влияний головного мозга.
• Клонус — высокая степень повышения сухожильно-мышечных рефлексов.
Клонус проявляется серией быстрых ритмичных сокращений отдельных
мышц, развивающихся спонтанно или в ответ на раздражение самой
мышцы либо ее сухожилия (примером может служить клонус мышц над­
коленника, стопы, кисти, подбородка).
• Синкинезии — непроизвольные содружественные мышечные сокращения
и движения, возникающие в парализованной конечности при осуществле­
нии произвольных движений другой конечностью или иной частью тела.
Синкинезии осуществляются при участии пирамидной системы, мозжеч­
ка, спинного мозга.
Периф ерические параличи и парезы
Причины периферического (вялого, атрофического) паралича или пареза:
—первичные (наследуемые или врожденные) поражения периферических
мотонейронов (клетки передних рогов спинного мозга, ядра черепных
нервов);
—вторичные (приобретенные) повреждения периферических мотонейронов.
Причины вторичных периферических параличей и парезов:
—дегенеративные изменения в нейронах (например, при боковом амиотро­
фическом склерозе);
—воспалительные процессы в нервной системе (например, при полиомиели­
те, энцефалите);
—интоксикации нейротропными ядами (например, ботулиническим, дифте­
рийным);
—механическая травма;
—нарушения нервно-мышечной передачи (например, при ботулизме, миасте­
ниях, действии ядов, токсинов, аминогликозидных антибиотиков и др.).
Проявления периферических парезов и параличей:
—гипотония (снижение мышечного тонуса); мышцы при этом на ощупь
дряблые, вялые;
—избыточность пассивных движений в парализованной конечности;
—гипо- или арефлексия (снижение или отсутствие сегментарных рефлек­
сов: сухожильных, надкостничных, кожных и др.);
—гипо- или атрофия мышц (формируется вследствие длительного бездей­
ствия мышц, а также в результате выпадений нейротрофических влия­
ний на них);
—дегенерация мышечных волокон с замещением их жировой и соедини­
тельной тканью;
—снижение возбудимости мышцы (развивается в связи с ее дистрофией;
этот феномен обозначается как реакция «перерождения» мышцы).
Экстрапирамидны е параличи и парезы
Причина их — поражение нейронов экстрапирамидной системы.
Проявления экстрапирамидных парезов и параличей:
—повышение тонуса мышц по ригидному типу. При этом отмечается при­
мерно одинаковое одновременное повышение тонуса сгибателей и раз­
гибателей, пронаторов и супинаторов;
—ригидность мышц (ригидный паралич);
—появление постуральных, позотонических рефлексов. Они наблюдаются
при изменении позы тела (например, нистагм глаз или головы при вра­
щении телом);
—каталепсия — длительное сохранение туловищем или конечностью приданного
ему (ей) положения, замедление темпа и ухудшение координации движений.
В отличие от центральных параличей, при экстрапирамидных не наблюда­
ется патологических рефлексов и выраженной гиперрефлексии.
М иастенические гипокинезии
К нервно-мышечным (миастеническим, синаптическим) гипокинезиям
относится миастения тяжелая псевдопаралитическая (туазШета §гау1з) и дру­
гие миастенические синдромы (в частности, Ламберта—Итона).
Причина миастений — нарушение синаптической передачи в холинергических
нервно-мышечных синапсах: от терминалей двигательных нервных к скелет­
ным мышечным волокнам.
Механизмы развития миастений:
—блокада постсинаптических холинорецепторов АТ к ним. 1§ фиксируются
на постсинаптической мембране мышечного волокна и тем самым пре­
пятствуют взаимодействию ацетилхолина с холинорецептором;
—снижение ответа мышечного волокна на ацетилхолин в связи с уменьше­
нием чувствительности (гипосенситизацией) их холинорецепторов.
Проявления миастений:
—мышечная слабость (миастения) разной степени выраженности;
—быстрая утомляемость мышц при физической нагрузке.
Гиперкинезии
Гиперкинез» — нейрогенное увеличение объема и количества непроиз­
вольных движений.
Гиперкинезы развиваются вследствие поражения нейронов различных
структур головного мозга (экстрапирамидной системы, таламуса, субталамического ядра, зубчатого ядра мозжечка, красного ядра, коры и их систем
связи).
С учетом различных критериев выделяют несколько типов гиперкинезий.
Они изображены на рис. 29.7.
-хорея;
-спастическая
-дрожание;
кривошея
-т и к
Рис. 29.7. Виды гиперкинезии
В зависимости от локализации пораженных структур мозга выделяют гипер­
кинезии:
—корковые;
—подкорковые;
—стволовые.
В зависимости от распространенности процесса различают:
—общие (генерализованные) гиперкинезы с вовлечением в процесс несколь­
ких или большинства групп мышц;
—местные (локальные) гиперкинезы, характеризующиеся непроизвольным
сокращением отдельных мышц или их волокон.
В зависимости от преобладания фазных (быстро сменяющихся) или тониче­
ских (медленных) компонентов сокращения различают:
—быстрые гиперкинезы;
—медленные гиперкинезы.
Бы стрые гиперкинезии
К быстрым гиперкинезам относят судороги, хорею, дрожания (тремор)
и тики.
• Судороги — внезапно возникающие, приступообразные или постоянные непро­
извольные сокращения мышц различной интенсивности, продолжительно­
сти и распространенности. Выделяют:
—клонические судороги — кратковременные и нерегулярные сокращения
отдельных групп мышц, следующие друг за другом через сравнительно
небольшие промежутки времени. Они возникают чаще всего в результа­
те чрезмерного возбуждения коры больших полушарий или поражения
структур пирамидной системы. Распространенные выраженные клони­
ческие судороги называют конвульсиями;
—тонические судороги — длительные (до нескольких десятков секунд)
мышечные сокращения, в результате которых происходит «застыва­
ние» туловища или конечностей в различных вынужденных положени­
ях. Развиваются при чрезмерном возбуждении подкорковых структур
и некоторых видах интоксикации (например, алкогольной, столбняч­
ной, окисью углерода). При столбняке может развиться опистотонус;
—смешанные (клонико-тонические, тонико-клонические). Наблюдаются
при коматозных и шоковых состояниях (например, при диабетической,
печеночной или уремической коме; ожоговом или анафилактическом
шоке).
• Хорея — беспорядочные, быстрые, неритмичные, насильственные сокраще­
ния различных групп мышц. Наблюдается при длительной ишемии мозга
(например, его сосудов), атеросклеротическом поражении, ревматическом
энцефалите, черепно-мозговых травмах. Может иметь наследственное
происхождение (например, хорея Гентингтона).
• Тремор — гиперкинез дрожательного типа. Характеризуется непроизволь­
ными, стереотипными ритмическими колебательными движениями тела
или его частей в результате повторяющихся сокращений и расслабле­
ний мышц. Возникает преимущественно при поражении ствола мозга.
Наблюдается при органических поражениях головного мозга (рассеянном
склерозе, болезни Вильсона—Коновалова, энцефалите, расстройстве кро­
воснабжения), экзогенной интоксикации организма (алкоголем, ртутью,
морфином).
• Тик — быстрые непроизвольные стереотипные сокращения мышцы или групп
мышц, обусловливающие насильственные движения (например, мигание,
прищуривание глаз, жестикуляция). Наблюдаются в основном при пора­
жении экстрапирамидной системы в результате энцефалита, интоксика­
ций, в том числе ЛС (например, при употреблении психофармакологиче­
ских средств), а также при некоторых психических расстройствах.
М едленные гиперкинезии
Медленные гиперкинезы представлены атетозом и спастической криво­
шеей.
• Атетоз — непроизвольные стереотипные, медленные червеобразные вычурные
движения, возникающие в результате одновременной длительной активации
мышц агонистов и антагонистов. Чаще всего поражаются дистальные отделы
конечностей, пальцы рук и стоп. Развиваются при поражении стриарной
системы (хвостатого ядра, скорлупы) при энцефалите, нарушениях мозго­
вого кровообращения, черепно-мозговых травмах, опухолях подкорковых
отделов головного мозга.
• Спастическая кривошея — деформация шеи и неправильное положение голо­
вы (наклон в одну сторону) в результате длительного нейрогенного сокраще­
ния — спазма мышц шеи. Нейрогенная кривошея наблюдается в результате
поражения головного мозга (например, отеке, кровоизлиянии, опухоли)
в области (еШопит сегеЬеШ, заднего мозга. Нередко бывает результатом
родовой травмы (ротационный подвывих I шейного позвонка) у детей.
Атаксии
Атаксии — локомоторные расстройства, характеризующиеся нарушением
пространственной и временной координации произвольных движений.
При этом сила мышц практически не изменена. Координация движений
достигается при взаимодействии различных структур: мозжечка, спинного мозга,
лобных отделов коры головного мозга, среднего мозга, таламуса, лабиринта.
Причины атаксии:
—поражение путей проприоцептивной чувствительности (с развитием сенси­
тивной атаксии);
—повреждение мозжечка (с развитием мозжечковой атаксии);
—поражение лобной и височной области коры головного мозга (с развитием
корковой атаксии);
—повреждение вестибулярного аппарата (с развитием вестибулярной атаксии).
Виды атаксий. По локализации основного поражения различают несколько
видов атаксий (рис. 29.8):
—сенситивную (заднестолбовая) — недостаточность или отсутствие аффе­
рентных сигналов от нервных окончаний в мышцах или сухожилиях,
предупреждающих о положении отдельных частей тела, степени сокра­
щений мышц, скорости их движений, о сопротивлении этим движени­
ям. Причина сенситивной атаксии — поражение задних столбов спинного
мозга, задних его корешков, зрительного бугра, периферических нервов.
Наблюдается сенситивная атаксия при сухотке спинного мозга, поли­
невритах, сирингомиелии, фуникулярном миелозе. При выраженной
сенситивной атаксии затруднено выполнение даже самых простых
движений, затрудняется ходьба (она становится беспорядочной и резко
ухудшается при выключении зрительного контроля передвижений);
—мозжечковую (наблюдается при поражении мозжечка и/или его прово­
дящих путей);
—корковую (результат повреждения нейронов лобной или височной зон
коры большого мозга);
—вестибулярную (лабиринтная). Возникает, например, при энцефалитах,
опухолях в области ствола мозга или IV желудочка.
| Виды атаксии в зависимости от локализации поражения мозга
________
5
Сенситивная
(заднестолбовая)
^
| Мозжечковая|
^ | Корковая|
__”
Т
__
| Вестибулярная|
Локализация основного поражения:
- задние столбы
-м о зж е ч о к;
- к о р а лобной
-ство л мозга;
спинного мозга; - проводящие
и/или затылочно- - область IV '
- задние корешки
пути мозжечка височной области
желудочка мозга
спинного мозга;
- зрительный бугор;
- периферические
нервы
Проявления атаксии:
—расстройство координации и равновесия тела в положении стоя и сидя (ста­
тическая форма);
—нарушение выполнения различных произвольных движений конечностями,
особенно руками (динамическая форма);
—расстройство координации движения преимущественно при стоянии и ходь­
бе (статико-локомоторная форма).
НАРУШЕНИЯ ЧУВСТВИТЕЛЬНОСТИ
Нейрогенные нарушения чувствительности, как «простой» (тактильной,
температурной, проприоцептивной, болевой), так и сложной (чувства лока­
лизации, дискриминации, стереогноза), имеют в своей основе повреждение
соматосенсорного анализатора.
Все виды чувствительности формируются при раздражении рецепторов
кожи, слизистых оболочек, мышц, суставов, сухожилий, внутренних органов.
Информация об этом передается по периферическим отросткам первичных
чувствительных нейронов (спинномозговых узлов и чувствительных нейронов
черепных нервов) и далее по их центральным отросткам к вставочным ней­
ронам или непосредственно в составе восходящих путей.
Типовые формы расстройств чувствительности
Нарушения чувствительности дифференцируют по нескольким критериям
(рис. 29.9).
Расстройства чувствительности
Нарушение
вида
чувствительности
Контактной!
▼
Дистантной
Нарушение
восприятия
интенсивности
раздражителя
▼
Анестезия
1
Гиперестезия
Интероцепторной|
Экстероцепторной I
Г ипестезия
Нарушение
адекватности
ощущения
вызвавшему его
раздражителю
г
Термалгия |
Полистезия
-
Парестезия -*~
Аллодиния
Рис. 29.9. Типовые формы расстройств чувствительности
По виду нарушенной чувствительности дифференцируют расстройства:
—контактной чувствительности (например, тактильной, болевой, темпера­
турной);
- дистантной чувствительности (например, нарушения зрительной, слухо­
вой, обонятельной).
По расположению поврежденных рецепторов выделяют нарушения:
- экстерорецепторной чувствительности (например, при поражении рецеп­
торов кожи и слизистых оболочек);
— интероцепторной чувствительности (например, при повреждении висце-
рорецепторов и проприорепепторов).
По нарушению восприятия интенсивности ощущения выделяются:
— анестезия — потеря чувствительности или отдельных ее разновидностей;
— гипестезия — снижение отдельных или всех видов чувствительности
(рис. 29.10);
— гиперестезия — чрезмерное повышение отдельных или всех видов чувстви­
тельности.
------- ■]Виды гипестезии, анестезии -------г
По уровню поражения
По «масштабу» поражения
сенсорной системы
сенсорной системы
1Г
1
1
1 Рецепторная| Проводниковая Слуховая |Тактильная| Болевая | Топоанестезия
1г
»Г
1>
.
| Центральная
Обонятельная 1Астереогнозия ( Другие
Рис. 29.10. Виды гипестезии, анестезии
Гипо- и анестезии
Виды гипо- и анестезии
• Рецепторная гипо- или анестезия. Она характеризуется:
—снижением;
—утратой чувствительности, соответствующей пораженному рецептору
(например, температурной, тактильной, зрительной, слуховой).
• Проводниковая гипо- или анестезия. При ней снижается или утрачивается
чувствительность при поражении:
— периферических нервных стволов (страдают все виды чувствительности
в области иннервации);
—левой или правой половины спинного мозга (синдром Броун-Секара);
—проводящих путей спинного или головного мозга — нарушаются различ­
ные виды чувствительности в зависимости от локализации поражения
(повреждение задних столбов спинного мозга сопровождается сниже­
нием или утратой проприоцептивной и тактильной чувствительности;
поражение волокон медиальной петли сочетается с утратой восприятия
скорости и направления движения конечностей, вибрации и тяжести
поднимаемого груза, раздельного ощущения прикосновения в разных
точках тела — дискриминационной чувствительности).
• Центральная гипо- или анестезия. Характеризуется снижением или утратой
чувствительности при поражении:
—нейронов постцентральной извилины коры большого мозга (на противополож­
ной стороне тела теряется дискриминационная чувствительность, ощуще­
ние положения конечности в пространстве, формы и массы предметов);
—теменной доли большого мозга (развивается синдром аморфосинтеза —
нарушения восприятия формы тела, его положения в пространстве;
при этом пациент не может самостоятельно одеться, причесаться,
побриться, он может даже «игнорировать» наличие противоположной
половины тела или патологических процессов в данном участке).
• Тотальная гипо- или анестезия. Наблюдается при поражении корешков
и нейронов спинномозгового узла, медиальной петли, внутренней кап­
сулы, задней центральной извилины. В результате утрачиваются все виды
чувствительности.
• Парциальная гипо- или анестезия. Развивается при повреждении задних
рогов, задних и/или боковых столбов спинного мозга, медиальных отделов
продолговатого мозга. При этой форме нарушаются отдельные виды чув­
ствительности, например:
—тактильной (ослабевают или утрачиваются ощущения, возникающие
при прикосновении к предмету или предмета к телу);
— болевой;
—топической (ослабевает или утрачивается чувство определения места
действия раздражителя);
—стереогнозической (отсутствует чувство восприятия предметов при их
ощупывании; при этом способность воспринимать отдельные их каче­
ства сохраняется);
—термической (снижена или утрачена температурная чувствительность);
— слуховой.
Причины парциальной гипо- или анестезии:
—гипо- или десенситизация рецепторов;
—уменьшение количества рецепторов на клеточных мембранах (например,
вследствие нарушения экспрессии генов, контролирующих их синтез);
—повреждение проводящих афферентных путей (например, в результа­
те диабетической полиневропатии; интоксикации алкоголем, ртутью,
свинцом; невритов);
—повреждение нейронов сенсорных систем (например, задних или боковых
столбов спинного мозга, таламуса, теменной доли, задней центральной
извилины).
Непосредственными причинами указанных изменений чаще всего являются:
—травмы нервной системы;
—дегенеративные изменения в ней (например, при сирингомиелии, про­
грессирующей невральной амиотрофии);
— опухоли головного и спинного мозга;
—расстройства мозгового кровообращения;
— врожденные нарушения развития проводящих афферентных путей и /
или нейронов сенсорных систем;
—энцефалиты.
Гиперестезии
Гиперестезии — повышение чувствительности к действию какого-либо
раздражителя.
В зависимости от распространенности («масштаба») поражения нервной
системы выделяют гиперестезию:
— тотальную;
— парциальную.
Причины гиперестезий
• Повышение чувствительности рецепторов (гиперсенситизация, рецепторная
гиперестезия). Наблюдается при патологии кожи и слизистых оболочек
(например, при ранах, ожогах, опоясывающем герпесе).
• Повышение возбудимости нейронов сенсорной системы (центральная
гиперестезия), главным образом — сенсорных полей коры мозга, гип­
покампа, ядер миндалевидного комплекса и др. Это происходит, напри­
мер, при неврозах, некоторых формах психических расстройств, энце­
фалитах.
Дизестезии
Дизестезии характеризуются неадекватными ощущениями по отношению
к вызвавшему его раздражителю.
Виды ди зестези й
• Термалгия — восприятие холодового и теплового воздействия как болевого.
• Полиестезия — ощущение действия множества раздражителей при воздей­
ствии одного реального фактора (например, ощущение жжения, покалыва­
ния и давления при уколе кожи иглой).
• Аллодиния — восприятие неболевого воздействия как болевого.
• Гиперпатия — чрезмерная боль, возникающая при действии различных раз­
дражителей, в том числе неболевых (например, поглаживание), сочета­
ющаяся с неспособностью точно локализовать их действие.
• Парестезия — тактильные неболевые, необычные по характеру ощущения
(в виде чувства онемения, одеревенения, ползания «мурашек», покалыва­
ния). Возникают без явного раздражителя. Наиболее частые причины —
ишемия тканей, охлаждение их, патологические процессы, поражающие
задние корешки спинного мозга (например, спинная сухотка — поздняя
форма нейросифилиса).
• Синестезия — вид иллюзорного восприятия, т.е. возникновение несколь­
ких ощущений при раздражении лишь одного органа чувств. Примерами
могут быть «цветной слух», «цветное зрение», «цветное обоняние».
При синестезии раздражение одного органа чувств (зрения, обоня­
ния, слуха), наряду со специфическим для него ощущением, вызывает
и другие, характерные для других органов чувств (например, ощущение
звука при восприятии света — «цветной слух»; ощущение разных цветов
при взгляде на черный рисунок — «цветное зрение»; ощущение окра­
шенных запахов — «цветное обоняние»). В основе данной разновидности
синестезий лежит иррадиация возбуждения с нервных структур одной
сенсорной системы на другую.
Виды синестезий:
— сегментарные (возникают в области иннервации данного сегмента спин­
ного мозга или черепных нервов: например, ощущение боли в области
левой лопатки при приступе стенокардии или инфаркте миокарда);
- проводниковые (воспринимаются в участках иннервации данного нерв­
ного ствола (например, ощущение боли в ампутированной конечно­
сти — фантомные боли).
В патологии синестезии проявляются ощущениями, возникающими за
пределами области действия раздражителя или развития патологического
процесса.
Общие механизмы расстройств чувствительности
Общие
рис. 29.11.
механизмы
расстройств
чувствительности
представлены
на
Рис. 29.11. Общие механизмы расстройств чувствительности
Расстройства чувствительности могут развиваться на уровне:
—рецепторов;
—проводящих путей;
—центральных нервных структур.
На уровне рецепторов наблюдаются изменения:
— их чувствительности (повышение/снижение порога чувствительности
рецепторов происходит, например, при повторном и/или длительном
воздействии раздражителя на них, в условиях гипоксии или гипотермии;
увеличении концентрации К+ во внеклеточной среде и/или внутрикле­
точного содержания № +);
— и/или количества рецепторов.
На уровне проводящих путей возможно торможение или полное блокирова­
ние проведения импульсации. Эти изменения развиваются при повреждении:
—нервных проводников; их повреждение чаще всего наблюдается при:
Оинтоксикации, например, алкоголем, ртутью, свинцом, солями тяже­
лых металлов);
о гиповитаминозах;
о СД;
о инфекционных, аллергических, иммуноагрессивных процессах;
о механических травмах нерва (при этом утрачиваются все виды чувстви­
тельности в области иннервации; на конечностях, например, возника­
ет дистальный тип расстройств чувствительность по типу «перчаток»
или «чулок»;
—задних корешков спинного мозга; их повреждение сопровождается рас­
стройством кожной чувствительности по сегментарному типу: образуют­
ся характерные «полосы» нарушений — круговые на туловище и продольные
на конечностях. Раздражение корешков спинного мозга сопровождается
болями и парестезиями;
—задних рогов спинного мозга; при их повреждении развиваются рас­
стройства чувствительности по сегментарному типу (как и при поражении
задних корешков). При этом развивается температурная и болевая ане­
стезия. Однако, в отличие от корешковых поражений, глубокая чувстви­
тельность сохраняется. Такие расстройства чувствительности называют­
ся диссоциированными;
—боковых столбов спинного мозга; их повреждение проявляется выпаде­
нием болевой и температурной чувствительности на противоположной
стороне и ниже места поражения. При поперечном повреждении поло­
вины спинного мозга развивается синдром Броун-Секара.
На уровне центральных структур сенсорной системы нарушения чувстви­
тельности возможны вследствие изменившегося порога чувствительности
нейронов и/или искаженного формирования ощущений. Эти изменения
наблюдаются, например, при повреждении:
—ядер таламуса; при этом происходит перекрестное снижение или выпа­
дение всех видов чувствительности (гемианестезия). Вследствие потери
мышечно-суставной чувствительности развивается контралатеральная
сенситивная атаксия. Патологические процессы в области зрительного
бугра нередко вызывают ощущения так называемых таламических болей
в проекции противоположной половины тела. Эти боли отличаются высо­
кой интенсивностью, разлитостью и резистентностью к аналгезирующим средствам;
—чувствительной зоны коры большого мозга (затылочная, теменная, височ­
ная, инсулярная и стриарная области, постцентральная извилина и др.);
патогенные агенты могут вызвать перекрестное ослабление или выпа­
дение соответствующих видов чувствительности (зрения, слуха, вкуса,
тактильных, температурных, болевых и др.). Например, при патологи­
ческих процессах в задней центральной извилине нарушается болевая,
температурная и частично тактильная чувствительность на ограничен­
ных участках лица, рук и др. Поражение верхней теменной доли при­
водит преимущественно к нарушению более сложных видов чувстви­
тельности: локализации конечностей, ощущения рельефа предмета,
мышечно-суставного чувства.
Боль
Боль — особый вид чувствительности, формирующийся под действием пато­
генного раздражителя.
Боль характеризуется субъективно неприятными ощущениями, а также суще­
ственными изменениями в организме, вплоть до серьезных нарушений его жиз­
недеятельности и даже смерти.
Значение боли
Боль имеет сигнальное и патогенное значение.
Сигнальное значение боли
Ощущение боли вызывается самыми различными агентами, но их объеди­
няет общее свойство — реальная или потенциальная опасность повредить орга­
низм. Болевой сигнал играет по меньшей мере двоякую роль:
—мобилизует организм на защиту от патогенного агента: например, боль
при повреждении ткани сочетается с развитием стрессорной реакции,
активацией фагоцитоза, пролиферацией клеток, изменениями крово­
обращения и т.п.; существенна также защитная поведенческая реакция
на боль, направленная либо на «уход» от действия повреждающего фак­
тора (отдергивание руки от повреждающего предмета), либо на ликвида­
цию его (например, на извлечение из кожи инородного тела);
—способствует охранительному ограничению функции затронутого болью
органа (например, болевое ощущение при инфаркте миокарда сопро­
вождается страхом смерти; это заставляет пациента значительно огра­
ничить двигательную активность, что, в свою очередь, существенно
снижает гемодинамическую нагрузку на поврежденное сердце).
Патогенное значение боли
Боль нередко является причиной и/или компонентом патогенеза раз­
личных болезней и болезненных состояний (например, боль в результате
травмы может вызвать шок и потенцировать его развитие; боль при неврите
обусловливает нарушение функции тканей и органов, развитие общих реак­
ций организма: повышение или снижение АД, нарушение функции сердца,
почек и т.п.).
Причины боли
Боль вызывают факторы различной природы:
—физические (например, механическая травма, повышенная или пони­
женная температура, высокая доза УФ, электрический ток);
—химические (например, попадание на кожу или слизистые оболочки
сильных кислот, щелочей, окислителей; накопление в ткани солей каль­
ция или калия);
— биологические (например, высокая концентрация кининов, гистамина,
серотонина).
Виды боли
Различают протопатическую и эпикритическую боль (или болевую чув­
ствительность).
Эпикритическая боль («быстрая», «первая» или «предупредительная»);
возникает в результате воздействия раздражителей малой и средней силы.
Свойства эпикритической боли приведены в табл. 29.1.
Протопатическая боль («медленная», «тягостная», «древняя») возникает
под действием сильных, «разрушительных», «масштабных» раздражителей.
Свойства протопатической боли перечислены в табл. 29.1.
Таблица 29.1. Сравнительная характеристика эпикритической и протопатической
боли
Свойство боли
Эпикритическая боль
Источник раздражителя
Кожа, слизистые обо­
лочки
Короткий
Латентный период
Продолжительность после Быстро прекращается
устранения раздражителя
Тип проводящего волокна Миелиновые, тип А
Низкий
Порог восприятия
Точная
Локализованность
Протопатическая боль
Внутренние органы
и ткани
Длительный
Долго сохраняется
Безмиелиновые, тип С
Высокий
Диффузная
Примечание. Характеристики разных типов нервных волокон приведены в статье
«Волокно нервное» (см. приложение «Справочник терминов»).
Только сочетанная — и протопатическая, и эпикритическая — чувствитель­
ность дает возможность тонко оценить локализацию воздействия, его характер
и силу.
Механизмы формирования боли
Принято различать две группы взаимосвязанных механизмов:
—механизмы формирования боли (с участием ноцицептивной системы);
—механизмы, контролирующие чувство боли (осуществляемые антиноцицептивной системой).
Чувство боли формируется на разных уровнях ноцицептивной системы —
от воспринимающих болевые ощущения рецепторов до проводящих путей
и центральных нервных структур.
Рецепторы, воспринимаю щ ие болевые раздражители
Считают, что болевые (ноцицептивные) раздражители воспринимаются сво­
бодными нервными окончаниями (они способны воспринимать воздействие раз­
ных агентов как болевые). Допускают существование и специализированных
ноцицепторов — свободных нервных окончаний, активизирующихся только
при действии ноцицептивных агентов (например, капсаицина).
Сверхсильное (в том числе разрушающее) воздействие на рецепторы других
модальностей (механо-, хемо-, терморецепторы и др.) также может вызвать
ощущение боли.
Алгогены — патогенное воздействие, вызывающее боль, — приводят
к высвобождению из поврежденных клеток ряда веществ (их нередко называ­
ют медиаторами боли), действующих на рецепторы. К медиаторам боли отно­
сят кинины (главным образом брадикинин и каллидин), гистамин (вызывает
чувство боли при его введении п /к даже в концентрации 1x10 18 г/мл), высо­
кая концентрация Н+, капсаицин, вещество Р, ацетилхолин, норадреналин
и адреналин в нефизиологических концентрациях, некоторые ПГ.
Пути, проводящ ие боль
К проводящим системам боли относят ряд структур.
Спинной мозг
Афферентные проводники боли проникают в спинной мозг через задние
корешки и контактируют с вставочными нейронами задних рогов. Считают,
что проводники эпикритической боли оканчиваются в основном на нейро­
нах пластинок I и V, а проводники протопатической боли — в роландовом
веществе (шЪзШпйа §е1аппош) пластинок III и IV. Малые нейроны «специа­
лизируются» на передаче болевой (от высокопороговых механорецепторов)
и температурной чувствительности.
В передаче болевых стимулов, действуя как возбуждающий нейромедиа­
тор в синапсах между центральными отростками чувствительных нейронов
спинномозгового узла и перикарионами нейронов спиноталамического пути,
участвует вещество Р. Блокирование секреции вещества Р и снятие болевых
ощущений происходят через рецепторы опиоидных пептидов, встроенных
в мембрану терминали центрального отростка чувствительного нейрона (при­
мер феномена пресинаптического торможения). Источник опиоидного пеп­
тида энкефалина — вставочный нейрон.
В спинном мозге возможна конвергенция возбуждения для разных видов
болевой чувствительности. Так, С-волокна, проводящие протопатическую
боль, могут контактировать с нейронами спинного мозга, воспринимающими
эпикритическую боль от рецепторов кожи и слизистых оболочек. Это приво­
дит к развитию состояния, которое обозначают термином «феномен сегментар­
ной (отраженной) кожно-висцеральной боли» (ощущение боли в участке тела,
отдаленном от истинного места болевой импульсации). Примерами иррадиа­
ции боли могут служить ощущения «ложной» боли:
—в левой руке или под левой лопаткой при приступе стенокардии
или при инфаркте миокарда;
—под правой лопаткой при прохождении (выходе) конкремента по желче­
выводящим путям;
—над ключицей при остром гепатите или раздражении париетальной брю­
шины;
—в паховой области при наличии конкремента в мочеточнике.
Возникновение указанных сегментарных кожно-висцеральных («отражен­
ных») болей обусловлено сегментарной структурой иннервации поверхности
тела и внутренних органов афферентами спинного мозга.
Восходящие пути спинного мозга
Проводники эпикритической боли переключаются на нейронах пластинок I
и V, перекрещиваются и восходят в таламус.
Проводники протопатической боли переключаются на нейронах задних
рогов, частично перекрещиваются и восходят в таламус.
Проводящие пути головного мозга
Проводники эпикритической боли проходят в стволе мозга экстралемниско-
вым путем. Значительная их часть переключается на нейронах ретикулярной
формации, а меньшая — в зрительных буграх. Далее формируется таламокортикальный путь, заканчивающийся на нейронах соматосенсорной и моторной
области коры.
Проводники протопатической боли также проходят экстралемнисковым путем
ствола мозга к нейронам ретикулярной формации. Здесь формируются «при­
митивные» реакции на боль: настораживание, подготовка к «уходу» от болевого
воздействия и/или устранению его (отдергивание конечности, отбрасывание
травмирующего предмета и т.п.). При этом нейроны ретикулярной формации
генерируют импульсацию с частотой 4—6 Гц. Эти импульсы оказывают акти­
вирующее влияние на кору и способствуют формированию интегративного
чувства боли. Далее пути распространяются к различным областям мозга:
к нейронам таламуса, гипоталамуса, миндалевидного комплекса и др. Это
обусловливает системный ответ организма на болевой стимул, включающий
вегетативный, двигательный, эмоциональный, поведенческий компоненты.
Центральные нервные структуры ощущения боли
Эпикритическая боль является результатом восхождения болевой импульса­
ции по таламокортикальному пути к нейронам соматосенсорной зоны коры
большого мозга и возбуждения их. Субъективное ощущение боли формиру­
ется именно в корковых структурах.
Протопатическая боль развивается в результате активации главным образом
нейронов переднего таламуса и гипоталамических структур.
Целостное ощущение боли у человека формируется при одновременном уча­
стии корковых и подкорковых структур, воспринимающих импульсацию о про­
топатической и эпикритической боли, а также о других видах воздействий. В коре
мозга происходит отбор и интеграция информации о болевом воздействии, чув­
ство боли превращается в страдание, формируется целенаправленное, осознанное
«болевое поведение». Цель такого поведения — быстро изменить жизнедеятель­
ность организма, чтобы устранить источник боли или снизить ее степень, предот­
вратить повреждение или уменьшить его выраженность и масштаб.
Болевые синдромы
Клинически значимыми синдромами боли являются таламическая боль,
фантомные боли и каузалгия.
Таламическая боль (таламический синдром)
Проявляется таламическая боль преходящими эпизодами сильных, труд­
нопереносимых, изнуряющих политопных болей. Ощущение боли сочетается
с вегетативными, двигательными и психоэмоциональными расстройствами.
Причиной таламической боли является, как полагают, повреждение ядер
таламуса и образование в них очагов усиленного патологического возбужде­
ния — генератора патологически усиленного возбуждения.
Фантомная боль
Характеризуется фантомный синдром болью в отсутствующей части
тела, чаще всего в конечностях. Диапазон болевых ощущений колеблется
от сильного зуда и жжения до мучительных, непереносимых ощущений.
Наблюдается синдром более чем у двух третей пациентов после ампутации
конечностей.
Причина фантомной боли — раздражение центральных концов перерезан­
ных при ампутации нервов. На них образуются утолщенные участки (ампу­
тационные невромы), содержащие переплетение (клубок) регенерирующих
аксонов. Раздражение нервного ствола или невромы (например, при надавли­
вании в области культи, сокращении мышц конечности, воспалении, образо­
вании рубцовой ткани) вызывает приступ фантомной боли.
Каузалгия
Проявляется каузалгия приступообразно усиливающейся жгучей болью
в области поврежденных нервных стволов (чаще всего тройничного, лицево­
го, языкоглоточного, седалищного). Различные воздействия (прикосновение,
тепло, холод), психоэмоциональный стресс провоцируют и/или усиливают
приступ каузалгии.
Причинами каузалгии могут быть:
—патологическое повышение чувствительности ноцицепторов в зоне повреж­
денных толстых миелинизированных нервных волокон;
—формирование очага усиленного возбуждения в различных участках про­
ведения болевого импульса.
Способствует развитию каузалгии выброс норадреналина, вещества Р, возмож­
но, других «медиаторов боли» окончаниями симпатической нервной системы,
которая активируется при любом повреждающем воздействии на организм.
Боли нейропатические и сом атические
Рассмотренные выше клинические синдромы (каузалгия, фантомные
и таламические боли) обусловлены повреждением структур нервной системы
(нейропатические боли).
Нейропатические боли следует отличать от соматических болей, возникающих
при повреждении кожи, мышц, внутренних органов, суставов (табл. 29.2).
Таблица 29.2. Отличия нейропатической и соматической боли
Соматическая боль
Нейропатическая боль
Причина
Повреждение нервной ткани
Повреждение поверхностных тканей,
мышц, органов
Болевой раздражитель
Идентифицируется с трудом
Выявляется легко
Окончание табл. 29.2
Нейропатическая боль
Соматическая боль
Локализованность боли
Плохая (диффузная; «миграция»
места ощущения боли)
Выраженная (определяется в месте дей­
ствия патогенного фактора)
Характер боли
Необычный (ранее не ощущав­
шийся) — «непереносимая»,
«нестерпимая», «ужасная», «всепо­
глощающая» боль — гиперпатия
Обычный (ощущавшийся ранее при раз­
личных повреждениях или болезнях)
Устранение боли наркотическими анальгетиками
Слабое
Выраженное (вплоть до полного пре­
кращения)
Антиноцицептивная систем а
Чувство боли контролируется нейрогенными и гуморальными механизма­
ми, входящими в состав антиноцицептивной системы (рис. 29.12).
Рис. 29.12. Компоненты антиноцицептивной системы
Нейрогенные механизмы антиноцицептивной системы обеспечиваются
импульсацией нейронов серого вещества вокруг желудочков мозга, покрыш­
ки моста, миндалевидного тела, гиппокампа, отдельных ядер мозжечка,
ретикулярной формации, которые образуют нисходящие пути, подавляющие
чувство боли.
Аналгезирующая импульсация от указанных структур тормозит поток вос­
ходящей болевой информации, по-видимому, на уровне синапсов в задних
рогах спинного мозга, а также ядер срединного шва продолговатого мозга
(в основном пис1еш гарНе тщпи$). Раздражение серого вещества вблизи желу­
дочков мозга уменьшает клинические проявления боли.
Гуморальные антиноцицептивные механизмы представлены следующими
системами:
—опиоидергической системой мозга;
—серотонинергической;
—норадреналинергической;
— ГАМК-ергической.
Каждая из указанных выше систем включает нейромедиаторы (эндорфины,
энкефалины, динорфин, серотонин, норадреналин, ГАМК), их рецепторы
и эффекторные механизмы аналгезии. Компоненты этих систем характеризу­
ются в статьях «Нейромедиаторы» и «Рецепторы» приложения «Справочник
терминов».
Нейрогенные и гуморальные механизмы антиноцицептивной системы
тесно взаимодействуют друг с другом. Они способны блокировать болевую
импульсацию на всех уровнях ноцицептивной системы — от рецепторов до ее
центральных структур.
Активацией антиноцицептивной системы объясняют также феномен
уменьшения боли при раздражении тактильных или холодовых рецепторов.
Это достигается, например, с помощью точечного массажа, поглаживания,
локальной гипотермии, акупунктуры.
НЕЙРОГЕННЫЕ РАССТРОЙСТВА ТРОФИКИ
Нервная система влияет на метаболизм (а через него — на характер
и интенсивность функционирования и пластических процессов) различных
органов и тканей через механизмы либо собственно иннервации (путем
регуляции функциональной активности и кровоснабжения иннервируемых
структур), либо нейротрофического контроля.
Концепция нейротрофического контроля органов и тканей заключается
в положении о взаимном регулировании функционального состояния не толь­
ко элементов нервной системы (через нейронные сети), но и иннервируемых
ими структур (например, крови или мышц) с помощью механизмов, отли­
чающихся от присущего нервной системе механизма (заключающегося в рас­
пространении ПД по аксонам
секреции нейромедиатора в синаптическую
щель -> взаимодействии нейромедиатора с его рецепторами на постсинаптической мембране -> на постсинаптическом электрогенезе).
Механизмы нейротрофического контроля
В рамках концепции нейротрофического контроля рассматривается
несколько возможных механизмов его осуществления (рис. 29.13). К числу
основных относят следующие:
- изменение импульсной активности в аксонах (частоты ПД, интервалов
между ними). Полагают, что эти импульсы играют как информацион­
ную, так и трофическую (нейротрофическую) роль;
- образование нейронами специализированных трофических факторов, кото­
рые секретируются в синаптическую щель и взаимодействуют с постсинаптическими структурами, регулируя в них характер и интенсивность
метаболизма и пластических процессов;
- изменение уровня функционирования постсинаптической структуры (на­
пример, регулируемая гипотрофия органа при снижении его функции
или гиперфункция сердца при активации симпатической нервной
системы).
Рис. 29.13. Механизмы влияния нервной системы на обмен веществ в клетках
Развитие денервационного синдрома после повреждения нерва или бло­
кады аксонного транспорта в нем является значимым следствием нарушения
этого механизма.
Нейродистроф ический проц есс
Нарушение трофической функции нервной системы составляет патогене­
тическую основу нейродистрофического процесса.
Нейродистрофический процесс может возникать как в периферических
органах и тканях, так и в самой нервной системе. В типичном варианте ней­
родистрофический процесс развивается при денервационном синдроме.
Денервационны й синдром
Проявления денервационного синдрома (на примере денервации скелет­
ной мышцы) изображены на рис. 29.14.
Рис. 29.14. Наиболее характерные расстройства в постсинаптических структурах
при нарушении аксонного транспорта
Для денервационного синдрома характерен ряд закономерных изменений
в ткани или органе, описанных ниже.
• Дисферментоз (изменение спектра ферментов в клетке, интенсивности
их биосинтеза и активности, появление или исчезновение изоферментов).
• «Эмбрионизация» обмена веществ (приобретение реакциями метаболизма
свойств и признаков, характерных для ранних этапов развития организ­
ма, например меньшая активность процессов окисления, доминирование
реакций анаэробного гликолиза, активация пентозного цикла).
• Ультраструктурные изменения клеточных элементов (главным образом, мем­
бран). При электронной микроскопии наблюдаются признаки набухания
и разрушения крист митохондрий, лабилизации мембран лизосом, нару­
шения селективной проницаемости плазмолеммы.
• Дистрофии и дисплазии различного характера (развиваются вследствие
нарушений экспрессии отдельных генов и расстройств метаболизма).
• Образование и действие аутоагрессивных 1§, Т-лимфоцитов, макрофагов
(в связи с нарушением структуры и спектра белков, а также других
их модификаций).
• Гиперсенситизация денервированных структур к недостающему нейромедиа­
тору (так, в скелетных мышечных волокнах увеличивается синтез рецеп­
торов ацетилхолина; рецепторы встраиваются в плазмолемму не только
области постсинаптической мембраны, но и по всей поверхности мышеч­
ного волокна).
Указанные (и другие изменения) обусловливают инертность механизмов гумо­
рального контроля, сужают диапазон компенсаторных возможностей денервированного органа, особенно в условиях его функциональной нагрузки или повреж­
дения. Подобные особенности наблюдаются и в трансплантированных органах
(в сердце, почках, печени) до периода восстановления их иннервации.
Существенно, что при денервации снижается также резистентность денервированного органа или ткани к повреждающим факторам: к инфекции,
механической травме, температурным и другим воздействиям.
Деаф ф ерентация
Нейротрофические расстройства возникают не только при денервационном
синдроме. Они развиваются при повреждении и афферентных структур нервной
системы. Так, деафферентация, вызванная деструкцией и/или дистрофически­
ми изменениями в чувствительном нерве, может привести к не менее выражен­
ным трофическим нарушениям в органе, чем его двигательная денервация.
Нейродистрофические процессы являются компонентом практически
всех форм патологии человека, обусловленных как функциональными рас­
стройствами, так и органическими повреждениями нервной системы. Они
проявляются не только изменениями функциональной активности органов,
но и изменениями их структуры (атрофия, изъязвления, малигнизация).
НАРУШЕНИЯ ВЫСШЕЙ НЕРВНОЙ ДЕЯТЕЛЬНОСТИ
Одной из наиболее распространенных форм патологии нервной системы
являются неврозы.
Термин «невроз» применяют для обозначения функциональных расстройств
нервной системы, т.е. нарушений высшей нервной деятельности.
По данным ВОЗ, заболеваемость неврозами в мире за последние 80 лет
возросла более чем в 20 раз.
Данные эпидемиологических исследований свидетельствуют не только
о большой медицинской, но и о социально-экономической значимости этой
проблемы, поскольку заболеваемость неврозами достигает 20—30%.
Неврозы относятся к так называемым болезням цивилизации. Широкую
их распространенность связывают с нарастающей урбанизацией населения,
информационными перегрузками, уменьшением доли физического труда в жизни
современного человека, с воздействием на него неблагоприятных социально­
бытовых факторов, многочисленных психотравмирующих ситуаций.
В клиническом аспекте невроз является психогенным состоянием и высту­
пает либо как самостоятельная нозологическая форма, либо как предболезненное состояние (предболезнь, пограничное состояние, синдром становле­
ния болезни), предшествующее различным соматическим и/или психическим
заболеваниям.
Исторически развитие учения о неврозах имеет три основных направления:
биологическое, личностно-психологическое и бихевиористское.
Биологическое. Оно исходит из доминирующей роли генетических измене­
ний, лежащих в основе неврозов. При этом недооценивают роль личностных
и психологических особенностей человека в возникновении невроза. С таким
подходом связан и так называемый принцип негативной диагностики невро­
зов. Он заключается в том, что к неврозам относят те расстройства функции
нервной системы, при которых отсутствуют изменения, выявляемые методами
клинической практики.
Личностно-психологическое. Это направление в учении о неврозах исходит
из предпосылки о личностно-психологической детерминированности воз­
никновения неврозов. Основной акцент делают на их психогенной приро­
де (связанной со стрессами, конфликтными ситуациями, экстремальными
состояниями). Однако при этом игнорируют роль соматических расстройств
в организме. Согласно личностно-психологической концепции, развитие
неврозов определяется особенностями личности человека, его неспособно­
стью разрешать различные психотравмирующие и конфликтные ситуации.
Бихевиористское. Представители этого направления рассматривают разви­
тие невротических состояний, беря за основу оценку особенностей поведения
человека в различных жизненных ситуациях.
В современных представлениях о неврозах указанные (и другие) тенденции
смыкаются.
В настоящее время неврозы рассматриваются как существенная часть пси­
хогенных состояний и болезней. Считается, что причиной невроза у человека
является психогенная травматизация личности в процессе психоэмоциональ­
ного стресса (как правило, негативного, повторного и/или затяжного). Это
приводит как к функциональным нарушениям в ЦНС, так и к определенным
микроструктурным изменениям в головном мозге (к деструкции мембран
шипикового аппарата дендритов, уменьшению количества рибосом в корко­
вых нейронах, дегенерации отдельных клеток гиппокампа, локальному нару­
шению микроциркуляции и др.).
Фундаментальный вклад в понимание неврозов внесли работы академи­
ка И.П. Павлова и его последователей. Исследуя неврозы в эксперименте
на животных, они сформировали ряд представлений об этой форме патологии
нервной системы.
Экспериментальные неврозы
С патофизиологической точки зрения невроз является типовой формой
патологии нервной системы.
Невроз возникает в результате перенапряжения и срыва ВИЦ пом влия­
нием воздействий, адекватность ответов на которые не обеспечивается ее
функциональными возможностями.
Патогенетическую основу неврозов составляют нарушения силы, подвижности
и уравновешенности основных нервных процессов — возбуждения и торможе­
ния, либо их столкновение («сшибка») в одно и то же (или близкое) время
и в одних и тех же структурах большого мозга.
Неврозы характеризуются расстройствами ВНД, развитием фазовых состоя­
ний в нервной системе, нейрогенными нарушениями вегетативных функций,
движения, чувствительности, трофики, а также снижением резистентности орга­
низма к различным эндо- и экзогенным патогенным агентам.
Экспериментальное воспроизведение неврозов базируется на едином принци­
пе — поставить перед животным нерешаемую (непосильную) для него задачу.
С этой целью применяются воздействия, вызывающие перенапряжение
и срыв возбудительного и/или тормозного процесса, нарушение их подвиж­
ности и уравновешенности или «сшибку» инстинктов альтернативной био­
логической значимости.
Перенапряжение и срыв процесса коркового возбуждения достигается при­
менением следующих воздействий:
—сильных безусловных раздражителей (например, болевого, светового,
звукового); они характеризуются большой интенсивностью, длительно­
стью или повторяемостью воздействия;
— сложных патогенных условных раздражителей (например, выработкой
условного рефлекса, сопровождающегося гипертензивной реакцией
на воздействия, которые следуют друг за другом в определенной после­
довательности, например световое, звуковое, тактильное);
—необычных раздражителей, имеющих биологически отрицательное значе­
ние (например, воздействие огня, взрывов и т.п.).
В результате указанных воздействий через определенное время (различное
у разных животных) развивается невротическое состояние с признаками преоб­
ладающего процесса торможения.
Перенапряжение и срыв тормозного процесса обеспечиваются в эксперимен­
те рядом методов:
— «отставлением подкрепления» (что обусловливает срыв процесса актив­
ного коркового торможения);
—выработкой тонких и сложных дифференцировок (это обеспечивает срыв
«дифференцировочного» торможения);
—отменой подкрепления (приводит к срыву «угасательного» торможения)
в ранее выработанных условных рефлексах.
Такими способами моделируют невротическое состояние с преобладанием
процесса возбуждения.
Перенапряжение и срыв подвижности основных корковых нервных процессов.
Этого достигают при:
—переделке сигнального значения разных условных раздражителей (напри­
мер, световой сигнал вместо ранее положительного подкрепления: полу­
чение пищи сопровождается последующим болевым воздействием);
—ломке сложившегося динамического стереотипа (серия последовательных
условных рефлексов).
Подобные воздействия обычно влекут за собой развитие невротических
состояний с патологической подвижностью нервных процессов.
«Сшибка» рефлексов-инстинктов взаимоисключающего (противоположного)
биологического значения
Осуществляется путем экстренного изменения качества подкрепляющего воз­
действия, например подачей электрического тока в пол кормушки в момент
пищевого подкрепления какого-либо сигнала или воздействием сильного
болевого раздражителя (биологически негативного) в момент полового акта.
Современные подходы к методам экспериментального воспроизведения невро­
зов у животных направлены на максимальное приближение к условиям их воз­
никновения у человека. К таким методам относятся:
— ограничение «рефлекса-инстинкта свободы» (например, насильственная
фиксация животного в станке);
—нарушение естественного суточного режима питания или светоритма, свя­
занных со сменой дня и ночи;
—изменение привычных стадно-иерархических или стадно-половых отноше­
ний (например, у обезьян подсадка в сложившийся прайд самца более
сильного и агрессивного конкурента);
—предварительная астенизация нервной системы (например, под влиянием
хронического шума, ионизирующей радиации, изоляции животного
от родителей в раннем детском возрасте).
Виды экспериментальны х неврозов
• Невроз с преобладанием процесса возбуждения. Развивается в резуль­
тате ослабления процесса торможения. Характеризуется постоянным
и неадекватным волнением, сочетающимся с агрессивностью и злобно­
стью животного. Этот вид нередко переходит в невроз тормозного типа
в связи с развитием запредельного торможения.
• Невроз с преобладанием процесса торможения. Является следствием ослаб­
ления процесса возбуждения. Характеризуется развитием пассивных обо­
ронительных реакций, депрессией и сонливостью животного.
• Невроз с патологической подвижностью основных корковых нервных про­
цессов. Развивается вследствие срыва процесса оптимальной смены воз­
буждения и торможения. Разновидности невроза с патологической под­
вижностью корковых нервных процессов:
— невроз с патологической инертностью. Характеризуется частым развитием
фобий;
— невроз с патологической лабильностью. Проявляется «суетливостью»,
незавершенностью действий, повышенной двигательной активностью;
— циркуляторный (циклический) невроз. Характеризуется хаотическим чере­
дованием перечисленных выше разновидностей невроза.
Роль особенностей высш ей нервной деятельности
в возникновении неврозов
Одни и те же экспериментальные воздействия могут вызывать различные
нарушения нервных процессов в высших отделах нервной системы. В боль­
шой мере это зависит от типа ВНД. В лаборатории И.П. Павлова была уста­
новлена зависимость вероятности возникновения и особенностей развития
невроза от особенностей ВНД.
Наиболее подвержен невротическим расстройствам слабый тип. Для этого
типа («меланхолик», по Гиппократу) характерна быстрая истощаемость воз­
будительного процесса, слабость внутреннего коркового торможения, пассив­
ность реакции на воздействие.
Это предопределяет возникновение невроза в результате срыва основ­
ных корковых нервных процессов с развитием торможения и формированием
пассивно-оборонительных реакций.
Холерик (сильный неуравновешенный тип; «безудержный», по И.П. Пав­
лову). Этот тип отличается сильным возбудительным процессом, слабым
корковым торможением, активными реакциями на раздражители.
Это обусловливает развитие невроза возбудительного типа с формированием
активно-поисковых реакций.
Флегматик (сильный уравновешенный инертный тип). Характеризуется
развитием невроза с патологической подвижностью нервных процессов.
Сангвиник (сильный уравновешенный подвижный тип). Наиболее устойчив
к воспроизведению неврозов в связи с высокой резистентностью к различного
рода патогенным агентам. Повышение силы раздражителя, «сшибка» инстинктов,
увеличение деятельности и повторение воздействий может привести к неврозу.
Проявления экспериментальны х неврозов
Расстройства ВНД. Они выражаются выпадением условных рефлексов, уве­
личением латентных периодов ответов на воздействия, трудностью или невоз­
можностью выработки новых условных рефлексов и, как следствие, — адек­
ватного приспособления к меняющимся условиям жизнедеятельности. Это
влечет за собой снижение адаптивных возможностей нервной системы и организ­
ма в целом, утрату индивидуальных черт реагирования, снижение «обучаемости»
животных новым навыкам.
Развитие так называемых фазовых состояний в нервной системе. Они харак­
теризуются качественной и/или количественной неадекватностью реагирова­
ния индивида на раздражители в зависимости от доминирующего в данный
момент фазового состояния.
Нарушение вегетативных функций. Этот признак является постоянным,
наиболее ранним и устойчивым проявлением неврозов. Изменение вегетатив­
ных функций, как правило, утрачивает свое адаптивное значение, становится
неадекватным раздражителю, не соответствующим сопутствующим локомо­
торным реакциям (например, развитие артериальной гипотензии и гипогли­
кемии при оборонительной реакции).
Расстройства движений. Они отличаются многообразием и выражаются
в форме различных гипер- и гипокинезов, атаксии.
Нарушения нервной трофики. Они проявляются различными дистро­
фиями, вплоть до появления эрозий и язв; изменениями иммуногенной и неспецифической реактивности организма (например, аллергиями
или диатезами).
Расстройства чувствительности. Это выражается развитием гипо- и гипере­
стезий, гиперпатий, парестезии, полистезии и других дизестезий.
Этиология неврозов
Невроз является реакцией личности на трудную, часто неразрешимую для нее
ситуацию.
В целом в основе возникновения неврозов находится невротический кон­
фликт, т.е. такое отношение личности к конкретной ситуации, которое делает
невозможным и «непосильным» ее рациональное решение.
Причина невроза — психическая травматизация личности.
Психотравмирующая ситуация оказывает патогенное воздействие
при наличии определенных условий, прежде всего особенностей личности.
Определяющей среди этих особенностей является гиперактуализация неблаго­
приятных воздействий и/или событий и придание им чрезмерной биологиче­
ской или социальной значимости.
Условия развития невроза
Выделяют три основных группы условий, способствующих или препят­
ствующих развитию неврозов: биологические, социальные и психогенные.
• Биологические факторы риска невроза:
—наследственная предрасположенность к неврозу. Отдельные невротиче­
ские состояния (например, панический синдром) чаще встречаются
у представителей одной генеалогической линии;
—пол (невроз чаще возникает у женщин);
—возраст (невроз чаще развивается в пубертатном и климактерическом
периоде);
—конституциональные особенности организма (к неврозам более склонны
астеники);
—заболевания, снижающие резистентность организма (перенесенные и теку­
щие).
• Социальные факторы риска возникновения невроза:
—особенности профессиональной деятельности (например, информацион­
ные перегрузки, однообразие трудовых операций);
—неблагополучное семейное положение;
—неудовлетворительные бытовые условия;
— особенности сексуального воспитания и др.
• Психогенные факторы риска развития невроза:
—личностные особенности (индивидуальный способ мышления, восприя­
тия, поведения и реагирования на воздействия у данного человека);
—психические травмы в детстве;
—психотравмирующие ситуации (например, тяжелая болезнь или утрата близ­
ких, служебные или «академические» трудности) и некоторые другие.
Формирование невроза определяется не только непосредственной
реакцией личности на патогенное воздействие, но и более или менее
длительным процессом анализа сложившейся ситуации. В медицине это
обозначают термином «идеаторная переработка» индивидом ситуации и ее
последствий, боязнью невозможности приспособиться к сложившимся
обстоятельствам.
Виды неврозов
Общепринятая классификация неврозов в настоящее время не разработана.
Традиционно выделяют три группы наиболее распространенных форм
неврозов: невроз навязчивых состояний, истерия, неврастения.
В современных классификациях болезней (например, в 10-м издании
Международной классификации болезней, МКБ-10, 1994) выделена группа
Р4, обозначенная как невротические, связанные со стрессом и соматоформные расстройства (рис. 29.15).
Невроз
навязчивых
состояний
Истерический
невроз
Неврастения
«Классические»
виды
Р40. ТревожноР44. Диссоциативные Р48.0. Неврастения'
фобические
(конверсионные)
расстройства
расстройства
Виды по
Р41. Другие тревожные Р45. Соматоформные
Г МКБ-10
расстройства
расстройства
(панический
синдром,
смешанные
расстройства)
Р42. Обсессивнокомпульсивные
расстройства
Рис. 29.15. Соотношение «классических» видов неврозов и нозологий по Между­
народной классификации болезней (МКБ-10)
Невроз навязчивых состояний
По МКБ-10 эти состояния отнесены к разделам «Р40. Тревожно-фобические
расстройства», «Р41. Другие тревожные расстройства», «Р42. Обсессивнокомпульсивные расстройства».
Причина невроза навязчивых состояний — диссоциация («конфликт»)
между желаниями, стремлениями, потребностями личности и невозмож­
ностью (!) их реализации по моральным или иным соображениям.
При этом в коре головного мозга формируется патологический очаг воз­
буждения. Обычно это происходит после одного из эпизодов, когда человек
забыл сделать что-то важное (выключить газ, закрыть дверь, накормить
ребенка и т.п.) или перенес состояние страха (высоты, остановки лифта, без­
защитности и т.д.).
Все разновидности навязчивых состояний характеризуются повторяющим­
ся чувством страха, боязни, фобии чего-либо и/или кого-либо — опре­
деленных предметов, деятельности, ситуаций. Начало невроза навязчивых
состояний формируется по механизму условного рефлекса. Позже условия
возникновения фобии расширяются.
Заключение о наличии фобии делается в том случае, когда это состояние
нарушает индивидуальную социальную и профессиональную жизнь.
Виды наиболее частых расстройств при неврозе навязчивых состояний:
—агорафобия (от древнегреч. а§ога — площадь для сбора граждан) — боязнь
находится вне дома, на открытом пространстве, например на открытой
городской площади или в большой комнате, в связи со страхом быть
подверженным опасности нападения или развития плохого самочув­
ствия и невозможностью получить своевременную помощь и т.п.);
— социальные фобии—повторяющая абсурдная боязнь, страх оказаться в затруд­
нительном или унизительном положении в обществе (например, во время
общественного выступления, чтения лекции, на экзамене, в гостях); нередко
это действительно приводит к неудаче и закрепляет фобию;
—обсессивно-компульсивные расстройства — навязчивые, повторяющие­
ся, «лезущие» в голову идеи, мысли, «приказы» совершить то или иное
действие. Человек сопротивляется этому, осознавая и нежелательность,
абсурдность, и бессмысленность такого действия, боясь его (например,
убийства любимого существа, родственника; постоянного мытья рук
из-за страха заразиться или испачкаться; бесконечной проверки себя:
закрыл ли дверь, выключил ли газ и т.п.);
— простые фобии — постоянные немотивированные страхи и/или стрем­
ление избегать ситуаций, которые могут вызвать к жизни эти опасения
(например, клаустро- или канцерофобия).
Истерический невроз
По МКБ-10, к истерическому неврозу относят состояния, включенные
в разделы «Р44. Диссоциативные (конверсивные) расстройства» и «Р45.
Соматоформные расстройства».
Указанные группы расстройств при истерическом неврозе характеризуются ими­
тацией (но не симуляцией!) пациентами болезней и богатыми соматоформными нару­
шениями. Последнее проявляется чрезмерным вниманием к своему здоровью,
необоснованной тревогой за него, убежденностью в наличии болезни, которая
в действительности отсутствует. При истерии, как правило, имеется «желание
болезни» и сильная внушаемость. Этим объясняется полиморфизм истерических
расстройств, их нарочитый, демонстративный и утрированный характер.
Причина истерического невроза — диссоциация («конфликт») между
завышенными требованиями личности к окружающим и невозможностью
их реализовать или достичь.
Последнее обусловлено недооценкой либо игнорированием реальных усло­
вий и/или требований других людей. При этом перенапряжение и истощение
корковых процессов способствует формированию в подкорковых структурах
«мобильных» очагов патологического возбуждения. Последние создают усло­
вия для развития сменяющих друг друга фазовых состояний (чаще парадок­
сальной, ультрапарадоксальной, реже наркотической, тормозной и др.) в коре
головного мозга.
Проявления истерии. Для истерического невроза характерна очень пестрая
и изменчивая симптоматика. Истерия — это постоянно меняющаяся мозаика
симптомов. Истерические расстройства являются защитной реакцией личности
в связи со сложившейся неразрешимой для нее ситуацией. Симптоматика исте­
рии может быть сведена к нескольким группам болезненных проявлений.
• Неадекватное поведение. Пациенты отличаются повышенной эффективно­
стью, впечатлительностью, внушаемостью и самовнушаемостью, неустойчи­
востью настроения, забывчивостью (диссоциативная амнезия). Истерические
эмоционально-аффективные расстройства сопровождаются театральностью
и наигранностью переживаний, их приуроченностью к определенным ситуа­
циям. Люди с истерическим неврозом характеризуются внешне «энергич­
ными», но в действительности поверхностными межличностными отноше­
ниями. Они стараются произвести впечатление весьма занятых, значимых,
влиятельных личностей, являющихся центром событий. На самом деле они
являются, как правило, персонами мелкими, невдумчивыми, суетливыми,
зависимыми от других людей. Некоторые из них угрожают покончить собой
(«суицидальный шантаж») и нередко пытаются это сделать реально.
• Вегетативные расстройства (например, гипо- или гипертензивные реак­
ции, одышка, приливы «горячей крови» к лицу, тахикардия, аритмия,
потливость, диспепсические расстройства).
• Двигательные расстройства. При истерии могут развиваться судорожные
припадки (однако без потери сознания и ушибов!), преходящие парезы
и параличи; возможна обычно кратковременная афония из-за паралича
голосовых связок и даже мутизм, что, однако, не огорчает пациента.
• Сенсорные нарушения. Истерический невроз нередко сопровождается пре­
ходящей слепотой, глухотой, потерей обоняния, вкуса, парестезиями.
• Сексуальные отклонения (например, импотенция, снижение либидо).
Большинство вегетосоматических нарушений при истерии носит психо­
генный характер. Это обозначается как диссоциативное, или конверсионное
расстройство — развитие нарушений жизнедеятельности организма на фоне
и под влиянием факторов психоэмоционального характера.
Неврастения
Неврастения считается наиболее распространенной формой невроза.
В МКБ-10 эта разновидность невроза обозначена как «Р48.0 Неврастения
(или синдром усталости)».
Причина неврастении — диссоциация («конфликт») между требованиями
к самому себе (как правило, завышенными) и невозможностью их осуще­
ствить. Это обусловливает перенапряжение и срыв процесса коркового воз­
буждения.
Проявления неврастении
• Вегетативные расстройства (например, нарушения ритма сердца, гипо-
или гипертензивные реакции, желудочно-кишечные расстройства, повы­
шенная потливость).
• Повышенные возбудимость, утомляемость и истощаемость нервной системы.
•Чрезмерная раздражительность, несдержанность, нетерпеливость («раздра­
жительная слабость»).
• Расстройства внимания, нарушение его концентрации.
• Сниженная работоспособность, вялость.
• Неустойчивость настроения, нередко — подавленность.
• Расстройства сна (нарушение засыпания, беспокойный сон, неприятные
сновидения).
• Сексуальные нарушения (например, снижение сексуального влечения,
импотенция).
Общ ие проявления невротических состояний
В процессе невротического состояния имеется закономерная последова­
тельность включения в структуру невроза различных систем и, как следствие,
формирование общих проявлений.
• Неадекватность вегетативных реакций воздействию (например, тахикардия,
аритмия, одышка, повышенная потливость, покраснение или побледне­
ние кожи и слизистых оболочек, гипо- и гипертензивные реакции, нару­
шения сна и аппетита, ощущение болей в сердце, возникающих в ответ
на воздействие, которому пациент придает особое значение).
• Развитие патологических сенсомоторных реакций (например, повышенная
чувствительность к различным внешним воздействиям или изменениям
в организме, суетливость, бессмысленные излишние движения, жестику­
ляция, преходящие парезы и параличи, неадекватная событию мимика).
• Частые аффективные реакции: бурное эмоциональное реагирование на воз­
действие или ситуацию (например, тревога, страх смерти, эмоциональное
напряжение, рыдания, брань). Пациент «не владеет своими чувствами,
чувства владеют пациентом».
• Интеллектуальный анализ болезненного состояния и принятие решения
о форме поведения, это обозначается как идеаторная переработка и выра­
ботка мер компенсации. Они направлены на преодоление сложившейся
ситуации и болезненных ощущений.
Указанная выше последовательность формирования проявлений в процес­
се развития неврозов выявляется у большинства пациентов. Такая же последо­
вательность, но с менее выраженными изменениями, наблюдается и при воз­
действии различных стрессорных агентов. Существенно, что и в онтогенезе
порядок включения реакций организма на различные воздействия носит ту же
последовательность: вначале наблюдаются вегетативные, а затем включаются
сенсомоторные, аффективные и идеаторные реакции.
С учетом указанных обстоятельств в клинической картине выделяют соот­
ветствующие фазы (стадии) развития невроза:
- вегетативных реакций;
- сенсомоторных реакций;
—аффективных реакций;
—идеаторной (ассоциативной, мыслительной, концептуальной) пере­
работки ситуации и выработки программы, помогающей преодолеть
болезненное состояние.
Вместе с тем имеются факты, свидетельствующие о том, что у пациента
могут одновременно появиться и вегетативные, и сенсомоторные, и аффек­
тивные, и идеаторные расстройства. В связи с этим правильнее говорить
не о стадиях или фазах невроза, а об общих компонентах его клинической
картины.
Понятие о «вегетоневрозе»
Один из общих, постоянных и ранних компонентов невроза — вегета­
тивные расстройства: разнообразные синдромы нарушения функций вну­
тренних органов и их физиологических систем (кровообращения, дыхания,
пищеварения, половой и др.). Они являются результатом центрогенных рас­
стройств, связанных с регуляцией деятельности органов нейрогенного генеза.
В клинической литературе эти расстройства обозначают различными терми­
нами: «нейроциркуляторная дистония», «вегетоневроз», «вегетодистония»,
«вегетативно-сосудистая дистония» и многими другими. Такое многообразие
терминов обусловлено объективной сложностью и неоднозначностью патоге­
неза и проявлений вегетативных нарушений при неврозах.
В общем виде эти расстройства характеризуются неадекватностью и раз­
нообразием реакций внутренних органов и их систем на различные раз­
дражители (см. раздел «Фазовые состояния»), рассогласованием отдельных
вегетативных компонентов и целостной реакции организма, диссоциацией
локомоторных и вегетативных реакций, утратой адаптивного значения веге­
тативных функций.
Указанные изменения при неврозах могут иметь перманентный, пароксиз­
мальный или сочетанный перманентно-пароксизмальный характер.
Наиболее частые проявления вегетоневрозов — боли в области серд­
ца, аритмии, эпизоды сердцебиения, тахипноэ или диспноэ, сосудистая
дистония (гипер- и/или гипотензивные реакции), диспепсические явления.
Развиваются эти изменения, как правило, на фоне слабости, повышенной
утомляемости, раздражительности, нарушений сна (бессонница, сонливость,
беспокойный сон).
Невротические состояния нередко предшествуют соматическим патоло­
гиям: ИБС, ГБ, язвенной болезни желудка и двенадцатиперстной кишки,
различным эндокринопатиям.
ЛИТЕРАТУРА
Пальцев М .А ., Аничков Н .М ., Лит вицкий П.Ф. Патология человека: учебник:
в 2 т. — М.: Медицина, 2009. — 1448 с.
Патофизиология / под ред. А. И. Воложина, Г. В. Порядина. — М .: Академия,
2006. - Т. 1-3. - 908 с.
Патофизиология / под ред. В.В. Новицкого, Е.Д. Гольдберга, О.В. Уразовой. — М.: ГЭОТАР-Медиа, 2009. — Т. 1, 2. — 1474 с.
Патофизиология. Задачи и тестовые задания: учебно-методическое посо­
бие / под ред. П.Ф. Литвицкого — М.: ГЭОТАР-Медиа, 2011. — 384 с.
Патофизиология: руководство к занятиям: учебно-методическое пособие /
под ред. П.Ф. Литвицкого. — М.: ГЭОТАР-Медиа, 2010. — 128 с.
СоиШ В.Е. РасНорЬу8ю1о§у Гог 1Ье НеаИЬ Рго^еззюп. — 31Ь Ей. — Е18еУ1ег,
2006. - 800 р.
СиуЮп А .С ., На11 ^ Е . Нитап. РЬу8ю1о§у апй МесЬашктк оГ 01$еазе. —
7 Ей. — 5аипс1ег$, 2009. — 737 р.
ЬНуШку Р.Р., Р и огккоу 8. V , Т еукоу Е.В. Ра1ЬорИуз1о1о§у. Сопс18е ЬесШгез,
Се818, сПп1со-ра1Ьор[1у81о]о81са1 зНиаНот апс! сНп1СО-1аЬога1огу ргоЫетз: 8Шс1еп1
тапиа1. — М.: ГЭОТАР-Медиа, 2012. — 448 р.
М сСапсе К .Ь ., НиеНгег 8.Е . РаИюрЬу8ю1о§у. ТЬе Вю1о§ю Ва818 Гог В 1$еа$е т
Ас1и11:8 апй СЫЫгеп. — 51Ь Ей. — Е18еу1ег, 2005. — 1779 р.
ПРИЛОЖЕНИЯ
СПРАВОЧНИК ТЕРМИНОВ
Составлен с дополнениями по следующим источникам: Англо-русский
медицинский энциклопедический словарь (81ес1тап'$ МесНса! Оюпопагу). —
М.: ГЭОТАР, 1995; М е т а т \\ЬЬз1ег’8 Ме<Иса1 Безк 01с1юпагу. — 8рпп§Пе1<±
Метат-\УеЪз1:ег 1пс., 1995; Нир://ул\'у/.Вп1:апгпса.сот; Кпр://\у\у\у.гиЬг1Соп.ги;
Энциклопедический словарь медицинских терминов. — М.: Советская энци­
клопедия, 1982—1984; 2000 болезней от А до Я. Справочник-путеводитель. —
М.: ГЭОТАР, 1998-2001; Внутренние болезни. - М.: ГЭОТАР-МЕД, 2001;
Медицинская микробиология. — М.: ГЭОТАР-МЕД, 2001; Гистология. — М.:
ГЭОТАР-МЕД, 2001; РзсЬугетЬе1 КНгшсНез УУоЯегЬисЬ, 259 АиЯа§е. — ВегНп —
1Че\у Уогк: \Уа11ег с!е ОгиуХег, 2002.
Через символ <=> приведены синонимы. В статьях и подстатьях применяет­
ся сокращение термина — первая его буква (например, в подстатьях к статье
«Анемия» вместо этого термина использовано сокращение «а.»).
Абеталипопротеинемия (р) и гипобеталипопротеинемия (5К) — наследуемые
расстройства, вызванные мутациями гена апоЛП В, главного апоЛП
хиломикронов и ЛПОНП (*200100, 2р23—р24, ген АРО В, множество
аллелей; также обнаружены дефекты в большой субъединице (СЕ) моле­
кулы микросомального белка-переносчика триглицеридов, * 157147).
При а. в крови нет р-ЛП, хиломикронов, ЛП с плотностью ниже 1,063
(ЛПНП и ЛПОНП), эритроциты имеют множественные шиповидные
выросты (акантоциты), развивается дефицит витаминов Е и А (резуль­
тат отсутствия ЛПНП, транспортирующих жирорастворимые витамины).
Нередки нарушения всасывания в кишечнике, а также (вследствие
демиелинизации аксонов) нарушения координации, атаксия, нистагм,
пигментная дегенерация сетчатки и отставание в умственном развитии.
Стеаторея возникает вследствие недостатка апопротеина В, опреде­
ляющего образование хиломикронов в клетках кишечника. Лечение.
• Специфической терапии нет. • Диета: продукты, богатые полиненасыщенными жирными кислотами из семейства линоленовой кислоты
и витамином Е (растительное масло различных сортов: подсолнечное,
кукурузное, хлопковое, соевое, льняное, конопляное), злаковые гру­
бого помола и бобовые. • Витамин Е (токоферола ацетат) в высоких
дозах (до 100 мг/кг/сут в несколько приемов внутрь) может уменьшить
выраженность неврологических проявлений. • Одновременно назна­
чают витамин А (ретинол) для профилактики гиповитаминоза А, так
как токоферол в высоких дозах может вызвать его развитие. <=> Бассена—
Корнцвейга синдром <=> Акантоцитоз (эритроцитов).
Абсцесс. 1. Ограниченное скопление гноя, возникающее при острой или хро­
нической очаговой инфекции и приводящее к тканевой деструкции в очаге
(нередко с перифокальным отеком) <=> гнойник. 2. Полость, возникающая
вследствие некротических процессов.
Авидность — интегральная характеристика силы связи между Аг и АТ, учиты­
вающая взаимодействие всех активных центров с эпитопами Аг.
Агаммаглобулинемия — отсутствие или резкое снижение уровня у-глобулинов
в сыворотке крови. А. — гетерогенная группа хронических заболеваний,
характеризующихся рекуррентными бактериальными инфекциями, часто
сочетается с различными аутоиммунными заболеваниями. Повышен риск
развития лимфом кишечника и рака желудка <=> гипогаммаглобулинемия
<=> иммунодефицит. • Брутона а. — сцепленная с хромосомой X агаммагло­
булинемия (*300300, X ^ 2 1 .2 -^ 2 2 , дефект гена АСМ Х1, кодирующего тирозинкиназу; ключевой регулятор развития В-клеток, не менее 50 аллелей; X,
также другие варианты): первичный иммунодефицит мальчиков, характе­
ризующийся сниженным содержанием (вплоть до отсутствия) циркулиру­
ющих В-лимфоцитов и соответствующим снижением 1§ всех изотипов (все
популяции Т-клеток в норме); выраженная восприимчивость к инфекциям
(в особенности опасны пневмонии и менингиты), вызванным пиогеиными
бактериями (прежде всего пневмококками и НаеторЫЫз т/1иещ ае)\ раз­
вивается вслед за снижением содержания трансплацентарно полученных
материнских АТ; необходимо постоянное введение антибиотиков и 1§ <=>
брутоновского типа а. <=> сцепленная с Х-хромосомой гипогаммаглобу­
линемия о сцепленная с Х-хромосомой гипогаммаглобулинемия ново­
рожденных <=> врожденная а. <=> Брутона болезнь. • Швейцарского типа а.
(*202500, р). Характерны: подверженность инфекциям любой этиологии,
гипоплазия тимуса (нет Т-лимфоидных клеток и телец Гассаля), гипо­
гаммаглобулинемия. Известны фенокопии вследствие внутриутробного
инфицирования вирусом краснухи. <=> Тяжелый комбинированный имму­
нодефицит типа I о алимфоцитарная а.
Агенезия (аплазия) — полное врожденное отсутствие органа или его части.
Агорафобия (от греч. а§ога — площадь для собраний граждан + -ркоЫа —
страх) — иррациональный страх находиться вне дома, боязнь открытого
пространства.
Агликогенозы — распространенная форма патологии углеводного обмена,
характеризующаяся существенным дефицитом или отсутствием гликоге­
на в клетках, гипогликемией, дистрофическими изменениями в тканях
и органах, молочнокислым ацидозом. А. — компонент различных гипогликемических состояний.
Агранулоцитоз — резкое уменьшение количества лейкоцитов [менее 1хЮ9/л
(нейтрофильных гранулоцитов менее 0,75хЮ9/л)], обусловливающее повы­
шенную восприимчивость к бактериальным и грибковым инфекциям.
Этиология • Ионизирующая радиация и лучевая терапия, химические
вещества (бензол, горчичный газ), инсектициды. • ЛС (могут приве­
сти к нейтропении и даже к а.): НПВС, антиметаболиты (метотрексат,
фторурацил, цитарабин), сульфаниламиды, барбитураты; ацетазоламид
(диакарб*), левамизол, тиамазол (мерказолил*), хлорамфеникол (левомицетин*), изониазид, триметоприм, хлорохин (хингамин*), алкилиру-
юшие вещества [циклофосфамид (циклофосфан*)], противоопухолевые
антибиотики (доксорубицин). • Аутоиммунные заболевания (например,
СКВ). • Генерализованные инфекции — брюшной тиф, паратиф, грипп,
корь, риккетсиозы, гепатиты. • Кахексия. • Генетическая предрасположен­
ность (см. «А. наследственный»). Типы а. Различают миелотоксический
(например, при воздействии ионизирующей радиации) и иммунный а.
Последний может быть обусловлен появлением аутоантител (например,
при СКВ) или АТ к гранулоцитам после приема ЛС (гаптенов), приобре­
тающих при попадании в организм после соединения с белком свойства
Аг. Протекает обычно остро. Летальность при а., вызванном приемом ЛС,
достигает 80%. См. также «Нейтропения». • Наследственный а. Характерные
признаки н. а. (нейтропений) — лихорадка, рекуррентные инфекции кож­
ных покровов и дыхательных путей. • Болезнь Костманна [*202700, в том
числе дефект рецептора колониестимулирующего фактора гранулоцитов
(138971, 1р35—р34.3, ген С 8ГЗЕ ), р, ассоциация с экспрессией НЬА-В12].
Тяжелые и часто смертельные рекуррентные инфекции кожных покро­
вов (фурункулез, рожистое воспаление) и дыхательных путей наблюдают
с первого месяца жизни. В костном мозге — задержка созревания нейтрофилов в стадии промиелоцита или юного миелоцита. Лечение — анти­
бактериальная терапия, антистафилококковый глобулин. Трансплантация
костного мозга — метод выбора. <=> Врожденная нейтропения <=> наследуе­
мый а. новорожденных. • Врожденная алейкия (ретикулярная дисгенезия,
*267500, р). Врожденный агранулоцитоз и лейкопения приводят к развитию
тяжелого иммунодефицита. Часто сочетается с гипоплазией вилочковой
железы. • Семейные нейтропении (прогноз хороший, несмотря на частые
инфекции, например фурункулез). О Доброкачественная семейная ней­
тропения (*162700, 9?). Хроническое течение, характерны язвенные пора­
жения слизистой оболочки ротовой полости, гиперпластический гингивит,
периодонтит, гиперглобулинемия. О Циклическая нейтропения (* 162800,
'Л). Циклические (15—35 дней) изменения содержания всех форменных
элементов крови. Характерны лихорадка, инфекции, язвенный стоматит.
• Синдром «ленивых» лейкоцитов (150550, спорадические случаи вследствие
новых мутаций). Лабораторно: внешне нормальные нейтрофилы с нор­
мальными показателями фагоцитоза и антибактериальной активности,
но с дефектами хемотаксиса и миграции. • Синдром Грисчелли [*214450,
15д21, ген М У 05А (М УН 12 , киназа тяжелой цепи миозина, 160777), р]
подобен по клинике и течению синдрому Чедиака—Штайнбринка—Хигаси.
• Хроническая гранулематозная болезнь. При недостатке в лизосомах 0 2
и Н20 2 снижена бактерицидная активность нейтрофилов, вследствие чего
фагоцитированные микроорганизмы (например, золотистый стафилококк)
размножаются внутриклеточно. Характерны рецидивирующие гнойные
инфекции. Заболевание нередко заканчивается летально. Диагноз под­
тверждают, обнаруживая в мазке периферической крови нейтрофилы,
не способные превращать нитросиний тетразолий в темно-синий формазан
(НСТ-тест). • Чедиака—Хигаси синдром (*214500, \<\А2Л—<\А2.2, ген СН51,
внутрилизосомный регулятор транспорта, р). Первичный дефект фагоци-
тоза, частичный иммунодефицит, гипогаммаглобулинемия, нейтропения,
тромбоцитопения. Характерен функциональный дефицит миелопероксидазы и торможение хемотаксиса. Патологические изменения гранул и ядер
всех типов лейкоцитов, тельца Деле. Проявления — светлая радужная
оболочка, альбинизм, возможна гиперпигментация кожи, гепатоспленомегалия, лимфаденопатия, анемия, тромбоцитопения, изменения в костях,
легких, сердце, а также психомоторные дефекты и предрасположенность
к инфекциям. <=> Чедиака—Ш тайнбринка—Хигаси аномалия (синдром,
болезнь).
Адгезия. 1. Формирование спаек — фиброзных тяжей между противополож­
ными серозными поверхностями в результате воспалительного процесса
или травмы. 2. Способность клеток избирательно прикрепляться друг
к другу или к элементам внеклеточного матрикса (см. также «Молекулы
адгезии»). Адгезионный контакт. Прикрепление клеток к молекулам а. вне­
клеточного матрикса реализуют точечные (фокальные) а. к. В образовании
а. к. участвуют трансмембранные рецепторы — интегрины, объединяющие
внеклеточные и внутриклеточные структуры. А. к. содержит также винкулин, а-актинин и другие белки. Характер распределения макромолекул а.
во внеклеточном матриксе (фибронектин, витронектин) определяет место
окончательной локализации клетки при перестройках тканевых структур
(например, при заживлении ран). Нарушения клеточной а. Клетки способ­
ны к перемещению только после дезинтеграции адгезионных контактов.
Подобную картину наблюдают при выселении злокачественных (транс­
формированных) клеток из опухоли и при их метастазировании, когда
отдельные трансформированные клетки отделяются от опухоли, мигриру­
ют и образуют новые опухолевые очаги (метастазы). Причиной этих ослож­
нений служат дефекты генов, кодирующих гликопротеины клеточной а.
Например, метастазы и разрастания карцином желудка, эндометрия и яич­
ника возникают вследствие нарушения а. опухолевых клеток, содержащих
мутантный ген Е-кадгерина, и экспрессии его дефектного продукта.
Аденозинтрифосфатаза (АТФаза) • Кальциевая (Са2+-насосы, Са2+-АТФазы) —
откачивают Са2+ из цитозоля против значительного концентрационного
градиента. Са2+-насос саркоплазматического ретикулума откачивает ионы
кальция из цитозоля не во внеклеточное пространство (как Са2+-АТФаза
плазмолеммы), а во внутриклеточные депо кальция (как правило, в замкну­
тые межмембранные объемы гладкой эндоплазматической сети, называемой
в скелетных мышечных волокнах и кардиомиоцитах саркоплазматической
сетью). • Натрий, калиевая. № +-, К +-насос — интегральный мембран­
ный белок, состоящий из 2 СЕ (каталитическая СЕ а и гликопротеин (3).
Участки связывания
расположены на цитоплазматической поверх­
ности а-СЕ, а К + — на наружной ее поверхности. При гидролизе одной
молекулы АТФ 3 № + выкачиваются из клетки, и 2 К + закачиваются в нее.
• Протонная и калиевая. С помощью этого фермента париетальные клетки
желез слизистой оболочки желудка участвуют в образовании соляной кис­
лоты (электронейтральный обмен внеклеточного К+ на внутриклеточный
Н+). Н+-, К +-АТФаза — гетеродимер (высокомолекулярная а-С Е + глико-
зилированная р-СЕ). р-СЕ — главный Аг, к которому при пернициозной
анемии и атрофическом гастрите в крови циркулируют АТ.
Аденокарцинома — злокачественная опухоль, происходящая и состоящая
из железистых или железистоподобных эпителиальных клеток <=> желези­
стый рак <=> железистая карцинома.
Аденома — доброкачественная опухоль, возникающая из железистого эпите­
лия и сохраняющая структурное сходство с исходной тканью.
Аденоматоз — наличие множественных аденом. • Семейный полиэндокринный а.
(СПЭА) — наличие функционирующей опухоли в двух и более эндокрин­
ных железах, чаще в островковой части поджелудочной железы и паращитовидной железе. Нередко сопровождается образованием пептических
язв желудка и повышением желудочной секреции. <=> Множественный
эндокринный а. о множественные эндокринные неоплазии. О СПЭА I
(5?, * 131100, 11я13) — опухоли передней доли гипофиза, островковых
клеток поджелудочной железы (инсулиномы) и околощитовидных желез.
❖СПЭА II (9?, #171400, 10я11.2, мутации онкогена К ЕТ) — феохромоцитома,
гиперплазия околощитовидных желез, медуллярная карцинома щитовид­
ной железы, Кушинга синдром, амилоидоз кожи, гипокальциемия, повы­
шенное содержание тирокальцитонина <=> синдром Сиппла. 0 СПЭА III (5К,
#162300, 10я11.2) — множественные невромы слизистой оболочки губ, век
и языка; телосложение аналогично такое же, как при синдроме Марфана,
также характерны аномалии скелета, медуллярная карцинома щитовидной
железы, феохромоцитома.
Адреналин — /-1-(3,4-дигидроксифенил)-2-(метиламино) этанол — секретаруется из хромаффинных клеток, только гуморальный фактор, в синап­
тической передаче не участвует, агонист адренорецепторов. Деградация а.
и других биогенных аминов происходит под влиянием моноаминоксидаз
и катехол-О-метилтрансферазы. В результате образуются экскретируемые
с мочой метанефрины и ванилилминдальная кислота, маркёры феохромоцитомы.
Адреноблокатор (антагонист адренорецепторов) — соединение, которое изби­
рательно блокирует или угнетает активность симпатических адренергиче­
ских нервов, препятствуя взаимодействию медиатора с адренорецепторами
без нарушения процесса образования медиатора и выделения его из нерв­
ных окончаний.
Адренолейкодистрофия — сфинголипидоз, характеризующийся сочетани­
ем лейкодистрофии и болезни Аддисона. Поражает преимущественно
мальчиков (частота 1:20 000 рождений). Проявляется в возрасте 5—12 лет
и заканчивается фатально. Адреномиелоневропатия — более мягкая форма —
проявляется в возрасте 15—30 лет. Биохимия и генетика. • р-Окисление жир­
ных кислот в пероксисомах последовательно катализируется ацил-КоАоксидазой (КФ 1.3.3.6), бифункциональным ферментом [с активностью
еноил-КоА-гидратазы (КФ 4.2.1.17) и 3-гидроксиацил-КоА-дегидрогеназы
(КФ 1.1.1.35)] и 3-кетоацил-КоА-тиолазой (КФ 2.3.1.16). Их дефекты
приводят к а. Также известны приводящие к а. мутации множества дру­
гих генов обмена жирных кислот (см. «Дефекты окисления жирных кис­
лот»). Проявления. Гиперпигментация кожи, гипогонадизм, спастическая
параплегия, генерализованная атаксия, нарушения речи и отставание
в умственном развитии, повышение содержания длинноцепочечных жир­
ных кислот в плазме крови. <=> Земмерринга—Крейтцфельда болезнь, меланодермическая лейкодистрофия, Шильдера бронзовая болезнь.
Адреномедуллин — мощный вазодилататор (52 аминокислотных остатка, выде­
лен из клеток феохромоцитомы, из семейства относящихся к кальцитониновому гену пептидов). Содержание а. в крови значительно увеличено
у больных артериальными гипертензиями.
Адреномиметик — вещество, стимулирующее адренорецепторы (медиатор —
норадреналин) о адреностимулятор <=> агонист адренорецепторов.
Адренохром — продукт окисления адреналина, имеет гемостатические свой­
ства. Применяют в виде стабильного производного — карбазохрома салицилата.
Адрессин — см. «Молекула адгезии МАСАМ1», «Маркёр, СЭ44».
Адъюванты (от лат. аф и чат — помогать) — вещества, введение которых
одновременно с Аг (или гаптеном) усиливает иммунный ответ. Другими
словами, а. — носитель, повышающий иммуногенность различных Аг
и гаптенов. Распространенные аа. — суспензии неорганических веществ,
на которых адсорбируется Аг. Классический пример — коллоидная суспен­
зия из убитых туберкулезных палочек, вазелина, ланолина, известная так
же, как полный адъювант Фрейнда.
Адъювантный (от лат. а й )т а т ) — вспомогательный (о методе, лечении, ЛС
и т.п.).
Азооспермия — см. «Бесплодие мужское».
Азуроцидин — катионный антимикробный белок с Мг 29 кД.
Акантоцит — эритроцит множественными шиповидными выростами.
Аквапорины (А(}Р) — семейство мембранных пор для воды. Идентифицировано
10 аа. АС^РЗ, АС>Р7 и АС)Р9 дополнительно проницаемы для глицерола
и других небольших молекул, а А(}Р0, АС>Р1, АС>Р2, А<ЗР4, АС>Р5 — толь­
ко для воды. Молекула а. состоит из двух половин — двух повторов трех
спиралей, являющихся зеркальным отражением друг друга, повернутым
на 180°. При складывании обеих половин а. формируется водная пора.
АС2Р1 появляется в эритроцитах после рождения практически одномомент­
но с формированием способности почки концентрировать мочу (вероятно,
АС2Р1 способствует регидратации эритроцитов, обезвоженных в гиперто­
нической среде капилляров, снабжающих кровью мозговую часть почки).
Экспрессия А<ЗР1 происходит в почке (проксимальные извитые канальцы
и тонкий отдел петли Генле) плода начиная со II триместра беременно­
сти. Полной экспрессии этот водный канал достигает после рождения,
что связывают со способностью почки концентрировать мочу. В тканях
глаза АС2Р1 обеспечивает гомеостаз внутриглазной жидкости. АС)Р2 экс­
прессируется только в собирательных трубочках почки. Активность этого
канала регулирует АДГ, увеличивая реабсорбцию воды из просвета трубо­
чек в межклеточное пространство. А(^РЗ — водный канал базолатеральных
мембран собирательных трубочек почки. Экспрессия А(}РЗ найдена также
в печени, поджелудочной железе, кишечнике, селезенке, простате. А()Р4
экспрессируется в клетках эпендимной выстилки сосудистого сплетения
желудочков и водопровода мозга, в синтезирующих АДГ нейросекреторных
нейронах гипоталамуса. Этот канал расценивают как осморецептор. А0Р5
принимает участие в формировании слезной жидкости, слюны, секретов
желез, находящихся в воздухоносных путях.
Акне — угри обыкновенные — стимулируемое андрогенами воспаление саль­
ных желез, приводящее к образованию комедонов, папул, воспалительных
пустул и (иногда) к рубцеванию; впервые появляются при половом созре­
вании и в подростковом возрасте. Почти у всех подростков имеются угри
различной степени выраженности. 15% подростков обращаются за меди­
цинской помощью. Чаще страдают мужчины.
Акроцефалия (башенный череп) — череп с высоким лбом, сглаженными над­
бровными и височными выступами вследствие преждевременного окосте­
нения венечного и затылочного шва.
АКТГ — см. «Проопиомеланокортин».
Активатор плазминогена тканевый. 1. См. «Недостаточность а. п.». 2. Продукт
генной инженерии, вызывающий лизис тромбов, которые закупоривают
коронарные артерии (используют в терапии инфаркта миокарда).
Активины — пептидные гормоны, вырабатываемые зернистыми клетками
фолликулов яичника и в плаценте, стимулируют секрецию ФСГ т у И г о .
Значение аа. для регуляции овариального цикла находится под вопросом.
Актиномикоз — хроническая инфекционная болезнь человека, вызываемая
А. йгаеШ, АгасИта ргорю т са. Характерно развитие хронических деструктив­
ных процессов и гранулем. Обычны поражения шейно-лицевой области,
грудной и брюшной полости <=> болезнь лучисто-грибковая.
Акупунктура — восточный метод лечения, заключающийся в прокалывании
тонкими иглами специфических зон с целью обезболивания при хирур­
гических вмешательствах для уменьшения болевого синдрома, а также
с лечебной целью <=> иглоукалывание о иглорефлексотерапия.
Алкалоз — форма нарушения КЩР, характеризующаяся сдвигом соотношения
между анионами кислот и катионами оснований крови в сторону увели­
чения катионов (рН >7,44). А. может быть респираторным (при начинаю­
щемся грамотрицательном сепсисе, отравлении салицилатами, поражении
ЦНС, тромбоэмболии легочной артерии, хронической сердечной и пече­
ночной недостаточности, неконтролируемой гипервентиляции при ИВЛ,
а также гипертиреозе и беременности) и метаболическим (при рвоте,
теряющей хлориды диарее, приеме диуретиков, передозировке гидрокарбо­
ната натрия, гиперкортицизме). • Метаболический а. характеризуется повы­
шением рН крови и увеличением концентрации бикарбоната. Этиология.
Увеличение уровня бикарбоната в плазме крови происходит в результате
повышенного его образования в желудке или в почках со снижением
почечной экскреции или в результате экзогенного применения бикарбона­
та или других щелочей. • Рвота. • Применение диуретиков и минералокор­
тикоидов. • Применение щелочей в виде бикарбоната натрия (например,
при кардиореанимации) или в виде органических ионов (в частности,
лактата, цитрата и ацетата, метаболизирующих в бикарбонат в печени).
• Быстрая коррекция гиперкапнии. Вслед за длительным респираторным
а. компенсаторно повышается образование бикарбоната почками. Если
затем (за счет ИВЛ) р С 0 2 артериальной крови резко снижается, развива­
ется преходящая гипербикарбонатемия и повышается рН крови (состоя­
ние, называющееся постгиперкапническим м. а.). Лечение. • Коррекция
основного заболевания. • Уменьшение выведения почками бикарбоната
путем увеличения объема внеклеточной жидкости с помощью растворов,
содержащих ЫаС1. • При м. а. с гипокалиемией (например, при чрезмер­
ной минералокортикоидной активности) — препараты калия (например,
калия хлорид). • При гиперкапническом м. а. — натрия или калия хлорид
(при гиповолемии) или аммония хлорид. • Респираторный а. характери­
зуется увеличением рН и снижением р С 0 2 крови. Этиология. Р. а. связан
с избыточным удалением С 0 2 через легкие. Причины: любые нарушения,
связанные с неадекватным увеличением частоты дыхания и выведения
С 0 2. • Возбуждение (истерическая гипервентиляция) — наиболее частая
причина р. а. • Отравление салицилатами. • Гипоксия. • Любое воспали­
тельное или объемное поражение легкого. • Патология ЦНС (инсульт, опу­
холь, инфекция, травма). • Грамотрицательная септицемия. • Печеночная
недостаточность. Лечение. Главная цель — коррекция основного заболева­
ния. В случаях тяжелого р. а. (рН >7,6) может возникнуть необходимость
в применении обогащенных С 0 2 дыхательных смесей или в управляемом
дыхании. О Острый р. а. Учащенное дыхание способствует избыточному
выведению С 0 2 легкими, что, в свою очередь, вызывает повышение рН
крови. При остром снижении р С 0 2 крови на каждые 10 мм рт.ст. уровень
бикарбоната плазмы снижается на 2 мэкв/л, а рН крови возрастает на 0,08.
0 Хронический р. а. В течение нескольких часов после острого снижения рС 02
артериальной крови уменьшается секреция Н+ в дистальной части нефрона,
что приводит к уменьшению бикарбоната плазмы крови. При хроническом
снижении рС 02 крови на каждые 10 мм рт.ст. уровень бикарбоната плазмы
уменьшается на 5—6 мэкв/л, а рН крови возрастает только на 0,02.
Алкаптонурия (гомогентизинурия) — врожденное нарушение обмена фенил­
аланина и тирозина [недостаточность гомогентизат-1,2-диоксигеназы
(*203500, КФ 1.13.11.5, ген АК11, Зя2, р)], характеризующееся экскрецией
гомогентизиновой кислоты с мочой. Обычно проявляется остеоартрита­
ми. При хранении мочи или добавлении щелочи моча темнеет (резуль­
тат образования продуктов полимеризации гомогентизиновой кислоты).
Характерен охроноз (например, голубоватая окраска ушей) и артриты.
Аллель — одна из двух или более альтернативных форм гена, каждая из кото­
рых характеризуется уникальной последовательностью нуклеотидов; аа.
занимают одинаковые позиции (локусы) в гомологичных хромосомах о
аллеломорф
аллельный ген.
Аллоантигены (изоантигены) — Аг конкретного индивидуума, обладающие
иммуногенностью по отношению к другим представителям этого вида,
но не к организму-донору трансплантата. Яркий пример изоантигенов —
групповые Аг крови, находящиеся на мембранах эритроцитов и других
клеток. Поскольку человек обладает естественными АТ к групповым Аг
крови, последние приобретают свойства сильных трансплантационных Аг.
Поэтому перед трансплантацией и гемотрансфузией необходимо опреде­
лить группу крови донора и реципиента.
Алопеция — стойкое или временное, полное или частичное выпадение (отсут­
ствие) волос на голове <=> облысение.
Альбинизм — врожденный дефицит или отсутствие пигмента в коже, волосах,
радужке и сетчатке глаза или только в радужке глаза за счет нарушения
обмена тирозина при синтезе меланинов. Клинически принято различать
генерализованные (кожно-глазные), изолированные (глазные) и смешан­
ные типы альбинизма. Известно не менее 10 отдельных форм альбинизма,
для каждой из которых идентифицирован пораженный ген. • Кожно-глазной
а. классифицируют по активности тирозиназы. ФТип 1 (ксантизм, желтый
альбинизм, *203100; у гомозигот нет активности тирозиназы, р) — тирозиназонегативный. Ребенок рождается мертвенно-бледным, затем посте­
пенно появляется желтая пигментация кожи и волос, выраженная глазная
патология. Известно не менее 60 аллелей. Некоторые фенотипы выделены
как отдельные формы типа 1. 0 Тип 2 [*203200, ген ОСА2 (Р , РЕИ, 0 1 5 8 1 2 ),
15д11.2—<\\2, р] — тирозиназопозитивный. Клинические проявления — аль­
бинизм, нистагм, снижение зрения. Лабораторно: нормальная активность
тирозиназы. 0 Тип 3 (#203290, ген ТУКР1, р) — мутация гена тирозиназозависимого белка 1 (115501); вероятно, аллельна типу 2. Клинические проявле­
ния — неполный альбинизм, нистагм; пигмент, присутствующий в сетчатке;
косоглазие. Лабораторно: нормальная активность тирозиназы.
Альвеолит фиброзирующий — общий термин для группы заболеваний, характе­
ризующихся диффузной воспалительной инфильтрацией альвеол, обрати­
мым интерстициальным пневмонитом, прогрессирующим до диффузного
легочного фиброза.
Альдостерон (11р, 21-дигидрокси-3,20-диоксо-4-прегнен-18-аль, мол. масса
360,45) — основной минералокортикоид. Другие стероиды надпочечни­
ка, расцениваемые как глюкокортикоиды (кортизол, 11-дезоксикортизол,
11-дезоксикортикостерон, кортикостерон), имеют и минералокортикоидную активность, хотя — сравнительно с а. — их суммарный вклад мал.
Регуляторы секреции а. Ангиотензин II — компонент системы ренин—
ангиотензины — главный регулятор синтеза и секреции а.; этот пеп­
тид стимулирует выброс а., тогда как атриопептин ингибирует синтез а.
Действие гипо- и гипернатриемии осуществляется через систему ренин—
ангиотензины — АДГ, эффекты К+ не зависят от содержания в крови Ыа+
и ангиотензина II: гиперкалиемия стимулирует секрецию а., гипокалие­
мия тормозит секрецию минералокортикоидов. ПГЕ, и ПГЕ2 стимулируют
синтез а., а ПГР1а и ПГР2а тормозят секрецию минералокортикоидов.
Травмы и стрессовые состояния увеличивают секрецию а. А. практически
не связывается с белками плазмы крови, по этой причине время его цир­
куляции в крови (время полужизни) не превышает 15 мин. А. из крови
удаляется печенью, где он трансформируется в экскретируемый почками
тетрагидроальдостерон-3-глюкуронид. Функция минералокортикоидов —
поддерживать баланс электролитов в жидкостях организма, осуществля­
ется посредством влияния на реабсорбцию ионов в почечных канальцах:
а. увеличивает реабсорбцию № + (задержка натрия приводит к увеличе­
нию содержания воды в организме и повышению АД) и экскрецию К+
(потеря калия вызывает гипокалиемию); а. увеличивает реабсорбцию С1",
бикарбоната и почечную экскрецию Н +. Рецептор а. — внутриклеточный
полипептид с Мг 107 кД, связывает а. (также глюкокортикоиды) и акти­
вирует транскрипцию генов. Дефекты рецептора влекут за собой развитие
псевдогипоальдостеронизма (задержка калия, потеря натрия, гипертензия
при нормальной или даже повышенной секреции а.).
Амавроз — обусловленная поражениями ЦНС полная слепота на один или оба
глаза при сохранности зрачковой реакции на свет.
Аменорея — отсутствие менструаций в течение 6 мес и более. А. — не само­
стоятельный диагноз, а симптом, указывающий на анатомические, био­
химические, генетические, физиологические или психические наруше­
ния. Частота вторичной а. — не менее 3%. Виды а. • Истинная: нет
циклических изменений в яичниках, эндометрии и во всем организме,
менструаций нет. Гормональная функция яичников резко снижена, поло­
вых гормонов для осуществления циклических изменений эндометрия
недостаточно. • Ложная: отсутствие периодического выделения крови
из влагалища при наличии циклических изменений в яичниках, матке
и во всем организме [например, сплошная девственная плева, атрезия
влагалища и шейки матки; кровь, выделяющаяся при менструациях, ска­
пливается во влагалище (гематокольпос), матке (гематометра), трубах
(гематосальпинкс)]. • Физиологическая: отсутствие менструаций перед
или сразу после менархе, во время беременности и лактации, после
менопаузы. 0 У многих девочек-подростков длительность а. составляет
2—12 мес в течение первых 2 лет после менархе. 0 Спонтанная менопауза
может возникать у женщин уже после 30 лет. • Патологическая: 0 пер­
вичная: отсутствие менструаций и других признаков полового созрева­
ния до 14 лет или отсутствие менструаций до 16 лет при наличии других
признаков полового созревания; 0 вторичная: отсутствие менструаций
в течение 3 циклов подряд с предшествующими нормальными менструа­
циями. Этиология. • Первичная а. 0 Поражение гонад: синдромы Тернера,
тестикулярной феминизации, резистентных яичников, аномалии развития
матки и яичников. 0 Внегонадная патология: гипопитуитаризм, гипогонадотропный гипогонадизм, задержка менархе, врожденная гиперплазия
надпочечников. 0 Нарушение проходимости входа во влагалище, влагали­
ща, шеечного канала и полости матки. • Вторичная а. О Психогенная а.
(стресс). 0 Гипоталамическая форма — а. на фоне похудания. ОГипоталамогипофизарная форма. <Н> Гиперпролактинемия — функциональная и орга­
ническая формы. ® Гипогонадотропная. ® Послеродовой гипопитуитаризм
(синдром Шиена). <Н) Прекращение приема пероральных контрацепти­
вов. ❖ ЛС: глюкокортикоиды, даназол, аналоги люлиберина, химиотера­
певтические препараты. 0 Декомпенсированные эндокринопатии: СД,
гипо- и гипертиреоз. 0 Надпочечниковая форма. ® Постпубертатный
адреногенитальный синдром. ® Вирилизирующая опухоль надпочечников.
О Яичниковая форма. ® Синдром истощения яичников. <Н
> Синдром реф­
рактерных яичников. ® Вирилизирующие опухоли яичников. О Маточная
форма. ® Синдром Ашермана. ® Специфический эндометрит. Генетические
аспекты. Существует множество наследуемых заболеваний, которые сопро­
вождаются а. Факторы риска: физические перегрузки, нарушения питания
(в том числе переедание и голодание), психоэмоциональный стресс.
Аменция (слабоумие, лишение разума) — форма помрачения сознания с пре­
обладанием растерянности, бессвязности мышления, речи и движений о
синдром аментивный.
Амилоид — аморфное, эозинофильное, гиалиноподобное вещество, отклады­
вающееся диффузно во внеклеточном пространстве в различных органах
и тканях при амилоидозе.
Амилоидоз — патология неясной этиологии (диспротеиноз), характеризу­
ющаяся внеклеточным накоплением амилоида в тканях и органах, что при­
водит к склерозу, атрофии, потере функции о амилоидная дистрофия.
Аминоацидемия — повышенное содержание в сыворотке крови одной
или нескольких аминокислот; возникает в результате наследственно обу­
словленных расстройств обмена аминокислот или нарушения дезамини­
рующей функции печени. Как правило (но не всегда), аминоацидемия
сопровождается аминоацидурией.
Аминоацидурия (аминокислотурия) — выведение повышенного количества
аминокислот с мочой или наличие в моче продуктов их обмена, в норме
в ней не содержащихся (например, КТ); развивается преимущественно
вследствие наследственных нарушений транспорта аминокислот через эпи­
телий почечных канальцев. Суммарная частота различных -урий (включая
фенилкетонурию) достигает (по некоторым, скорее заниженным, оценкам)
1:200 новорожденных. Лечение проводят индивидуально (в зависимости
от конкретного дефекта). Общие принципы: • диета с пониженным содер­
жанием белка; • обильное питье; • ощелачивание мочи с целью предупре­
дить образование камней. • Б-пеницилламин (в рефрактерных случаях
при цистинурии); • никотинамид внутрь (при болезни Хартнапа); • литотомия (по показаниям). Профилактика. Серьезные и нерешенные пробле­
мы — доступная сеть генетического консультирования и тотальный скри­
нинг новорожденных (как и ЭКГ для идентификации врожденных дефектов
ССС). • Болезнь Хартнапа — см. «Триптофанурия». • Гомоцистинурия —
расстройство обмена метионина, характеризующееся выделением гомоцистина с мочой, задержкой умственного развития, эктопией хрусталика,
редкими светлыми волосами, вывернутыми наружу коленями, тенденцией
к судорожным реакциям, анемией, явлениями тромбоэмболии (ИБС)
и жировым перерождением печени. Существует несколько наследствен­
ных (р) форм заболевания. Этиология. • Недостаточность цистатион(он)
р-синтетазы. • Дефект метаболизма витамина В,2 (277400). • Недостаточность
N (5,10)-метилентетрагидрофолатредуктазы. • Избирательная мальабсорбция витамина В12 (261100, синдром Имерслунд-Гресбека, 10р12.1; МСА1).
0 Цистатион(он)-р-синтетазынедостаточность (*236200, КФ 4.2.1.22, 2\(\22.Ъ,
ген СВ8, 9 дефектных аллелей, р). Клинические проявления — эктопия хру­
сталика, высокий риск развития инфаркта миокарда, умственная отсталость,
психиатрическая патология, марфаноидное телосложение, остеопороз,
возможен панкреатит. Лабораторно: г., метионинурия. О Недостаточность
метилкобаламина (*236270). Развивается зависимая от витамина В]2 гомоцистинурия с мегалобластной анемией и тяжелой умственной отсталостью.
Лабораторно: г., гипометионинемия, содержание в крови фолиевой кисло­
ты и витамина В]2 нормальное. ОДвухосновная а. — наследственный дефект
транспорта лизина, орнитина и аргинина (но не цистина). Выделяют
два типа заболевания. Тип I (*222690): диарея, выраженное отставание
в умственном развитии, непереносимость фенотиазинов. Тип II (222700,
14^11.2, ген ЬР1) изучен у финнов: для гомозигот характерна гипераммониемия, непереносимость белка (лизинурическая).
Амины — азотсодержащие органические соединения — продукты замещения
одного или нескольких атомов водорода в молекулах аммиака или гидро­
окиси аммония на органические радикалы; многие аа., применяемые в про­
мышленности (особенно ароматические, например анилин, фенилгидразин
и др.), токсичны для человека. • Биогенные аа. — продукты ферментатив­
ного декарбоксилирования некоторых аминокислот; многие б. а. обладают
высокой биологической активностью (гистамин, тирамин, серотонин,
катехоловые аа.). • Катехоловые аа. синтезируются из тирозина по цепочке:
тирозин (превращение тирозина катализируется тирозингидроксилазой,
геном 77/, 191290, 11р15.5, КФ 1.14.16.2) —» ДОФА (ДОФА-декарбоксилаза,
КФ 4.1.1.28) -> дофамин (дофамин-р-гидроксилаза, см.) -» норадреналин
(фенилэтаноламин-/У-метилтрансфераза, ген Р И М Т , 171190,
с(22,
КФ 2.1.1.28) -» адреналин. Мутации этих генов приводят к блокированию
синтеза соответствующих продуктов и накоплению субстратов. Рецепторы
к. а. — адренергические, рецепторы дофамина — дофаминергические.
Амиотрофия: • Кугельберга—Веландера а. (р, *253600) — медленно прогрес­
сирующая атрофия мышц в проксимальных отделах конечностей, обу­
словленная поражением мотонейронов передних рогов спинного мозга;
проявляется слабостью, непроизвольными сокращениями отдельных групп
мышц. Начало заболевания — между 2-м и 17-м годом жизни <=> а. псевдомиопатическая <=> дистрофия мышечная прогрессирующая с фибрил­
лярным подергиванием о Кугельберга—Веландера болезнь <=> юношеская
(ювенильная) проксимальная мышечная а. • Наследственная невральная
а. — см. «Болезнь Шарко—Мари—Тута».
Амниоцентез — прокол амниотического мешка с целью получения амниоти­
ческой жидкости.
Амплификация — увеличение числа копий определенного фрагмента ДНК.
Анаплазия — стойкая дедифференцировка клеток злокачественной опухоли
с изменением их структуры и биологических свойств.
Анасарка — распространенный отек подкожной клетчатки, крайняя степень
выраженности отеков при различных заболеваниях.
Анафилаксия — острая системная реакция сенсибилизированного организма
на повторный контакт с Аг, развивающаяся по типу I аллергических реак­
ций и проявляющаяся острой периферической вазодилатацией. Крайнее
проявление анафилаксии — анафилактический шок.
Ангидроз — отсутствие потоотделения.
Ангиогенин — белок, стимулирующий рост кровеносных сосудов в нормаль­
ных и трансформированных тканях (см. также «Белок катионный»),
Ангиопоэтины. 1. а.-1 — проангиогенный фактор, опосредующий мезенхимно­
эндотелиальные взаимодействия при развитии сердца и сосудов, обла­
дает антиапоптозным действием в отношении эндотелиальных клеток
и стабилизирует структуру сосудов. 2. а.-2 — антагонист а.-1 — участвует
в перестройке сформированных сосудов во взрослом организме, а также
экспрессируется в эндотелии сосудов опухолей перед их регрессией.
Ангиография — рентгенологическое исследование кровеносных и лимфатиче­
ских сосудов после введения в них контрастного вещества. А. — инвазивная
процедура.
Ангиома — доброкачественная опухоль, развившаяся из кровеносных (гемангиома) или лимфатических (лимфангиома) сосудов.
Ангиопластика — пластическая операция на сосудах. • Баллонная а. — расши­
рение просвета артерии с помощью специального раздуваемого баллона,
который подводят к месту сужения сосуда. • Коронарная а. — см. «Дилатация
баллонная и стентирование». • Стентирование — см. «Дилатация баллонная
и стентирование». • Чрескожная транслюминальная коронарная а. — рас­
ширение суженного атеросклеротическим процессом участка коронарной
артерии миниатюрным баллоном под большим давлением при визуальном
контроле во время ангиографии. Параллельно с расширением просвета
коронарной артерии в последнее время проводят стентирование (имплан­
тацию в место сужения стентов — тончайших проволочных каркасов,
предотвращающих рестеноз).
Ангиоретикулема — доброкачественная опухоль, состоящая из большого коли­
чества капилляров и межсосудистой ретикулярной ткани; возникает в моз­
жечке, реже в продолговатом и спинном мозге.
Ангиотензины — биологически активные полипептиды, образующиеся из ангиотензиногена и повышающие АД в результате сужения кровеносных сосу­
дов. • А. I — неактивная форма а., предшественник а. II — декапептид,
образующийся из ангиотензиногена под действием ренина. • А. II — актив­
ная форма а. — октапептид, образующийся из а. I под действием ангиотензинпревращающего фермента (АПФ). Этот процесс осуществляется
в основном в легких (примерно 50% образующегося а. II), в плазме крови
и интерстициальной ткани почек (около 10—20% а. II). А. II инактивирует­
ся под влиянием ангиотензиназ. А. II — мощный вазоконстриктор (за счет
сокращения ГМК артериол) и стимулятор синтеза альдостерона. • АПФ
(р, * 106180, КФ 3.4.15.1, ген Б С Р 1, \7 ц 2 2 —с(24) участвует в конверсии а.
I в а. II и катаболизме функционального антагониста а. II — брадикинина — двух пептидов, регулирующих тонус сосудов и пролиферацию ГМК.
На подавлении активности этого фермента основано действие ингибито­
ров АПФ. • А. III — гептапептид, производное а. II, имеет сходное с ним
действие, но меньше влияет на кору надпочечников.
Аневризма — локальное расширение просвета кровеносного сосуда или сердца
вследствие патологических изменений их стенки или аномалий развития.
Наибольшее клиническое значение имеет аневризма аорты.
Анемия — любое состояние, при котором количество эритроцитов, содержание
НЬ и Н1 снижены по сравнению с нормой. Это относится к концентрации
переносящего кислород материала в определенном объеме крови (в отличие
от общего количества при олигоцитемии, олигохромемии и олигемии).
• Апластическая а. — а., характеризующаяся выраженно сниженным образо­
ванием эритроцитов и НЬ. Обычно ассоциируется с гранулоцитопенией
и тромбоцитопенией из-за гипоплазии или аплазии красного костного мозга
<=> гипопластическая а. • В12-дефицитные а. развиваются вследствие дефици­
та витамина В12 (суточная потребность 1—5 мкг). В большинстве случаев
сочетается с фундальным гастритом и ахлоргидрией. О Пернициозная а. —
аутоиммунное заболевание с образованием АТ к париетальным клеткам
желудка или внутреннему фактору Касла, однако существуют В,2-дефицитные
а. алиментарного генеза. В12-дефицитные а. могут быть врожденными
или приобретенными. Этиология. • Витамин В12-дефицитная наследственная
а. О Фундальный гастрит (тип А). ® АТ к париетальным клеткам желудка.
® Иммунные нарушения (выработка АТ к внутреннему фактору Касла).
• Другие В12-дефицитные а. О Вегетарианская диета без дополнительного
приема витамина В12. О Гастрэктомия. О Синдром приводящей петли.
О Инвазия лентецом широким. О Синдром мальабсорбции. О Хронический
панкреатит. О Хронический алкоголизм. О ЛС (бигуаниды, фенилбутазон,
аминосалициловая кислота, пероральные контрацептивы). Генетические
аспекты. Существует ряд генетически разнородных форм пернициозной а.
• Классическая пернициозная а. взрослых (170900, предположительно $К)
с нарушением всасывания витамина В|2. • Пернициозная а. у подростков
с полигландулярным аутоиммунным синдромом (*240300, 2^22.3, р).
• Пернициозная а. ювенильная с относительной недостаточностью всасыва­
ния витамина В]2 и протеинурией (*261100, синдром Имерслунд-Гресбека,
10р12.1, МСА1, р). При этой мегалобластной а. возможны мальформации
мочевыделительного тракта и протеинурия. • Врожденная пернициозная а.
(*261000, хр. 11, мутация гена С1Р, р) вследствие отсутствия секреции гастромукопротеина при нормальных кислотности желудочного сока и морфоло­
гии слизистой оболочки. Характерна экспрессия НЬА-БК2, НЬА-ОЯ4.
Патоморфология. • Костный мозг — мегалобластный тип кроветворения,
повышенное содержание железа, гиперсегментированные нейтрофилы.
• Желудок — фундальный гастрит, гипертрофия слизистых оболочек клеток,
атрофия париетальных и главных клеток, характерен клеточный атипизм.
• Спинной мозг — дегенерация миелина задних и боковых столбов, дегене­
ративные изменения ганглиев задних корешков (фуникулярный миелоз).
• Дегенерация периферических нервов. Клиническая картина определяется
дефицитом витамина В|2. • Общие признаки а. — слабость, одышка, тахи­
кардия, бледность, шум в ушах и др. • Фуникулярный миелоз (парестезии,
снижение вибрационной чувствительности, атрофии мышц, полиневрит,
патологические рефлексы). • Нарушение координации (положительные
проба Ромберга и пальценосовая проба). • Психические нарушения (спутан­
ность сознания, депрессия, деменция). • Со стороны ЖКТ — атрофический
глоссит (малиновый «лакированный» язык), гепатоспленомегалия, анорек­
сия. • Кожа — гиперпигментация, пурпура, витилиго. Лечение. Общая так­
тика. • Лечение необходимо проводить пожизненно. • Режим амбулаторный.
• Эндоскопическое обследование каждые 5 лет для исключения рака желуд­
ка или по показаниям. • Диета с повышенным содержанием белка.
Лекарственная терапия. Витамин В|2 (цианокобаламин) — по 100 мкг п/к еже­
дневно в течение 1 нед, затем 1 раз в неделю в течение 1 мес и далее 1 раз
в месяц в течение всей жизни. Течение и прогноз благоприятны при своев­
ременном и адекватном лечении витамином В12. Признаки поражения ИНГ,
могут сохраняться, если лечение пациента было начато через 6 мес и позже
после начала заболевания о злокачественная а., Аддисона—Бирмера болезнь,
Бирмера болезнь, Адцисона-Бирмера а. • Гемолитическая а. — общее назва­
ние а., вызванных разрушением эритроцитов. 0 Аутоиммунная гемолитическая
а. характеризуется развитием усиленного гемолиза в результате выработки
АТ к собственным неизмененным Аг эритроцитов. А. носит приобретенный
характер. В 50% случаев причина синтеза аутоантител не известна. Существуют
факторы, предрасполагающие к возникновению аутоиммунного гемоли­
за, — неопластические процессы (лейкоз, множественная миелома, лимфома, тимома), диффузные заболевания соединительной ткани, вирусная
инфекция (например, гепатит), микоплазменная инфекция, прием Л С
(метилдопы, антибиотиков в высоких дозах). Патогенез. Пусковой меха­
низм — выработка АТ к собственным эритроцитам. При разных формах
характер АТ и их специфичность различны, что и определяет клиническую
картину заболевания. Различают два типа АТ — тепловые (реагирующие
с эритроцитами при температуре тела не менее 37 °С) и холодовые (реаги­
рующие с эритроцитами при температуре менее 37 °С). Образующийся
комплекс эритроцит—АТ поглощается макрофагами селезенки (внесосудистый гемолиз). Другая форма гемолиза (внутрисосудистый) возникает
в результате агглютинации эритроцитов при незначительном воздействии
холода. Последствия гемолиза — а., желтуха, активация свертывающей
системы крови. Также аутоиммунные АТ могут вырабатываться против тром­
боцитов, вызывая тромбоцитопению. Лечение. • Режим — стационарный.
• Глюкокортикоиды назначают, устанавливая диагноз, для подавления иммун­
ного ответа. • Спленэктомия. Неэффективность лечения глюкокортикоидами, выявляемая по нарастанию а., процента ретикулоцитов, требует прове­
дения спленэктомии уже в самом начале болезни. • Иммунодепрессанты.
При неэффективности спленэктомии или наличии противопоказаний к ней
назначают циклоспорин, антилимфоцитарный глобулин, а при неэффек­
тивности — циклофосфамид, азатиоприн, метотрексат и др. • Заместительная
терапия. Трансфузии эритроцитов проводят по строгим показаниям. Очень
часто переливаемые эритроциты либо разрушаются макрофагами селезенки,
либо вызывают выработку АТ. Чтобы избежать провоцирования аутоиммун­
ных реакций, отмытые эритроциты переливают при температуре тела 37 °С.
0 Микроангиопатическая гемолитическая а. — вторичная или врожденная а.,
развивающаяся вследствие сужения или обструкции мелких кровеносных
сосудов, что приводит к механическому повреждению эритроцитов
при их взаимодействии с эндотелием сосудов. Этиология. • Артериальная
гипертензия. • Хронические заболевания почек. • Протезы сердечных кла­
панов. • Гемолитико-уремический синдром. • ДВС. • Врожденная микроангиопатическая гемолитическая а. (276850, р): полиморфная симптоматика,
возможно вовлечение ЦНС; возникает вследствие отложения фибрина
или образования тромбоцитарных тромбов в артериолах и капиллярах боль­
шинства органов; характерны тромбоцитопения, эпизоды лихорадки и петехиальной сыпи, гломерулопатия, мегакариоцитоз (костный мозг). Лечение
этиотропное и общеукрепляющее. • Гиперхромная а. — общее название а.,
характеризующихся высоким цветовым показателем. • Гипопластическая
а. — прогрессирующая нормохромная нормоцитарная гипорегенераторная
а., возникающая вследствие сильного угнетения, неадекватного функциони­
рования красного костного мозга; как правило, не сочетается с лейкопени­
ей и тромбоцитопенией. Если процесс персистирует, может развиться апластическая а. 0 Врожденная г. а. (синдром Дайемонда-Блекфэна) — аллельный
вариант а. Фанкони (*205900, р). От а. Фанкони ее отличают раннее появ­
ление а. (как правило, в первые месяцы жизни), отсутствие нейтропении
и тромбоцитопении. Диагностика: макроцитоз, ретикулоцитопения, повы­
шенное содержание НЬЕ Лечение: • Преднизолон. Эффективен примерно
у 50%. • Переливание эритроцитарной массы. 0 Приобретенная г. а. (транзиторная эритропения детей, эритробластопения) проявляется несколько
позднее, чем синдром Дайемонда-Блекфэна. Протекает обычно остро
(с возможным развитием гемолитических и апластических кризов), реже —
хронически. Причины: предполагают длительное ингибирующее действие
вирусов (парвовирусов) на эритропоэз. Диагностика: ретикулоцитопения,
уровень НЬР в норме, концентрация железа и общая железосвязывающая
способность снижены. Через 2—4 нед многие пациенты выздоравливают
без лечения. 0 Другие формы приобретенной г. а. ® Заболевания почек, пече­
ни и щитовидной железы могут привести к угнетению эритропоэза.
<Е> Гипопластическая а. может сопровождать бактериальные и вирусные
инфекции. Так, парвовирус часто вызывает а. (увеличено содержание рети­
кулоцитов и концентрация железа в крови). Эти а. обычно протекают легко,
но возможны острые гемолитические кризы; при этом костный мозг
не в состоянии компенсировать повышенное разрушение эритроцитов (раз­
вивается апластический криз). Лечение симптоматическое. Для молодых
пациентов перспективна трансплантация костного мозга. • Гипохромная а. —
общее название а., характеризующихся низким цветовым показателем.
• Дизэритропоэтическая а. (от греч. ёуз- означает нарушение, расстройство +
эритропоэз) — общее название группы а., характеризующихся качественным
нарушением эритропоэза. • Железодефицитная а. (ЖДА, сидеропеническая
а.) — гипохромная микроцитарная гипорегенераторная (у детей гиперрегенераторная) а., возникающая вследствие абсолютного снижения ресурсов
железа в организме (как правило, при хронической потере крови или недо­
статочном поступлении железа извне). Частота. ЖДА наблюдают у 10-30%
взрослого населения. Наиболее распространенная форма а. (80-95%).
У женщин ЖДА возникает значительно чаще, чем у мужчин. По разным
оценкам, до 20% женщин страдают ЖДА. Этиология. • Хроническая потеря
крови (например, при желудочно-кишечных или маточных кровотечениях).
• Алиментарные факторы — недостаточное поступление железа в организм.
• Нарушение всасывания железа в ЖКТ: 0 резекция желудка и/или кишеч­
ника; 0 гипоацидный (анацидный) гастрит, гастродуоденит; 0 синдром
мальабсорбции. • Повышенная потребность организма в железе (например,
у грудных детей, в подростковом возрасте, при беременности, при глистных
инвазиях). • Опухоли (например, гипернефрома, рак мочевого пузыря).
• Другие причины (пароксизмальная ночная гемоглобинурия, гемосидероз
легкого). Клиническая картина. • Общие симптомы (утомляемость, слабость,
раздражительность, апатия, бледность кожных покровов и слизистых обо­
лочек). При тяжелой форме возникает одышка, тахикардия, артериальная
гипотензия, головная боль, головокружение, парестезии. • Специфические
симптомы: 0 ангулярный стоматит; 0 койлонихия; Фатрофический глоссит;
0 дисфагия; 0 извращение аппетита (пристрастие к мелу, извести, глине,
углю, зубному порошку или льду). Возрастные особенности. • ЖДА часто
развивается у грудных детей, находящихся на искусственном вскармлива­
нии. • Приблизительно 60% всех случаев ЖДА наблюдают у пациентов
старше 65 лет. • У пожилых людей ЖДА может привести к обострению ИБС
с последующим развитием левожелудочковой недостаточности. Беременность.
ЖДА часто развивается в период беременности без коррекции питания
железосодержащими добавками. Лабораторные исследования. • Золотой стан­
дарт — окрашивание аспирата костного мозга для определения содержания
железа. • Лучший неинвазивный метод — выявление сниженного содержа­
ния ферритина в сыворотке крови. • При исследовании мазка перифериче­
ской крови у больных обычно выявляют гипохромную микроцитарную а.,
анизоцитоз, пойкилоцитоз. Мазок может быть и нормальным. 0 При незна­
чительной а. или острой потере крови средний эритроцитарный объем
и среднее содержание НЬ в эритроцитах могут быть в норме. • Уровень НЬ —
менее 120 г/л, но у пациентов с повышенным уровнем НЬ до заболевания
(курильщики, лица с хронической гипоксемией) а. может развиться
при наличии и более высоких показателей НЬ. Таким образом, при исполь­
зовании стандартных критериев а. необходимо проводить исследования,
направленные на выявление скрытой а. Лечение. Диета. • Следует ограни­
чить употребление молока до 0,5 л/сут (для взрослых). Молоко и другие
молочные продукты нужно исключить полностью за 2 ч до приема железо­
содержащих препаратов. • Необходимо уделять пристальное внимание коли­
честву потребляемого белка и железосодержащих пищевых продуктов (мяс­
ные блюда, бобовые). • Чтобы уменьшить вероятность развития запора
у пациентов, получающих заместительную терапию препаратами железа,
в рационе рекомендуют увеличить содержание растительной клетчатки.
Лекарственная терапия. • Железа сульфат 300 мг 3 раза в сутки внутрь между
приемами пищи (обеспечивает поступление в организм 180 мг чистого желе­
за в сутки; при приеме препарата во время еды всасывание железа снижает­
ся на 50%). При адекватной терапии через 7 сут в крови наблюдают ретикулоцитоз; через 2 нед уровень НЬ возрастает (обычно на 0,7—1 г/нед).
Отсутствие лечебного эффекта (или слабый эффект) свидетельствует о про­
должающемся кровотечении, сопутствующей инфекции или злокачествен­
ном новообразовании, недостаточной дозе препарата или (очень редко)
мальабсорбции железа. Содержание НЬ достигает нормальных показателей
в течение 2 мес лечения. Препарат следует принимать в течение 6 мес
(но не более). 0 При непереносимости сульфата железа можно назначить
или железа лактат, или железа фумарат. Течение и прогноз благоприятны
при своевременной диагностике ЖДА и адекватной терапии железосодер­
жащими препаратами. • Кули а. — большая талассемия. • Макроцитарная
а. — гемолитическая а., при которой средний размер циркулирующих эри­
троцитов больше, чем в норме о а. гемолитическая несфероцитарная
наследственная « Д а й к а —Янга врожденная гемолитическая а. • Мегалобластная тиаминчувствительная а. (*249270, 1я23.2—я23.3, ген ТКМА, р) сочетает­
ся с СД и нейросенсорной тугоухостью. Проявления: мегалобластная а.,
чувствительная только к тиамину, кольцевидные сидеробласты, СД, нейросенсорная тугоухость, дисфония, прогрессирующая атрофия зрительных
нервов, транспозиция внутренних органов, генерализованная отечность,
врожденные септальные пороки сердца. Лабораторно: аминоацидурия, недо­
статочность а-кетоглутаратдегидрогеназы, накопление железа в митохон­
дриях эритробластов. См. также «Недостаточность дигидрофолатредуктазы»,
«Недостаточность метионинсинтетазы». • Миелофтизная а. — нормохромная
нормоцитарная а., возникающая при замещении нормального костного
мозга патологической тканью. Этиология: • лимфома, • лейкоз, • множе­
ственная миелома, • туберкулез, • гранулематоз, • метастазы злокачествен­
ной опухоли в костный мозг. Диагностика. Появление незрелых лейкоцитов
и ядросодержащих эритроцитов в периферической крови в непропорцио­
нально большом количестве по сравнению с тяжестью. Пункция костного
мозга и трепанобиопсия подтверждают диагноз. Лечение этиотропное и сим­
птоматическое о миелопатическая а., лейкоэритробластоз. • Микроцитарная
а. — а., при которой средний размер циркулирующих эритроцитов меньше,
чем в норме о а. гемолитическая микросфероцитарная. • Нормохромная
а. — общее название а., при которых цветовой показатель находится в пре­
делах нормальных значений. • Нормоцитарная а. — общее название а.,
при которых размер эритроцитов находится в пределах нормальных значе­
ний. • Пернициозная а. (не рекомендуется) — см. «Анемия В,2-дефицитная».
• При недостаточности эритропоэтина а. — нормохромная (иногда гипохромная) нормоцитарная гипорегенераторная а. Н. э. возникает при ХПН, когда
возрастает содержание в крови конечных продуктов азотистого обмена
и клиренс креатинина меньше 45 мл/мин. Выраженность а. обычно зависит
от тяжести почечной недостаточности (наиболее тяжело протекает при пер­
вичном поражении клубочкового аппарата). Этиология и патогенез. • Синтез
эритропоэтина подавляется высоким содержанием уремических токсинов
(при а. в результате хронических заболеваний этот эффект опосредован
цитокинами), что приводит к подавлению эритропоэза. • Снижена чувстви­
тельность костного мозга к эритропоэтину. • Срок жизни эритроцитов уко­
рачивается (незначительный гемолиз). • Возможен алиментарный дефицит
фолиевой кислоты или железа. • Уремический геморрагический диатез: воз­
можны кровоизлияния в плевру, перикард, мозг, о При повреждении эндо­
телия сосудов почек (злокачественная артериальная гипертензия, узелковый
периартериит, острая ишемия) возникает микроангиопатическая гемолити­
ческая а. с фрагментацией эритроцитов и тромбоцитопенией (у детей про­
текает остро в виде гемолитико-уремического синдрома). Лечение этиотропное и патогенетическое. • Рекомбинантный эритропоэтин — препарат
выбора. • Гемотрансфузии. • При хронических, инфекционных и онкологиче­
ских заболеваниях а. (как правило, нормохромные нормоцитарные гипорегенераторные а., в 40% случаев — гипохромные микроцитарные). Эти а.
занимают второе место по частоте после ЖДА. Этиология и патогенез.
• Укорочение срока циркуляции зрелых форм эритроцитов в крови.
• Нарушение утилизации железа (нарушается его высвобождение из депо).
• Относительная недостаточность эритропоэтина (эффект опосредован пода­
влением его продукции цитокинами — р- и у-ИФН). • Измененная реакция
костного мозга на эритропоэтин. • Серповидно-клеточная а. — наиболее
часто регистрируемая наследственная гемоглобинопатия, характеризующая­
ся умеренно выраженной хронической гемолитической а., рецидивирующи­
ми острыми болевыми кризами и повышенной восприимчивостью к инфек­
ционным заболеваниям (главным образом 81гер 1ососсш рпеитотае) вследствие
образования патологического НЬ8. Заболевание диагностируют у 1% афроа­
мериканцев, часто встречают в Азербайджане и Дагестане. Этиология.
• На молекулярном уровне. Дефект гена НВВ (*141900, 11р15.5, классическая
мутация: НЪ8 образуется в результате замены валина на глутаминовую кис­
лоту в положении 6 р-цепи молекулы НЬ, также другие мутации, '.К). В веноз­
ном русле НЬ8 полимеризуется с формированием длинных цепей, изменяю­
щих форму эритроцитов (становятся серповидными). • На клеточном уровне.
Серповидные эритроциты повышают вязкостиь крови, вызывают стаз; соз­
дают механическую преграду в мелких артериолах и капиллярах, приводя
к тканевой ишемии (с чем связаны болевые кризы). Кроме того, серповид­
ные эритроциты менее устойчивы к механическим воздействиям, что при­
водит к их гемолизу. Проявления: умеренная желтуха, трофические язвы
в области лодыжек, отставание в физическом развитии (особенно у мальчи­
ков), приапизм, предрасположенность к апластическим и гемолитическим
кризам, болевые кризы, спленомегалия, холелитиаз, аваскулярные некрозы,
язвы ног, остеонекроз с развитием остеомиелита. Лечение. • При острых
болевых кризах средней степени тяжести в амбулаторных условиях — ненар­
котические анальгетики (ибупрофен, парацетамол). • При острых болевых
кризах в стационарных условиях — наркотические анальгетики парентераль­
но. • Необходимо учитывать возможность развития дегидратации и ацидоза.
• При развитии инфекции до получения результатов бактериологического
исследования нужно назначить антибиотик, активный в отношении 5{гер{ососсиз рпеитотае и НаеторкИш 1п/1иепще. • Поддерживающая терапия —
трансфузии отмытых или размороженных эритроцитов, а также антикоагу­
лянтов. • Трансплантация костного мозга. Осложнения. • Асептический
некроз головки бедренной кости и другие некротические осложнения.
• Сепсис. • Цереброваскулярные расстройства с неврологическими прояв­
лениями. • Кардиомегалия. • Ретинопатия. • Гемосидероз. Течение и прогноз.
А. радикально неизлечима. На втором десятилетии жизни количество кризов
уменьшается, но осложнения возникают чаще. Некоторые пациенты умира­
ют в детстве от некротических осложнений или сепсиса. Большинство боль­
ных доживают в среднем до 50 лет <=> дрепаноцитоз, менискоцитоз,
серповидно-клеточный синдром, менискоклеточная а., менискоцитарная а.
• Сидеробластная а. (сидероахрестическая а.) — гипохромная микроцитарная
гипорегенераторная а. вследствие нарушения утилизации внутриклеточного
железа для синтеза НЬ, несмотря на нормальное или повышенное содержа­
ние железа в митохондриях эритробластов. В результате в костном мозге
увеличивается количество сидеробластов — нормобластов с характерным
кольцевидным расположением гранул железа вокруг ядра (кольцевидные
сидеробласты). Классификация. • Наследственная сидеробластная а. (*301300,
Хр11.21, мутации гена А1Л82, X) обусловлена аномалией метаболизма пиридоксина (витамин В6, кофермент для синтеза 5-аминолевулиновой кисло­
ты) — недостаточностью синтетазы 8-аминолевулиновой кислоты. А. обыч­
но проявляется в подростковом возрасте. Массивные дозы пиридоксина
обеспечивают частичную коррекцию а. 0 Отдельно выделяют врожденную
форму (*205950), резистентную к лечению пиридоксином; повышено содер­
жание свободного эритроцитарного копропорфирина, а содержание свобод­
ного эритроцитарного протопорфирина снижено (недостаточность копропорфириногеноксидазы). • Приобретенные сидеробластные а. возникают
под действием веществ, ингибирующих ферментные системы синтеза про­
топорфирина [(например, свинец, алкоголь, изониазид, циклосерин, хлорамфеникол (левомицетин*)], при опухолевых заболеваниях (лимфограну­
лематоз, множественная миелома), аутоиммунных заболеваниях
(ревматоидный артрит). • В 50% случаев — идиопатическая форма. У 10%
пациентов с идиопатической сидеробластной а. развивается острый лейкоз.
Лечение. • Исключение провоцирующих факторов (ЛС, алкоголь). • Всем
пациентам следует пробно назначить пиридоксин в больших дозах (в боль­
шинстве случаев, исключая почти все наследственные формы, состояние
не улучшается). Аналогичен и эффект андрогенов. • Гемотрансфузии.
• Фанкони а. — тип рефрактерной а., характеризующийся панцитопенией,
гипоплазией костного мозга и врожденными аномалиями (низкорослость,
микроцефалия, гипогенитализм, косоглазие, дефекты развития скелета,
почек, микрофтальмия, умственная отсталость). Для а. Ф. существует 4 груп­
пы комплементации, т.е. не менее 4 генов, мутация любого из которых ведет
к развитию апластической панцитопении 4 типов (все р). Встречается
как семейная болезнь (р, *227650 и *227660) <=> панцитопения Фанкони о
врожденная панцитопения. • Фолиеводефицитная а. возникает, если концен­
трация фолиевой кислоты в сыворотке крови снижается менее 4 нг/мл.
Часто возникает у алкоголиков. Этиология. • Недостаточное поступление
фолиевой кислоты (суточная потребность — 50 мкг, у детей и беременных —
в 2—3 раза выше). • Нарушение всасывания фолиевой кислоты в кишечни­
ке. • Повышенная потребность в фолиевой кислоте (например, при бере­
менности, злокачественных новообразованиях). • Длительный прием ЛС
(триметоприм, метотрексат, сульфасалазин, пероральные контрацептивы,
противосудорожные средства). • Хронический алкоголизм. Проявления.
Кроме общих симптомов а. (бледность кожных покровов, тахикардия, арте­
риальная гипотензия и т.д.), характерны атрофический глоссит, анорексия,
неустойчивый стул и незначительная желтуха (за счет непрямого билируби­
на). Неврологические нарушения отсутствуют. Лечение. • При мегалобластной а. необходимо сначала исключить наличие дефицита витамина В]2
(пернициозную а.), так как в этом случае прием фолиевой кислоты приведет
к улучшению показателей крови, но не окажет влияния на выраженность
неврологических нарушений. • Отказ от провоцирующих а. ЛС. • За­
местительная терапия фолиевой кислотой (1—5 мг/сут). Беременность и
фолиевая кислота. Не менее 70% дефектов нервной трубки, развивающихся
у эмбриона и плода, можно предупредить, если принимать фолиевую кис­
лоту в период предполагаемого зачатия и во время беременности.
• Эритропоэтиновой недостаточности а. — см. «Анемия при недостаточности
эритропоэтина».
Анергия — полное отсутствие реакций организма на любые раздражители.
В частности, для милиарного туберкулеза характерна туберкулиновая анер­
гия, и потому отрицательные кожные пробы не имеют дифференциально­
диагностического значения.
Анеуплоидия — измененный набор хромосом, в котором одна или несколько
хромосом из обычного набора или отсутствуют, или представлены допол­
нительными копиями.
Анизокория — неравные диаметры зрачков правого и левого глаза.
Анизоцитоз — появление эритроцитов аномальных размеров, при этом клет­
ки диаметром более 9 мкм называются макроцитами, диаметром менее
6 мкм — микроцитами. Макроцитоз в сочетании с анемией и появлением
мегалобластов наблюдают при дефиците фолата и витамина В12, при ряде
наследственных заболеваний (например, оротовой ацидурии, синдроме
Леша—Нихена). Макроцитоз без появления мегалобластов — признак забо­
левания печени, наблюдаемый, например, при злоупотреблении алкого­
лем, гипотиреоидизме. Эритроциты диаметром менее 6 мкм (микроцитоз)
появляются в крови при гемоглобинопатиях и талассемии.
Аниридия — отсутствие радужной оболочки.
Анкилостомидоз — гельминтоз, вызываемый Апсу1о$юта с1иос1епа1е. Характе­
ризуется эозинофилией, анемией, диспепсией, истощением. При длитель­
ном течении у детей — постоянно вздутый живот, задержка физического
и умственного развития о унцинариоз.
Аномалия Эбштейна — врожденный порок сердца в виде смещения створок
трехстворчатого клапана в правый желудочек, клапаны деформирова­
ны и прилежат к перегородке. Обычно расширено фиброзное кольцо
и полость правого желудочка о Эбштейна болезнь.
Анорексия — отсутствие аппетита при наличии физиологической потребности
в питании, обусловленное нарушениями деятельности пищевого центра.
• Неврогенная а. — упорное стремление к похуданию путем целенаправ­
ленного длительного самоограничения в еде, обусловленное страхом перед
ожирением и прибавлением массы тела. Наблюдают вторичные эндо­
кринные, обменные нарушения и функциональные расстройства. Часто
приводит к опасному для жизни истощению. Частота: 1% женщин и 0,1%
мужчин, чаще в возрасте 13-20 лет. Течение и прогноз: 40% больных выздо­
равливают, у 30% — состояние улучшается, в 30% случаев болезнь при­
нимает хроническую форму. 6% больных погибают вследствие истощения
или самоубийства <=> анорексия нервно-психическая, анорексия нервная.
Анофтальмия — врожденное отсутствие глазного яблока.
Антацид. 1. Агент, нейтрализующий кислоту. 2. Вещество, подавляющее секре­
цию соляной кислоты в желудке (например, ранитидин). 3. ЛС щелочного
характера, снижающие кислотность желудка (например, натрия гидрокар­
бонат, магния оксид, кальция карбонат, алюминия гидроксид*). Указанные
вещества связывают фосфаты, находящиеся в желудке и кишечнике,
и выводят их с калом.
Антесистолия — преждевременное возбуждение желудочков по отношению
к возбуждению предсердий. На ЭКГ проявляется укорочением интерва­
ла Р -0 ,. Встречается при синдромах Вольфа-Паркинсона-Уайта и КлеркаЛ еви—Кристеско.
Антиген (Аг) — вещество, несущее признаки генетически чужеродной инфор­
мации и вызывающее в организме развитие специфических иммунологи­
ческих реакций, т.е. при контакте с иммунной системой индуцирующее
состояние чувствительности и/или резистентности к инфекциям или ток­
синам после латентного периода. Аг можно определить как молекулу, кото­
рую иммунокомпетентные клетки распознают как чужеродную (не свою).
Молекула Аг взаимодействует с АТ или рецептором Т-лимфоцитов. Если
В-лимфоциты распознают свободную молекулу Аг, то Т-лимфоциты —
фрагмент Аг на поверхности других клеток. Взаимодействие с тканями
и/или АТ сенсибилизированного организма можно продемонстрировать
т у/уо или т VIIго. Молекулярная масса Аг имеет существенное значение.
Вещества с массой более 5—10 кД — сильные иммуногены. Исключение —
нуклеиновые кислоты, обладающие большой молекулярной массой,
но слабой (по сравнению с белками) иммуногенностью. Растворимость —
важное условие для проявления иммуногенности Аг. Нерастворимые белки
(например, кератины) не могут находиться в коллоидной фазе и не вызы­
вают иммунных реакций. Специфичность Аг. По способности специфично
взаимодействовать с АТ выделяют несколько типов Аг: видовые, груп­
повые, гетерогенные, аллоантигены. • НЬА — см. «а. главного комплек­
са гистосовместимости человека (НЬА)». • Австралийский Аг — первый
идентифицированный Аг вируса гепатита В; выделен из крови австра­
лийского аборигена, поэтому этот Аг также называют австралийским.
• Аллоантигены (изоантигены) — Аг конкретного индивидуума, обладающие
иммуногенностью по отношению к другим представителям этого вида,
но не к организму-донору трансплантата. Яркий пример изоантигенов —
групповые Аг крови, существующие на мембранах эритроцитов и других
клеток. Поскольку человек обладает естественными АТ к групповым Аг
крови, последние приобретают свойства сильных трансплантационных Аг.
Поэтому перед трансплантацией и гемотрансфузией необходимо опреде­
лить группы крови донора и реципиента. У микроорганизмов имеются
собственные изоантигены, так же известные, как типоспецифичные Аг.
Например, по составу полисахаридных Аг пневмококки подразделяются
на типы I, II, III и т.д., а возбудители ботулизма — на типы А, В, С, О и т.д.
• Видовые аа. представлены антигенными детерминантами, имеющимися
у особей одного вида. Так, отдельные штаммы микроорганизмов могут
содержать внутривидовые Аг, по которым их разделяют на серологические
варианты (серовары). • Гетерогенные (перекрестно реагирующие) Аг пред­
ставлены антигенными детерминантами, общими для организмов разных
таксономических групп. Характерный представитель — полисахаридный
Аг Форссмана, содержащийся в эритроцитах кошек, собак, овец и почке
морских свинок. У человека типичные перекрестные Аг представлены
КЬ-системой эритроцитов: КН-Аг человека перекрестно агглютинируют АТ
к эритроцитам обезьян Масасш гкезш. Известны общие Аг эритроцитов
человека и палочки чумы, вирусов оспы и гриппа. Перекрестно реагирующие
Аг могут блокировать способность Аг-распознающих клеток идентифициро­
вать чужеродные структуры. Например, сходство Аг эритроцитов группы
О и чумной палочки затрудняет распознавание последних иммунной систе­
мой; во многом это обусловливает высокую смертность от чумы. • Главного
комплекса гистосовместимости человека (Н1А) лейкоцитарные а. — систе­
ма обозначения продуктов генов по крайней мере четырех сцепленных
локусов (А, В, С и О) шестой хромосомы. Идентифицировано более 60
аллелей, большинство из которых располагается в локусах, отвечающих
за продукцию Н1А-А и НЬА-В о а. главного локуса <=> общий а. лейко­
цитов о главного комплекса а. • Групповые Аг представлены антигенными
детерминантами, обусловливающими внутривидовые различия у особей
одного вида, что позволяет разделять их на группы. • Изоантигены — см.
«Антиген. Аллоантигены». • Карциноэмбриональный а. — гликопротеин
гликокаликса эмбрионального энтодермального эпителия. Не экспрес­
сируется клетками взрослых особей, за исключением клеток некоторых
опухолей (при этом Аг содержится и в сыворотке больных) « онкофетальный а. • Неполный а. — см. «Гаптен». • Органо- и тканеспецифические аа.
Многие ткани организма человека имеют большое количество собствен­
ных Аг. Их идентификация позволяет различать клетки отдельных тканей,
а также определять изменения в клетках при дифференцировке и транс­
формации. • Патологические Аг. Некоторые воздействия могут вызывать
изменения клеточных молекул (в первую очередь белков), придавая им
антигенные свойства. Например, под действием излучения или высокой
температуры в организме образуются так называемые лучевые или ожого­
вые Аг. Нормальный набор клеточных Аг может изменяться в результате
злокачественной трансформации, что приводит к появлению аномальных
Аг. Такие опухолевые Аг (или онкоантигены) — важные маркёры злокаче­
ственного роста; их выявление — один из важных методов диагностики
опухолей. • Полный а. Значительная часть Аг способна запускать иммун­
ные реакции, выступая в последующем в качестве мишени, в отношении
которой эти реакции происходят. Такие иммуногены известны как пол­
ные Аг. Некоторые Аг имеют малые размеры и простое строение, тогда
как другие представляют собой крупные и сложные молекулы, содержат
множество эпитопов, каждый из которых распознает различные рецепторы
лимфоцитов и/или АТ. • Простатоспецифический а. (176820, ^ 1 3 , калликреиновая протеаза, экспрессию регулируют андрогены) — маркёр рака
предстательной железы. Умеренное повышение характерно для диффузной
гиперплазии предстательной железы, резкое повышение — для рака пред­
стательной железы. • Экзогенные аа. подвергаются эндоцитозу и расщепле­
нию в антигенпредставляющих клетках. Далее фрагмент Аг, содержащий
антигенную детерминанту (эпитоп) в комплексе с молекулой М НС клас­
са II, транспортируется к плазматической мембране антигенпредставляющей клетки, встраивается в нее и предъявляется С Э 4+ Т-лимфоцитам.
• Эндогенные аа. — продукты собственных клеток организма. Чаще всего
это вирусные белки, которые синтезируются клетками хозяина, инф ици­
рованными вирусом, и аномальные белки опухолевых клеток. Их анти­
генные детерминанты предъявляются С Б 8 + Т-лимфоцитам в комплексе
с молекулой М НС класса I.
Антидот — препарат, нейтрализующий яд или подавляющий его действие.
Антиидиотип — антигенсвязывающий фрагмент АТ, специфически распозна­
ющий идиотип — антигенсвязывающий фрагмент АТ, которые образуются
при антигенной стимуляции. Антиидиотипические АТ присоединяются
в том же гипервариабельном участке идиотипического АТ, связывающего
Аг. Антиидиотипические АТ образуются в норме и участвуют в регуляции
иммунного ответа.
Антикоагулянты — ЛС, тормозящие процесс свертывания крови [например,
гепарин натрия, этил бискумацетат (неодикумарин*), фениндион (фенилин*)]. • Антикоагулянт волчаночный — см. «Антитела антифосфолипидные», «Синдром антифосфолипидный».
Антимонголоидный разрез глаз — наружные углы глаз располагаются ниже
внутренних.
Антистрептогиалуронидаза — АТ, образующиеся в организме при стрептокок­
ковой инфекции и обладающие способностью нейтрализовать стрептогиалуронидазу. Выявление а. в сыворотке крови имеет диагностическое
значение.
Антистрептолизин — АТ, ингибирующ ие или предотвращающие эффекты
стрептолизина О, который вырабатывается р-гемолитическими стрепто­
кокками группы А. Концентрация а. в сыворотке часто увеличивается
во время или после стрептококковой инфекции, и сравнительные титры
могут иметь диагностическое и прогностическое значение.
Антитело (АТ) — эффекторные молекулы гуморального иммунитета. Синтез АТ
запускают Аг, поступающие в организм извне (при инфекциях, вакцинации,
действии ксенобиотиков) или образующиеся эндогенно. Как правило, АТ
специфически взаимодействует с комплементарным Аг. Существуют, однако,
АТ, взаимодействующие с Аг-детерминантами, общими для различных Аг.
Такие АТ известны как перекрестно реагирующие, или гетероспецифичные.
АТ существуют в миллионах разновидностях, и каждая молекула имеет уни­
кальный участок связывания Аг-детерминанты. В большинстве случаев АТ
представлены сывороточными гликопротеинами, мигрирующими в составе
медленной фракции у-глобулинов при электрофорезе белков сыворотки
крови. Поэтому для обозначения сывороточной фракции АТ иногда приме­
няют термин «у-глобулины» (а также «иммуноглобулины», 1§). АТ образуют
одну из основных фракций белков крови, составляя 20% массы общего белка
плазмы. АТ устойчивы к действию слабых кислот и щелочей, а также к нагре­
ванию до 60 °С. Структурная единица АТ — мономер — молекула цилиндри­
ческой формы, состоящая из двух идентичных тяжелых Н-цепей (от англ.
кеауу — тяжелый) и двух идентичных легких Ь-цепей (от англ. Иф1 — легкий).
Тяжелые и легкие цепи 1§ состоят из аминокислотных остатков и соединены
дисульфидными ( - 8 —8—) связями. В цепях различают вариабельную,
или У-область (от англ. тпоиз — разный), и константную, или С-область
(от англ. сопз(ап( — постоянный). У-область у разных АТ варьирует. У-области
Ь- и Н-цепей образуют Аг-связывающий центр (активный центр АТ, паратоп),
или РаЬ-фрагмент (от англ. /га%теп( — фрагмент + аШщеп ЫпсИщ — связыва­
ющий Аг). Константная область молекулы называется Гс-фрагментом (от
англ. _/га§теп1 сгуШ1Н?,аЫе — фрагмент кристаллизации). В месте соединения
РаЬ- и Рс-фрагментов расположена шарнирная область, позволяющая
Аг-связывающим фрагментам разворачиваться для более тесного контакта
с Аг. Гены, кодирующие синтез известных классов 1§, расположены в хромо­
сомах 2, 14 и 22. См. также «Иммуноглобулины». Ко-АТ характерны для пре­
имущественно кожной формы СКВ и фотосенсибилизации. Анти-Ьа АТ — а.
к протеину в составе РНК. Анти-ИЧР АТ — а. к полипептидам рибонуклеопротеидов. Анти-Ко АТ — а. к РНК-полимеразе. Анти-8ш АТ — а. к полипеп­
тидам коротких ядерных РНК. • Антинуклеарные АТ — а., обнаруживающие
сродство к клеточному ядру. Находят в сыворотке крови при СКВ, РА, коллагеновых болезнях (также у части родственников больных) и примерно у 1%
здоровых лиц. • Антифосфолипидные АТ (спектр аутоантител к клеточным
фосфолипидам, например к кардиолипину). Термин «волчаночный антикоа­
гулянт» относится к группе АТ (преимущественно 1§С) с ингибиторным
эффектом по отношению к коагуляционно активным т л>Иго фосфолипидным
компонентам в тестах изучения свертывания крови. Встречается при СКВ,
аутоиммунной тромбоцитопении. • Моноклональные а. — а., продуцируемые
клоном или генетически гомогенной популяцией гибридомных клеток.
Гибридомные клетки специально клонируются для получения клеточных
линий, вырабатывающих специфические АТ. А., конъюгированные с радио­
нуклидами (1311, 90У, 67Си), используются для лечения ряда болезней (в осо­
бенности лимфом и лейкозов). • Неполные а. содержат один Аг-связывающий
центр и поэтому одновалентны (например, АТ, вырабатываемые при бруцел­
лезе). Второй Аг-связывающий центр у подобных 1§ экранирован различными
структурами либо обладает низкой авидностью. Н. АТ функционально дефек­
тны, так как они не способны агрегировать Аг. Н. АТ могут связывать эпито­
пы Аг, препятствуя контакту с ними полных АТ; поэтому их также называют
блокирующими АТ. • Полные а. (в частности, 1§М, 1§С) вызывают агрегацию
Аг, видимую невооруженным глазом (например, реакция агглютинации).
• Антителообразование. • Генерация разнообразия АТ. Кроме различных классов
1§ между молекулами АТ существуют аллотипические, изотопические и штиотипические различия. Аллотипы. Аллотипические детерминанты расположены
на легких и тяжелых цепях 1§, генетически детерминированы и строго инди­
видуальны для каждого организма. Их образование обусловлено различиями
небольших аминокислотных последовательностей в константных участках
тяжелых и легких цепей в результате незначительного полиморфизма генов,
кодирующих их синтез. Аллотипические различия не влияют на функцио­
нальные свойства молекул АТ и характеризуются моногенным (менделевским)
наследованием. Идиотипы. Идиотипические детерминанты определяют инди­
видуальную характеристику конкретного АТ и соответствуют его Аг-связывающим участкам. Все молекулы 1§, продуцируемые отдельным лимфоцитом
и его потомками (т.е. клоном плазматических клеток), несут один и тот же
идиотип и обозначаются термином «моноклональные АТ». Изотипы. Изотипические детерминанты носят видовые признаки и идентичны у всех пред­
ставителей одного вида. По их структуре различают классы и подклассы
тяжелых цепей и варианты легких цепей. Образование изотопических детер­
минант обусловлено более существенными структурными различиями в соста­
ве цепей, влияющими на функциональные свойства АТ. • Динамика антителообразования. На скорость образования АТ влияет ряд факторов: доза Аг
(сила Аг-воздействия), частота Аг-стимуляции и состояние иммунной систе­
мы индивида (т.е. его иммунный статус). Если организм впервые встречается
с Аг, то развивается первичный иммунный ответ, а при повторном контакте —
вторичный ответ. Первичный ответ. Появлению АТ предшествует латентный
период продолжительностью 3—5 сут. В это время происходит распознавание
Аг и образование клонов плазматических клеток. Затем наступает логарифми­
ческая фаза, соответствующая поступлению АТ в кровь; ее продолжитель­
ность — 7—15 сут. Постепенно титры АТ достигают пика, и наступает стацио­
нарная фаза продолжительностью 15—30 сут. Ее сменяет фаза снижения титров
АТ, длящаяся 1—6 мес. В основу пролиферации клеток-продуцентов АТ зало­
жен принцип селекции. В динамике антителообразования титры высокоаф­
финных АТ постепенно нарастают: после иммунизации аффинность АТ к Аг
постоянно увеличивается. Первоначально образуются 1§М, но постепенно
их образование уменьшается и начинает преобладать синтез 1§С. Так как пере­
ключение синтезов от 1§М к 1§С не меняет идиотипа АТ (т.е. его специфич­
ность по отношению к конкретному Аг), то оно не связано с клональной
селекцией. Особенности первичного ответа — низкая скорость антителообра­
зования и появление сравнительно невысоких титров АТ. Вторичный ответ.
После антигенной стимуляции часть В- и Т-лимфоцитов циркулирует в виде
клеток памяти. Особенности вторичного иммунного ответа — высокая ско­
рость антителообразования, появление максимальных титров АТ и длительное
(иногда многолетнее) их циркулирование. Основные характеристики вторич­
ного ответа: • образование АТ индуцируется значительно меньшими дозами
Аг; • индуктивная фаза сокращается до 5—6 ч; • среди АТ доминируют 1§0
с большой аффинностью, пик их образования наступает раньше (3—5 сут);
• АТ образуются в более высоких титрах и циркулируют в организме длитель­
ное время. Основные функции АТ. АТ через Аг-связывающие центры взаимо­
действуют с различными Аг. Тем самым АТ предотвращают инфицирование
или элиминируют возбудитель либо блокируют развитие патологических
реакций, активируя при этом все системы специфической защиты. Активация
комплемента. АТ (1§М и 1§С) после связывания с Аг (микроорганизм, опухо­
левая клетка и др.) активируют систему комплемента, что приводит к уни­
чтожению этой клетки путем перфорации ее клеточной стенки, усиления
хемотаксиса, хемокинеза и иммунного фагоцитоза. Антителозависимая цито­
токсичность. Опсонизируя Аг, АТ стимулируют их разрушение цитотоксическими клетками. Аппарат, обеспечивающий распознавание мишеней, —
рецепторы к Рс-фрагментам АТ. Разрушать опсонизированные мишени
способны макрофаги и гранулоциты (например, нейтрофилы). Антитоксический
эффект. АТ могут связывать и тем самым инактивировать бактериальные ток­
сины. Нейтрализация. Взаимодействуя с рецепторами клетки, связывающими
бактерии или вирусы, АТ могут препятствовать адгезии и проникновению
микроорганизмов в клетки организма-хозяина. Опсонизация (иммунный фаго­
цитоз). АТ (через РаЬ-фрагменты) связываются с клеточной стенкой микро­
организма; Рс-фрагментом АТ взаимодействует с соответствующим рецепто­
ром фагоцита. Это опосредует последующее эффективное поглощение
фагоцитом образовавшегося комплекса. Циркулирующие иммунные комплексы.
АТ связывают растворимые Аг и образуют циркулирующие комплексы,
с помощью которых Аг выводится из организма, преимущественно с мочой
и желчью.
а 1-Антитрипсин — гликопротеин, ингибитор протеазы сыворотки крови чело­
века. Синтезируется в печени, генетически полиморфен. Известно более 30
аллелей. См. также «Недостаточность антитрипсина».
Антитромбины — общее название веществ, содержащихся в плазме крови
и являющихся физиологическими антагонистами тромбина (а. разрушают
его). Наибольшее значение имеет антитромбин III (1д23—д25, ген АТЗ),
поскольку на избирательном связывании этого вещества основано антикоагулянтное действие гепарина натрия.
Антрациклины — противоопухолевые антибиотики (например, доксорубицин,
даунорубицин, митоксантрон).
Аплазия. 1. Отсутствие дифф еренцировки опухолевых клеток (атипизм).
Выделяют четыре степени а. (или злокачественности), различающиеся
по степени дифференцировки и числу митозов. 2. Полное врожденное
отсутствие органа или его части (агенезия).
Апноэ — временная остановка дыхания. Постанестетическое а. — см.
«Недостаточность ферментов, недостаточность холинэстеразы».
Аполипопротеин (апоЛП) — белковая часть ЛП. См. «Дефекты аполипопротеинов».
Апоптоз (от греч. арор1оыз — опадание листьев) — программированная (регу­
лируемая) гибель клетки путем деградации ее компонентов (включая
конденсацию хроматина и фрагментацию Д Н К ) с последующим фагоци­
тозом макрофагами; а. наблюдается при морфогенезе органов, удалении
аутореактивных клонов иммунокомпетентных клеток, регуляции числен­
ности пролиферирующих клеточных популяций, повреждении генома
клеток. Контроль а. происходит по двум путям. «Внешний» (рецептор­
ный) путь запускает агонист рецептора смерти (например, /'ах-лиганд,
Ф Н О -а). Опосредованная лигандом олигомеризация рецептора приводит
к активации каспазы-8. «Внутренний» (митохондриальный) путь: боль­
шинство других проапоптозных стимулов инициирует активацию каспазы-9, что опосредует А раМ . Эти стимулы действуют на митохондрии,
из которых выделяется цитохром С. Вместе с А раМ и каспазой-9 цитох­
ром с формирует комплекс активации (апоптосому). Каспаза-8 и каспаза-9 активируют эффекторные каспазы (например, каспазу-3), которые
участвуют в протеолизе и вызывают а. Аномально повышенная устой­
чивость (резистентность) клеток к а. значима в патогенезе пороков раз­
вития, аутоиммунных нарушений и злокачественных новообразований
вследствие подавления процесса гибели дефектных и мутантных клеток
(например, при аутоиммунном лимфопролиферативном синдроме угнетен
а. лимфоцитов вследствие мутации гена, кодирующего гликопротеин Раз).
Аномально повышенная гибель клеток путем а. сопровождает острые забо­
левания (инфекции, ишемические повреждения), а также ряд хронических
патологий (нейродегенеративные заболевания, СПИД).
Апоптосома — молекулярный комплекс активации апоптоза, включает молеку­
лу АраГ-1, цитохром С, выделяющийся из митохондрий в ответ на действие
проапоптозного сигнала, и каспазу-9.
Апоферритин — белок, связывающий железо в виде комплексного соединения
гидроокиси железа и фосфорной кислоты (ферритина); обеспечивает вса­
сывание в кишечнике и депонирование железа в организме; содержится
в селезенке, печени и слизистой оболочке кишечника.
Арахнодактилия («паукообразные» пальцы) — узкая длинная ладонь с длин­
ными пальцами.
Аритмия сердца — нарушение формирования импульса возбуждения или его
проведения по миокарду, проявляю щ ееся обычно нарушением ритма
сердечных сокращений. См. также «Блокады сердца», «Брадикардии»,
«Тахикардии», «Экстрасистолы». • Дыхательная а. с. — синусовая а. с.,
при которой происходит увеличение ЧСС во время вдоха и ее уменьше­
ние во время выдоха. Этот вид а. с. связан с изменением тонуса блуж­
дающего нерва. Наиболее часто возникает в детском и юношеском воз­
расте <=> Херинга феномен. • Желудочковая а. с. — гетеротопный ритм
сердца, при котором водитель ритма расположен в миокарде желудочков.
• Мерцательная а. с. — фибрилляция предсердий с полной нерегулярно­
стью интервалов между сердечными сокращениями и силой сокращений
желудочков сердца <=> полная а. с. <=> «бред» сердца. О Мерцательная брадисистолическая а. с. — мерцательная а. с., протекающая с нормальной
или сниженной ЧСС и отсутствием дефицита пульса. О Мерцательная тахисистолическая а. с. — учащение сердечных сокращений более 90 в минуту
в покое, сопровождающееся дефицитом пульса <=> тахиаритмия мерцатель­
ная. Примечание. В России часто применяют термин «мерцательная арит­
мия», в котором объединяются два различных состояния — фибрилляция
предсердий и трепетание предсердий. • Реперфузионная а. — а., возникаю­
щая после возобновления кровообращения в глубоко ишемизированной
части миокарда, одно из опасных осложнений тромболитической терапии
(возможно появление брадикардии, полной АВ-блокады). • Синусовая
а. — а., обусловленная колебаниями автоматической активности синусно­
предсердного узла; чаще всего связана с изменениями парасимпатической
регуляции. Разница в интервалах Р—Р у здоровых людей обычно не пре­
вышает 0,15 с. Физиологические колебания частоты синусового ритма
связаны с дыханием (дыхательная а.). Синусовая а. наиболее выражена
в юношеском возрасте, у тренированных спортсменов, а также у пациен­
тов с неврозами, нейроциркуляторной дистонией. ЭКГ-идентификация.
• Нерегулярный ритм — неодинаковые интервалы К—К. Колебания про­
должительности интервалов К—К, превышающие 0,16 с. • Наличие зубца Р
перед каждым комплексом. Проявления: уменьшение ЧС С на вдохе, уча­
щение на выдохе. Лечения не требуется о нерегулярный синусовый ритм.
Лечение а. Антиаритмические препараты. • Класс I — блокаторы быстрых
натриевых каналов, разделяются на подклассы в зависимости от влия­
ния на длительность ПД и эффективны й рефрактерный период (интер­
вал <2—Т). 0 Подкласс 1а. ЛС вызывают удлинение ПД и эффективного
рефрактерного периода. ® Хинидин. ® Прокаинамид (новокаинамид*).
ФПодкласс 1Ь. ЛС укорачивают ПД и слабо влияют на эффективный реф ­
рактерный период. ® Лидокаин. ® Тримекаин. ® Ф енитоин (дифенин*).
0 Подкласс 1с. ЛС слабо влияют на ПД и эффективный рефрактерный
период. ® М орацизин (этмозин*). ® Этацизин*. ® Л аппаконитина гидро­
бромид (аллапинин*). <8> Пропафенон. • Класс II — р-адреноблокаторы
(ограничивают симпатическое воздействие на сердце, угнетают синусо­
вый автоматизм, замедляют проведение через предсердно-желудочковое
соединение). 0 Пропранолол (анаприлин*). 0 Атенолол. 0 Метопролол.
• Класс III — препараты, равномерно удлиняющие все фазы реполя­
ризации и ПД. 0 Амиодарон. 0 Бретилия тозилат. 0 Соталол. • Класс
IV — антагонисты кальция (замедляя медленный кальциевый ток, угне­
тают предсердно-желудочковую проводимость и автоматизм синусно­
предсердного узла). 0 Верапамил. 0 Дилтиазем. Немедикаментозное лече­
ние. • Хирургическое (пересечение дополнительных проводящих путей,
устранение очага эктопической активности и др.). • Радиочастотная
катетерная аблация (разруш ение) А В -соединения, аномальных путей
проведения и аритмогенных очагов. • И мплантация электрокардиости­
мулятора. • И мплантация портативных кардиовертеров-дефибрилляторов
(автоматических устройств, мониторирующих сердечный ритм и нанося­
щих разряд электрического тока при появлении фибрилляции желудочков
или желудочковой тахикардии).
Арренобластома — редкая, чаше доброкачественная опухоль яичника. Харак­
теризуется наличием структурных компонентов яичка (канальцев с эпители­
альными клетками и стромальными клетками, подобными гландулоцитам).
Может вызывать маскулинизацию <=> андробластома <-> гинандробластома
<=> арренома <=> маскулинома <-> тубулярная аденома.
Артериит височный (187360) — панартериит с некрозом средней оболочки
и содержащими многоядерные гигантские клетки гранулемами в височных
артериях, артериях сетчатки и головного мозга. Развивается у лиц старше­
го возраста и может проявляться общими симптомами, а также тяжелой
двусторонней головной болью, внезапной потерей зрения <=> краниальный
а. <=> гигантоклеточный а. <=> темпоральный а. <=> а. гранулематозный о
мезартериит генерализованный гранулематозный <=> Хортона—М агата—
Брауна синдром <=> Ш мидта—Варбурга синдром.
Артериография — рентгеноконтрастное исследование артерии или артериальной
сети. • Коронарная а. — рентгенологическое исследование венечных артерий
сердца после заполнения их контрастным веществом <=> коронарография.
Артрогрипоз — множественные врожденные контрактуры суставов.
Асбестоз — профессиональный пневмокониоз, развивающийся в результате
систематического вдыхания пыли асбеста. Асбест — канцероген.
Асистолия — отсутствие сокращений сердца.
Аскаридоз — гельминтоз из группы кишечных нематодозов, вызываемый
аскаридами (обычно А зсат ЫтЪпсоШез), характеризующийся в ранней
стадии явлениями аллергии, а в поздней — диспепсическими явлениями
и осложнениями при проникновении гельминтов в другие органы, а также
в результате закупорки или спазма кишечника.
Аспергиллез — микоз, вызванный грибами рода Азрег§И1из.
Астма бронхиальная. Характеризуется повторными, остро развиваю щ ими­
ся приступами удушья (ощущение нехватки воздуха и/или невозможно­
сти сделать вдох). Эта форма патологии выявляется у 2—4% населения.
Выделяют атопический и неатопический (инфекционно-аллергический)
варианты астмы. Причины атопического варианта — неинфекционные
агенты (пыль, пыльца, химические вещества, компоненты шерсти живот­
ных, чешуи рыбы, корма для рыб и др.). Причины неатопического вариан­
та — микроорганизмы, вызывающие хронические инфекции носоглотки,
трахеи, бронхов и легких. В основе патогенеза при обоих вариантах брон­
хиальной астмы лежат механизмы развития аллергии типа I, II, III и IV.
Асцит — скопление избытка серозной жидкости (транссудат) в брюшной
полости <=> гидроперитонеум.
Атаксия-телеангиэктазия — см. «Синдром Луи-Бар».
Ателектаз — отсутствие газа в части или во всем легком вследствие недоста­
точного растяжения альвеол или транспорта газа из них.
Ателия — отсутствие сосков.
Атеросклероз (от греч. а1кеге — кашица, $к1его$ — твердый) — патологический
процесс, приводящий к изменению стенки артерий в результате накопле­
ния липидов, образования фиброзной ткани и формирования бляшки,
сужающей просвет сосуда. А. — системное заболевание, поражающее арте-
рии эластического (аорта и ее ветви) и мышечно-эластического (артерии
сердца, головного мозга и др.) типа. При этом во внутренней оболочке
артериальных сосудов формируются очаги липидных, главным образом
холестериновых, отложений (атероматозные бляшки), что вызывает про­
грессирующее сужение просвета сосудов вплоть до их полной облитера­
ции. А. — главная причина заболеваемости и смертности в России, СШ А
и большинстве стран Запада. • При хронической, медленно нарастающей
облитерации клиническую картину а. определяет степень недостаточности
кровоснабжения органа, питаемого пораженной артерией. • Возможна
острая окклюзия просвета артерии тромбом и /и ли содержимым распав­
шейся атероматозной бляшки, что ведет к образованию очагов некроза
(инфаркт) или гангрены органа или части тела, расположенного (ой)
в бассейне пораженной артерии. • Наиболее подвержена атеросклероти­
ческому повреждению область бифуркации сонной артерии, коронарные
артерии и брюшной отдел аорты. Частота. 150:100 000 в возрасте 50 лет.
Последствия а. — главная причина смертности. Преобладающий возраст —
пожилой. Преобладающий пол — мужской (5:1). Этиопатогенез. Теория
повреждений и накопления основана на признании повреждающего дей­
ствия различных факторов риска (см. «Факторы риска») на эндотелий сосу­
дов. Начинается пролиферация ГМК и миграция макрофагов в сосудистую
стенку. Через поврежденный эндотелий во внутреннюю оболочку сосуда
проникают липиды и холестерин, формирующие атероматозную бляшку.
Атероматозная бляшка приводит к стенозу сосуда, индуцирует актива­
цию тромбоцитов и формирование тромбов, что влечет за собой ишемию
и /и ли некроз пораженного органа. Генетические аспекты. Семейная пред­
расположенность к а. связана с наследованием факторов риска (исключая
курение и прием пероральных контрацептивов, см. также «Дефекты аполипопротеинов»). Факторы риска. • Курение. • СД. • Артериальная гипер­
тензия. • Ожирение. • Гиперхолестеринемия (отношение Л П Н П к ЛПВП
более 5:1). • Гипертриглицеридемия. • Гиподинамия. • Инсульты и забо­
левания ССС в семейном анамнезе. • Прием пероральных контрацепти­
вов. Патоморфология. • Степень I — доклинический период заболевания.
На неизмененной внутренней оболочке артерий обнаруживают единичные
липидные пятна и полоски (липоидоз). • Степень II — слабо выраженный а.
На неизмененной внутренней оболочке артерий — липоидоз и единичные
мелкие фиброзные и атероматозные бляшки. • Степень III — значительно
выраженный а. Кроме липоидоза в артериях на утолщенной волнистой
и деформированной внутренней оболочке — большое количество мелких
и крупных, сливающихся фиброзных и атероматозных бляшек, атерокальциноз. • Степень IV — резко выраженный а. На утолщенной и дефор­
мированной бугристой внутренней оболочке артерий — многочисленные
фиброзные и атероматозные бляшки с кальцинозом и изъязвлениями.
Модели. • Моделирование а. на животных (преимущественно на кроликах)
ранее проводили, скармливая значительное количество липидов. В настоя­
щее время патогенез заболевания изучают на мышах с модифицирован­
ными генами. 0 Дефект апоЛП Е: развивается значительное поражение
стенки сосудов, но состав ЛП в крови не такой, как у человека. О Дефект
рецепторов ЛГТНГТ (ген Ю Ь К ) и отсутствие апоЛП В-48 (ген АРОВ, иска­
женный процессинг): значительно увеличено содержание холестерина
Л П Н П , у гомозигот много выше, чем у гетерозигот. О Дефект рецепторов
ЛГТНП (ген ЬВЬК) и гиперэкспрессия апоЛП В-48 (ген АРОВ): значительно
увеличено содержание холестерина Л П Н П и триглицеридов. Клиническая
картина варьирует в зависимости от преимущественной локализации и рас­
пространенности процесса и в большинстве случаев определяется прояв­
лениями и последствиями ишемии ткани или органа. Лабораторные иссле­
дования. • Гиперхолестеринемия. • Гипертриглицеридемия. • Повышение
Л П Н П и ЛПОНП . • Снижение ЛПВП. Лечение. Режим амбулаторный
до развития осложнений. Диета №10с. • Жиры: общее количество — менее
30% общей энергетической ценности пищи; животные жиры с высоким
содержанием насыщенных жирных кислот — менее 7%. • Углеводы —
50—60%, повышение содержания растительной клетчатки (фрукты, овощи).
• Белки — 10—20%. • Холестерин — менее 200 мг. • Соль — 1650—2400 мг.
• Регулярное употребление незначительных доз алкоголя может повысить
уровень ЛПВП. Физическая активность. Ф изические упражнения по мень­
шей мере по 30 мин 3 раза в неделю, активный образ жизни. Лекарственная
терапия. • Основные гиполипидемические средства. 0 Статины (ингибиторы
З-гидрокси-З-метил-глутарил-КоА-редуктазы): флувастатин, ловастатин.
правастатин или симвастатин снижают концентрацию ЛН О П , ЛПНП.
холестерина. Большинство больных семейной гиперхолестеринемией рези­
стентны к статинам. При резистентности к статинам, сопутствующей
триглицеридемии, статины сочетают с другими гиполипидемическими
средствами. 0 Анионообменные смолы (секвестранты желчных кислот
вызывают снижение концентрации Л П Н П и холестерина. Рекомендуюназначать при умеренном повышении уровня Л П Н П , а также женщинам
в предменопаузном периоде; в более тяжелых случаях применяют комбина­
ции препаратов с другими гиполипидемическими средствами, в частноспсо статинами. 0 Никотиновая кислота вызывает снижение к о н ц ен тр ат1г
холестерина и триглицеридов и повышает уровень ЛПВП. 0 Фибраты (геу фиброзил) снижают концентрацию триглицеридов и Л П ОНП и повышаю*
ЛПВП. Поскольку фибраты не снижают содержания Л П Н П , их не относ —
к препаратам с наибольшей эффективностью. 0 Пробукол по 500 мг 2 раз;
в сутки умеренно снижает концентрацию Л П Н П и (!) ЛПВП. Осложнен*,
обусловливают половину всех смертных случаев и треть смертных слу­
чаев у людей в возрасте 35-65 лет. • Стенокардия. • И нфаркт миокар и
• Симптоматическая вазоренальная артериальная гипертензия. • С ердечна
недостаточность. • Инсульт. • Нарушения сердечного ритма. • Х П ':
• Расслаивающаяся аневризма аорты. • Артериальные тромбоз и эмбол;
• Внезапная смерть. Прогноз неопределенный. Трудоспособность загсит от функциональной сохранности органов и систем с пораженны
артериями. Устранение факторов риска и повышение культурного уроЕ
населения (как показывает опыт США) могут существенно снизить по*
затели смертности. См. также «Недостаточность липаз», «Недостаточн: .
лецитин-холестерин-ацилтрансферазы», «Дефекты аполипопротеинов»,
«Гиперлипидемия», «Гиперхолестеринемия». Примечание. При недостаточ­
ности белка-переносчика эфиров холестерина (* 118470, 16я21, ген СЕТР,
р) существенно замедляется развитие возрастных атеросклеротических
изменений и увеличивается продолжительность жизни (дефект гена рас­
пространен у японцев). Лабораторно: низкое содержание Л П Н П и тригли­
церидов, высокий уровень ЛПВП.
Атопия — общее название аллергических болезней, в развитии которых значи­
тельную роль отводят наследственной предрасположенности к сенсибили­
зации: например, поллинозы, аллергический ринит, крапивница.
Атрезия — полное отсутствие канала или естественного отверстия.
Атриопептин — см. «Фактор натрийуретический».
Атрофия. 1. Уменьшение массы и объема клеток, тканей и органов вследствие
гибели тканевых элементов, уменьшения пролиферации клеток, ишемии,
сдавления, недоедания, снижения функционирования органа, наруше­
ния гормональной регуляции метаболизма, мутаций. 2. Форма адаптив­
ной реакции на воздействие повреждающего фактора. Для а. характерно
уменьшение размеров клетки. А. клеток обычно сочетается с уменьшением
количества клеток в рассматриваемой популяции, а также с ухудшенным
пополнением утерянных клеток. Эти причины приводят к уменьшению
объема органа, истончению тканевых образований (например, кожи, сли­
зистых оболочек). При атрофии в лизосомах часто наблюдается образова­
ние липофусцина — богатого липидами бурого пигмента, образующегося
из накапливаемого в лизосомах материала. 3. Класс нозологических форм
(в том числе наследственных). • Лебера зрительного нерва наследственная
а. (#535000) — дегенерация зрительного нерва с быстрым развитием цен­
тральной скотомы, чаще встречается у мужчин; может развиться в любом
возрасте <=> Лебера а. зрительного нерва <=> Лебера синдром.
АТФаза — см. «Аденозинтрифосфатаза».
Аутизм — склонность к самоизоляции, отгороженность от реального мира
и утрата связей с ним, погружение в мир личных переживаний. А. обуслов­
лен дисфункцией мозга генетической этиологии. Пренатальные осложне­
ния, синдром ломкой Х-хромосомы (как и другая генетическая патология),
краснуха во время беременности, фенилкетонурия, менингит и энцефалит
считаются предрасполагающими факторами.
Аутоантигены. Некоторые Аг при определенных условиях способны проявлять
аутоантигенные свойства и индуцировать синтез аутоантител. Такие а. раз­
деляются на врожденные и приобретенные. • Врожденные аа. Некоторые
ткани организма обладают антигенными свойствами и запускают иммун­
ные реакции в собственном организме. К ним относятся головной мозг,
глаз (передняя камера, роговица, хрусталик, сетчатка, стекловидное тело),
семенные канальцы яичек, фолликулы щитовидной железы, подкожная
жировая клетчатка, волосяные луковицы, рубцовая ткань. В норме Аг
этих органов находятся вне иммунного надзора (так называемые иммунопривилегированные области организма). При повреждении этих органов
возможен контакт аа. с иммунокомпетентными клетками и развитие ауто­
иммунных реакций. • Приобретенные аа. Способностью запускать аутоим­
мунные реакции обладают ткани, находящиеся в зоне иммунного надзора
и изменяющие свои антигенные свойства под различными воздействиями
(ЛС, переохлаждение, вирусные и бактериальные инфекции).
Аутоантитело — АТ, аффинное к какой-либо из тканей организма, в котором
АТ образуется.
Аутосенсибилизация — повышение чувствительности организма к аллергенам
собственных тканей (аутоаллергены).
Аутосома — любая неполовая хромосома. В диплоидном наборе человека
имеется 22 пары аутосом.
Аутотрансплантат — орган или ткань, предназначенные для пересадки на дру­
гую часть тела.
Афакия — отсутствие хрусталика.
Ахондрогенез — карликовость, для которой характерны различные деформа­
ции костей конечностей, нормальных размеров или увеличенный череп,
короткое туловище, задержка окостенения в нижних отделах позвоночника
[р, И].
Ахондроплазия — нарушение энхондрального остеогенеза длинных трубчатых
костей; вариант хондродистрофии [у некоторых больных найдена мута­
ция (глицин замещен аргинином в позиции 380 рецептора фактора роста
фибробластов 3), 5К с полной пенетрантностью; гомозиготы погибают
в плодном периоде], приводящий к очевидной при рождении карликовости
с короткими конечностями, но нормальным туловищем и относительной
макроцефалией.
Ацетилхолин — см. «Нейромедиаторы».
Ацидоз — форма нарушения КЩР, характеризующаяся сдвигом соотношения
между анионами кислот и катионами оснований в сторону увеличения
анионов (при этом рН <7,37). Различают метаболический, почечный
канальцевый и респираторный а. • Метаболический а. характеризуется
снижением рН крови и уменьшением концентрации бикарбоната плазмы
вследствие потерь бикарбоната или накопления других кислот, кроме
угольной (например, молочной). Типы и причины м. а. М. а. с увеличенной
анионной разницей. • Кетоацидоз. Усилено образование КТ (кетокислот).
Кетоацидоз развивается как осложнение СД, длительного голодания, зло­
употребления алкоголем. • Молочнокислый а. ввиду замедленной доставки
кислорода к тканям усиливает образование лактата с сопутствующим тяже­
лым м. а. Характерный признак многих состояний, сочетающихся с низкой
перфузией тканей (например, ш ок и сепсис). • Почечная недостаточность.
Накопление органических и неорганических анионов, связанное со сни­
жением скорости клубочковой фильтрации, приводит к увеличению ани­
онной разницы при тяжелой почечной недостаточности. • Отравление
салицилатами, метанолом, этиленгликолем может привести к накоплению
органических кислот (например, МК). М. а. с нормальной анионной раз­
ницей (гиперхлоремический м. а.). • Потеря бикарбоната почками.
О Проксимальный канальцевый а. характеризуется сниженной проксималь­
ной канальцевой реабсорбцией бикарбоната, приводящей к чрезмерной
экскреции бикарбоната с мочой. Причины: цистиноз, СКВ, множествен­
ная миелома, отравление тяжелыми металлами, болезнь Уилсона и нефро­
тический синдром. О Дистальный канальцевый а. Причины: отравление
тяжелыми металлами, применение амфотерицина В, СКВ, обструктивная
уропатия, синдром Ш егрена и другие состояния, сопровождающиеся
гиперглобулинемией. О Гиперкалиемический почечный канальцевый а.
Гиперкалиемия, в частности, сочетающаяся с гипоренинемическим гипоальдостеронизмом, характеризуется снижением экскреции аммиака, умень­
ш енным образованием бикарбоната и неспособностью нейтрализовать
нелетучие кислоты с помощью буферных систем. О Потеря органических
анионов. При диабетическом кетоацидозе вследствие потери кетонов
с мочой уменьшается содержание метаболических предш ественников
бикарбоната, так как кетоны метаболизируют в печени с использованием
водородных ионов в различных окислительно-восстановительных реакци­
ях цикла трикарбоновых кислот. Ф И нгибирование карбоангидразы.
Диуретик ацетазоламид (диакарбА) и мафенид (применяют местно при лече­
нии ожогов) ингибируют карбоангидразу и уменьшают проксимальную
канальцевую реабсорбцию бикарбоната. • Потеря бикарбоната через Ж КТ
(диарея, фистула поджелудочной железы, уретеросигмоидостомия).
• Применение минеральных кислот. Гиперхлоремический м. а. развивается
при использовании соляной кислоты или любых ее метаболических пред­
шественников, включая хлорид аммония, гидрохлорид аргинина, хлорид
кальция (только при приеме внутрь). Проявления: м. а. обычно связана
с основным заболеванием. При рН крови менее 7,2 может снижаться сер­
дечный выброс. А. иногда сопровождается резистентностью к сосудосужи­
вающему действию катехоламинов, приводящей к развитию артериальной
гипотензии. Если дыхание учащается в ответ на снижение рН сыворотки,
появляется дыхание Куссмауля. Диагностика. • Электролиты сыворотки.
Снижение бикарбоната и непостоянные величины содержания хлорида
в зависимости от того, сопровождается ли а. нормальной или увеличенной
анионной разницей. • Анализ газового состава артериальной крови.
Снижение уровня бикарбоната с компенсаторным уменьшением р С 0 2
крови. При чистом метаболическом ацидозе р С 0 2 должно быть равно
концентрации бикарбоната, умноженной на 1,5 плюс 6—10 мм рт.ст.
Отклонение от этого значения предполагает наличие осложнения в виде
респираторной дисфункции (показатель р С 0 2 ниже предсказываемого
позволяет предположить первичный респираторный алкалоз; значение
р С 0 2 выше ожидаемого свидетельствует в пользу нарушения функции
дыхания центрального генеза, что приводит к неадекватной задержке С 0 2).
Лечение. При метаболическом ацидозе с рН крови ниже 7,2 — введение
натрия бикарбоната (натрия гидрокарбоната) в/в (44—88 мэкв в 5% раство­
ре глюкозы или 0,45% растворе № С1) до достижения значения рН, равно­
го 7,2 (концентрации бикарбоната плазмы 8—10 мэкв/л), с одновременным
устранением причины а. • Необходимое количество бикарбоната можно
вычислить по формуле. Приблизительное количество бикарбоната натрия,
необходимое для того, чтобы концентрация бикарбоната плазмы повыси­
лась от 6 до 10 мэкв/л, равно: 4 м экв/л х 0,5 х масса тела в кг. Применяя
этот способ расчета, необходимо повторно измерять уровень бикарбоната
плазмы и рН крови. • Недооценка потребностей в бикарбонате может воз­
никнуть при продолжающейся потере бикарбоната (проксимальный
канальцевый а.) или довольно быстром образовании органической кисло­
ты с потреблением вводимого бикарбоната в буферных реакциях (молоч­
нокислый а.). При проксимальном канальцевом а. потребность в бикарбо­
нате составляет 2—4 мг/кг/сут. • Осложнения инфузии раствора натрия
гидрокарбоната: перегрузка объемом, особенно при сердечной и почечной
патологии, гипернатриемия, гипокалиемия, алкалоз. • Почечный канальце­
вый а. (ПКА) — группа расстройств, характеризующихся нарушением
окислительных механизмов в почечных канальцах при сохранных функци­
ях клубочкового аппарата, что приводит к метаболическому а. Эти рас­
стройства подразделяются на три типа. 0 ПКА-1: классический, дисталь­
ный, гипокалиемический (вследствие неспособности поддерживать ионный
градиент Н +). 0 ПКА-2: проксимальный (вследствие снижения реабсорб­
ции бикарбоната), реабсорбция в дистальных канальцах не нарушена.
❖ ПКА-4: генерализованный, дистальный, гиперкалиемический (вслед­
ствие первичного или вторичного дефицита альдостерона или резистент­
ности к нему). Преобладающий возраст: ПКА-1 и -2 — преимущественно
у детей, ПКА-4 — преимущественно у взрослых. Этиология. • ПКА-1: лекар­
ственный (амфотерицин В, препараты лития, НПВС); синдромы, протекаю­
щие с гипергаммаглобулинемией, цирроз печени, пиелонефрит. • ПКА-2:
лекарственный [тетрациклин, ацетазоламид (диакарб*), сульфаниламиды,
соли тяжелых металлов], амилоидоз, множественная миелома. • ПКА-4: волчаночная и диабетическая нефропатия, нефросклероз вследствие артериаль­
ной гипертензии, тубулоинтерстициальные нефропатии, болезнь Аддисона,
острая надпочечниковая недостаточность. Факторы риска: гиперпаратиреоз,
ХПН. Клиническая картина. • Апатия при ПКА-1 и ПКА-2. • Рвота. • Полиурия.
• Дегидратация. • Слабость вследствие потери калия. • ДН, гипервентиляция
как компенсаторная реакция на метаболический ацидоз. • Нарушения поход­
ки и боли в костях вследствие нарушения метаболизма кальция при ПКА-2.
Лечение. Тактика ведения. Адекватная медикаментозная коррекция ацидоза.
Диета. Ограничение поваренной соли. Лекарственная терапия: натрия бикар­
бонат (натрия гидрокарбонат). • Респираторный а. характеризуется снижением
рН крови и повышением р С 0 2 крови (более 40 мм рт.ст.). Этиология. Р. а.
связан с уменьшением способности выделять С 0 2 через легкие. Причины: все
нарушения, угнетающие функцию легких и клиренс С 0 2. • Первичное пора­
жение легких (альвеолярно-капиллярная дисфункция) может привести
к задержке С 0 2 (обычно в качестве позднего проявления). • Нервно-мышечные
поражения. Любая патология дыхательной мускулатуры, приводящая к затруд­
нению вентиляции (например, псевдопаралитическая миастения), может
вызывать задержку С 0 2. • Патология ЦНС. Любое тяжелое повреждение
ствола мозга может сочетаться со снижением вентиляционной способности
и задержкой С 0 2. • Лекарственно-обусловленная гиповентиляция. Любой
препарат, вызывающий выраженное угнетение ЦНС или функции мышц,
может стать причиной развития р. а. Патогенез. • Острая задержка С 0 2
приводит к повышению р С 0 2 крови с минимальным изменением содер­
жания бикарбоната в плазме. При повышении р С 0 2 на каждые 10 мм рт.ст.
уровень бикарбоната плазмы возрастает примерно на 1 мэкв/л, а рН крови
снижается примерно на 0,08. При остром респираторном ацидозе концен­
трации электролитов сыворотки близки к норме. • Хронический респира­
торный ацидоз. Через 2—5 дней наступает почечная компенсация: уровень
бикарбоната плазмы равномерно повышается. Анализ газового состава
артериальной крови показывает, что при повышении р С 0 2 на каждые
10 мм рт.ст. содержание бикарбоната плазмы возрастает на 3—4 мэкв/л,
а рН крови уменьшается на 0,03. Клинические проявления. • Различные
симптомы генерализованного угнетения ЦНС. • Сердечные нарушения:
снижение сердечного выброса, легочная гипертензия — эффекты, которые
могут обусловить критическое замедление притока крови к жизненно важ­
ным органам. Лечение. • Терапия основного заболевания. • Дыхательная
терапия. р С 0 2, превышающее 60 мм рт.ст., может быть показанием к ИВЛ
при выраженном угнетении ЦНС или дыхательной мускулатуры.
Ацидурия метилмалоновая — наследственное заболевание, характеризующее­
ся задержкой психического и физического развития, наличием в моче
метилмалоновой кислоты, метаболическим кетоацидозом. По патогенезу
различают недостаточность метилмалонил-КоА-мутазы, мевалонаткиназы, метилмалонил-КоА-рацемазы и дефекты метаболизма кобаламина.
См. также «Метионинсинтетаза» в статье «Недостаточность ферментов».
• Мевалонаткиназа (*251170, КФ 2.7.1.36, 12д24, ген МУК, р). Клинические
проявления: многочисленные дефекты Ц Н С , значительное отставание
в росте и развитии, анемия, гепатоспленомегалия, катаракта, мевалоновая
ацидемия, мевалонатацидурия. • Метилмалонил-КоА-мутаза (*251000, КФ
5.4.99.2, 6р21.2—р12, ген МСМ, множество дефектных аллелей, группа ком­
плементации/им?; р). Заболевание: В12-резистентнаям. а. (от бессимптомной
до обусловливающей летальный исход сразу после рождения). Клинические
проявления: эпизоды метаболического кетоацидоза, задержка психиче­
ского и физического развития, различные неврологические проявления,
нейтропения, остеопороз. Лабораторно: непостоянная гиперглицинемия,
в моче — метилмалоновая кислота и кетоны с длинной углеродной цепью.
Витамин В12 для коррекции бесполезен. • Метилмалонил-КоА-рацемаза
(251120, метилмалонил-КоА-эпимераза, КФ 5.1.99.1, р;). Клинически — см.
выше «Мевалонаткиназа». • Дефекты метаболизма кобаламина. Различают
следующие дефекты синтеза В12-коферментов [аденозин- и метилкобаламины): сЫА, сЫВ, сЫС, сЬЮ, сЫЕ, сЬ //(витам ин В12-чувствительные, все р)];
недостаточность метилмалонил-КоА-мутазы — витамин В 12-резистентная
(тШ) форма метилмалоновой ацидурии.
Багассоз — диффузное гранулематозное поражение легочной паренхимы,
индуцированное ингаляцией Аг спор термофильных актиномицетов.
Возникает при контактах с остатками переработки сахарного тростника.
Базофилы составляют 0—1% общего числа лейкоцитов в циркулирую ­
щей крови. В крови б. находятся 1—2 сут. К ак и другие лейкоциты,
при стимуляции могут покидать кровоток, но их способность к аме­
боидному движению ограничена. Продолжительность жизни и судьба
в тканях не известны. • Специфические гранулы б. довольно крупные (0,5—
1,2 мкм), окрашиваются метахроматически (от красновато-фиолетовых
до интенсивно-фиолетовых). В гранулах содержатся различные ферменты
и медиаторы. К наиболее значимым из них можно отнести гепаринсульфат
(гепарин), гистамин, медиаторы воспаления (например, медленно реаги­
рующий фактор анафилаксии 8К5-А, фактор хемотаксиса эозинофилов
ЕСР). • Функция б. Активированные б., покидая кровоток, мигрируют
в очаги воспаления и участвуют в аллергических реакциях. Активация
и дегрануляция б. происходит при попадании в организм аллергена и опо­
средована 1§Е. Б. имеют высокоаффинные поверхностные рецепторы
к Рс-фрагментам 1§Е. 1§Е синтезируют плазматические клетки при попа­
дании в организм Аг (аллергена). Молекулы 1§Е присоединяются к базофилу (формируется комплекс 1§Е—базофил). При повторном попадании
Аг (аллергена) он связывается двумя и более молекулами 1§Е на поверх­
ности б., что приводит к дегрануляции последнего — быстрому экзоцитозу
содержимого гранул. Параллельно образуются метаболиты арахидоновой
кислоты.
Бактериофаг — бактериальный вирус, обнаруживаемый во всех группах бак­
терий. Содержит РН К или ДНК. Связь с бактерией-хозяином специфич­
на. В случае умеренного фага может быть генетически близким и носит
название вида, группы или штамма бактерий, по отношению к которому
он специфичен <=> вирус бактериальный.
Баланс кислотно-щелочной — см. «Нарушения обмена жидкости и электро­
литов», «Ацидоз», «Алкалоз».
Барьер гематоэнцефалический — механизм, селективно контролирующий
проникновение большинства ионов и макромолекулярных соединений
из крови в ткань мозга. Образован базальной мембраной и непрерывным
эндотелием капилляров, клетки которого соединены обширными плотны­
ми контактами. Сходные капилляры обнаружены в сетчатке глаза, радуж­
ной оболочке, внутреннем ухе и периферических нервах.
Белок. • АраГ-1 (арорШ к рго1еа$е-асШайп%/ас(ог 1) — апоптозный активирую­
щий каспазу фактор 1, связывает и активирует каспазу-9, активирует сам
себя в ходе конформационных изменений, индуцируемых взаимодействием
с АТФ и цитохромом С. Активированные молекулы АраГ-1 вместе с каспазой-9 и другими белками формируют белковый комплекс, называющийся
апоптосомой, который поддерживает каспазу-9 в активном состоянии.
• Вс1-2 — семейство белков-регуляторов апоптоза, существующих как анта­
гонисты (Вс1-2, Вс1-Х]_, Вс1-\у, ВО-1, Вга§-1, Мс1-1, А1), или агонисты
апоптоза (Вах, Вак, Вс1-Х3, Вай, ВШ, В1к, Нгк). М ногие из б. семейства
и сам Вс1-2 расположены в наружной митохондриальной мембране. Вс1-2
обнаружен также в ядерной мембране и эндоплазматическом ретикулуме.
• С — см. «Протеин С». • Са2+-связывающие бб. (например, кальмодулин,
щелочная фосфатаза, т-белок в микротрубочках, тропонин С, кальсеквестрин, кальретикулин и др.). Во внутриклеточных кальциевых депо (напри­
мер, внутри цистерн эндоплазматической сети) эти бб. непрочно связаны
с Са2+; • сёс (сей йтзюп сус1е) бб. индуцируют митоз. • сйк (сусИп ёерепйеШ
ргогет ктазё) — циклинзависимые протеинкиназы; наряду с циклинами
регулируют клеточный цикл, переключая его с одной фазы на другую
(с С , на 8 или с С 2 на М). • С-реактивный б. (СРБ) — р-глобулин сыво­
ротки крови пациентов с некоторыми воспалительными, дистрофическими
и опухолевыми заболеваниями. Не являясь специфическим АТ, осаждает
т VI(го С-углевод, присутствующий во всех типах пневмококков. • 8 —
см. «Протеин 8». • Бенс-Джонса б. — высокотермостабильный б. (легкая
цепь 1§), обнаруживаемый в моче пациентов с множественной миеломой
и иногда с другими гематологическими болезнями. • Катионные бб. (напри­
мер, нейротоксин из эозинофилов, эозинофильный катионный белок,
панкреатическая рибонуклеаза, ангиогенин) обладают нейротоксической,
гельминтотоксической и рибонуклеолитической активностью. • Острой
фазы воспаления бб. — б. плазмы крови, включая С-реактивный б., связы­
вающий маннозу б., компонент амилоида Р, а,-антитрипсин, фибриноген,
церулоплазмин. Содержание о. ф. в бб. увеличивается в ответ на ИЛ-1, -6,
-11. • Тироксинсвязывающий б. — плазменный а-глобулин, обладающий
высокой аффинностью к Т4; Т3 связывается с этим белком менее прочно <=>
тироксинсвязывающий глобулин <=> тироксинсвязывающий преальбумин.
• Холекальциферолсвязывающий б. — белок плазмы крови, связывающий
витамин Б <=> витамин Б-связы ваю щ ий белок.
Бесплодие. • Женское б. — неспособность к зачатию в детородном возрасте.
В 95% случаев у здоровой женщ ины, желающей иметь ребенка, беремен­
ность наступает в течение 13 мес. Терминология. • Абсолютное б. — бере­
менность исключена полностью (отсутствие или крайняя степень гипопла­
зии матки, отсутствие яичников, пороки развития половых органов и др.).
• Относительное б. — женщина, живущая половой жизнью без применения
противозачаточных средств, никогда не беременела. • Инфертильность —
бесплодие, обусловленное невынашиванием. • Стерильность — неспособ­
ность зрелого организма производить потомство. Факторы, обеспечивающие
наступление беременности. • Сперматогенез (мужской фактор). • Овуляция
(яичниковый фактор). • Взаимодействие шеечной слизи и спермы (шеечный
фактор). • Целостность эндометрия, нормальные размеры и форма полости
матки (маточный фактор). • Проходимость маточных труб и анатомиче­
ские взаимоотношения их с яичниками (трубный фактор). • Осеменение
(коитальный фактор). Основные причины. • Приблизительно в 15% случа­
ев причина б. остается невыясненной. • Аномалии развития (например,
агенезия или поперечная перегородка влагалища, заращение девственной
плевы, аномалии матки). • Воспалительные заболевания органов малого
таза (ВЗОМТ): эндометрит, сальпингит, оофорит, миометрит, перитонит.
До 60% случаев женского б. вызвано нарушением проходимости маточных
труб в результате ВЗОМТ, и 50% всех эктопических беременностей возни­
кает в результате повреждения труб после ВЗОМТ. • Туберкулезный саль­
пингит обычно развивается на фоне легочного туберкулеза. Среди больных
ВЗОМТ туберкулез диагностируют у 11%, среди больных с нарушениями
менструального цикла — у 8,4%, среди больных б. — у 18%. • Синдром
Ашермана. • Синдром гиперстимуляции яичников — увеличение яичников
с множественными фолликулярными кистами, большим кистозным жел­
тым телом и отеком стромы. Может развиться после лечения гормонами.
• Гипотиреоз. • Гирсутизм. • Диэтилстильбэстрол т и (его. • Синдром лютеинизации неовулировавшего фолликула — преждевременная лютеинизация
предовуляторного фолликула без овуляции с циклическими изменениями
секреции прогестерона и несколько запоздалой секреторной трансфор­
мацией эндометрия; основное клиническое проявление — б. • Синдром
поликистоза яичников — склерокистозная патология яичников, обычно
проявляющаяся гирсутизмом, ожирением, нарушением менструаций, б.
и увеличением яичников; вызвана врожденной или приобретенной недо­
статочностью ферментных систем (превращение андрогенов в эстрогены
с повышением уровня первых). • Недостаточность пролактина (264110,
р). П ризнаки — послеродовая недостаточность лактации, нерегулярные
менструации, б., отсутствие секреции пролактина после стимуляции фенотиазинами. • Синдром резистентных яичников. Признаки — аменорея, б.,
нормальное развитие вторичных половых признаков, макро- и микро­
скопически не измененные яичники и высокий уровень гонадотропинов;
наблюдают у женщин моложе 35 лет. • Синдром Тернера. • Шеечная
слизь, препятствующая проникновению сперматозоидов, часто бывает
причиной б. Плохое качество шеечной слизи может быть результатом
неадекватного действия эстрогенов или инфекции. Поэтому для лечения
можно назначить малые дозы эстрогенов или антибиотики. При неэф­
фективности проводимой терапии применяют внутриматочное осеменение
или экстракорпоральное оплодотворение с последующей имплантацией
эмбриона. • Эктопическая беременность в анамнезе. Приблизительно 40%
женщин не могут забеременеть повторно. Из 60% вновь забеременевших
женщин у 12% повторно возникает внематочная беременность, а у 15—20%
происходит самопроизвольный аборт. • Эндометриоз. У 30—40% женщин
с эндометриозом регистрируют б. Эндометриоз у женщ ин при б. обнару­
живают с помощью лапароскопии в 15—20% случаев. Лечение может быть
хирургическим или медикаментозным, основано на характере нарушения
или нарушений, вызывающих бесплодие. Примечание. Для успешного
оплодотворения яйцеклетка должна встретиться со сперматозоидом в тече­
ние суток после овуляции. Из практических соображений время, в течение
которого овулировавшая яйцеклетка может быть оплодотворена, оцени­
вают в 72 ч. • Мужское б. — неспособность мужчины к оплодотворению
вследствие расстройств сперматогенеза, эрекции или эякуляции. В 1 мл
эякулята в норме содержится около 60 млн сперматозоидов. В женских
половых путях они сохраняют способность к оплодотворению до 6 сут.
Примерно 200 из них достигают воронки маточной трубы, где обыч­
но происходит встреча сперматозоида с яйцеклеткой. Сперматозоиды,
не участвующие в оплодотворении, удаляются из женских половых путей
или перевариваются фагоцитами. Терминология. • Олигозооспермия —
20 млн и менее сперматозоидов в 1 мл эякулята. • Азооспермия — полное
отсутствие сперматозоидов в семенной жидкости. • Первичная тестику­
лярная инфертильность характеризуется недостаточным сперматогенезом
(гипосперматогенез), его отсутствием (асперматогенез) или дефектами
сперматогенеза (например, блокадой на разных этапах). • Вторичное муж­
ское б. развивается в различных ситуациях (например, обструкция семявыносящих путей, недостаточность гипоталамических и/или гипофизарных
гормонов). Этиология. • Неподвижные сперматозоиды. О При синдромах
Картагенера и неподвижных ресничек сперматозоиды не передвигаются,
хотя такие мужчины потенциально фертильны. В этих случаях проводят
оплодотворение т жХго с последующим введением концептуса в матку.
❖Дефектная акросома (102530, 'Л, также X или полигенное) — отсутствие
способности к оплодотворению вследствие нарушенного формирования
акросом сперматид и сперматозоидов. По разным оценкам, эта патоло­
гия обусловливает до 15% мужского б. Сперматозоиды имеют головку
округлой формы, они подвижны, но невозможна акросомная реакция.
0 Азооспермия. Причины — дефекты сперматогенеза при мутациях гена
одного из факторов азооспермии (*415000, фактор 1 азооспермии, Уя 11, ген
А2Р, *400000, Уд, фактор 2 азооспермии, ген А2Р2, У-сцепленное насле­
дование), лучевое поражение, непроходимость выводящих путей мужской
половой системы. 0 Олигоспермия. Уменьшенный объем эякулята (<1 мл)
вследствие патологии яичек и придатков, семенных пузырьков, желез
(предстательной, Купера, Литтре). • Обратимое мужское б. при длитель­
ном приеме ряда ЛС и воздействии нелекарственных химических веществ.
0 Спиронолактон. 0 Сульфасалазин. 0 Антибластомные средства. 0 Кокаин.
0 Марихуана. 0 Химические вещества, применяемые в производстве и быту.
• Синдром Клайнфелтера среди мужчин, страдающих б., наблюдают с часто­
той 1:9. • Синдром мужского б. (#308370, мутации андрогенового рецептора):
азооспермия. Коррекция мужского фактора б. • Медикаментозная: коррекция
факторов, лежащих в основе нарушений (например, заболеваний щитовидной
железы, избытка пролактина, нарушений питания и др.). • Искусственное
осеменение спермой донора. • Хирургическая коррекция: восстановительная
операция после стерилизации. • Хирургическое лечение варикоцеле.
Билирубин — красный желчный пигмент, находящийся в виде натриевой
(растворимой в желчи) или кальциевой (нерастворимой в желчи) соли.
Продукт восстановления биливердина, образуется в результате нормально­
го и патологического разрушения эритроцитов. • Конъюгированный б. — см.
«Билирубин прямой». • Неконъюгированный б. — см. «Билирубин непря­
мой». • Непрямой б. — фракция сывороточного билирубина, не соеди­
нившаяся в клетках печени с глюкуроновой кислотой (назван так потому,
что реагирует с диазореактивом Эрлиха только после добавления эти­
лового спирта) <=> неконъюгированный б. <=> несвязанный б. <=> свобод­
ный б. • Прямой б. — фракция сывороточного билирубина, соединившаяся
в гепатоцитах с глюкуроновой кислотой с образованием диглюкуронида
билирубина (назван так потому, что напрямую реагирует с диазореактивом
Эрлиха) «• связанный б. о конъюгированный б. • Свободный б. — см.
«Билирубин непрямой». • Связанный б. — см. «Билирубин прямой».
Биопсия — прижизненное взятие небольшого объема ткани для микроскопи­
ческого исследования с диагностической целью. • Аспирационная б. В опу­
холь вводят тонкую иглу, клетки засасывают в иглу и помещают на пред­
метные стекла. • Инцизионная б. Удаляют поверхностную или доступную
часть опухоли. Проводят с целью установления диагноза перед началом
лечения. • Пункционная б. В опухоль вводят толстую иглу и берут столбик
ткани. Поскольку при п. б. берут гораздо больше ткани, чем при аспирационной, то более вероятны осложнения (например, кровотечение), но обра­
зец больше и диагноз точнее. • Хориона б. — процедура, осуществляемая
на 7—11-й неделе беременности с целью получить клетки для пренатальной
диагностики. • Эксцизионная б. Полностью удаляют небольшую отдельную
опухоль без широкого поля здоровых тканей; применяют, когда локальное
удаление не осложнит лечение.
Бластомикоз (общее название) — глубокие и системные микозы, вызываемые
дрожжевыми и дрожжеподобными грибами. Характеризуются развитием
хронических гнойных гранулем. Типичный представитель — ШазЮтусех
ёет аШ й к. Заболевание начинается как респираторная инфекция, рас­
пространяясь затем преимущественно в легкие, кости, кожу.
Блефарофимоз — короткая и узкая глазная щель.
Блокада сердца — патологическое замедление или полное прекращение про­
ведения импульса от синусно-предсердного узла на предсердия, предсердно­
желудочковый узел и нижележащие отделы проводящей системы. Высокие
степени б. с. характеризуются брадиаритмией, которая может вызвать голо­
вокружение, обмороки и внезапную смерть. Продолжительность зубцов
и интервалов ЭКГ превышает нормальные величины; характерно несоот­
ветствие между ритмом предсердий и ритмом желудочков. Классификация.
• Межпредсердная блокада. • Синоатриальная блокада. • АВ-блокада I сте­
пени. • АВ-блокада II степени. • АВ-блокада III степени. • Внутрижелудочковые блокады (блокады ветвей и ножек пучка Гиса). Этиология.
• Атеросклероз коронарных артерий. • Инфаркт миокарда. • Митральные
пороки сердца, вызывающие гипертрофию предсердий. • Интоксикация
сердечными гликозидами. • Лечение хинидином и другими антиаритмическими препаратами. • Гиперкалиемия. • Кардиомиопатии. • Ревмокардит.
• Эссенциальная артериальная гипертензия. • Гипотиреоз. • Сифилис.
• Протезирование сердечных клапанов. • Алкогольная интоксикация.
Проявления. Для б. с. характерны брадиаритмии, сочетающиеся с головокру­
жением или обмороками вследствие снижения сердечного выброса. Лечение.
Лекарственная терапия (подавление парасимпатических влияний и стимуля­
ция адренорецепторов). • Атропин — 0,5 мг в/в каждые 3 -5 мин (или каж­
дые 3 -4 ч) до общей дозы 2 мг. • Изопреналин (изадрин*) — по 2 ,5 -5 мг
под язы к 3—6 раз в сутки или 1—2 мг в 200—400 мл 5% раствора глюкозы
в/в капельно со скоростью 2 мкг/мин; затем дозу титруют до достижения
ЧСС 60-70 в минуту. • 5 мл 0,05% раствора в 200-400 мл 0,9% раствора №С1
в/в капельно со скоростью 10—20 капель в минуту. Хирургическое лечение.
При неэффективности атропина и тяжелом общем состоянии — временная
или постоянная электрокардиостимуляция. • Альтернирующая б. с.
Чередование периодов нормальной проводимости и периодов ее истощения
(проявляется более или менее регулярным чередованием нормальных желу­
дочковых комплексов ЭКГ с аберрантными или идиовентрикулярными).
• Арборизационная б. с. Нарушение перехода возбуждения с конечных раз­
ветвлений проводящей системы сердца на сократительный миокард (напри­
мер, при диффузных поражениях миокарда); проявляется на ЭКГ расшире­
нием желудочкового комплекса. • Атриовентрикулярные б. АВ-блока­
да — частичное или полное нарушение проведения возбуждения от
предсердий к желудочкам; возникает на разных уровнях проводящей систе­
мы сердца (область предсердно-желудочкового узла, предсердно­
желудочковый пучок, обе его ножки или правая ножка и оба разветвления
его левой ножки одновременно) <=> предсердно-желудочковая б. с. О АВ
неполная б. с. — замедление проведения возбуждения от предсердий к желу­
дочкам и/или выпадение отдельных комплексов сокращений желудочков.
О АВ полная б. с. Полное прекращение проведения возбуждения от пред­
сердий к желудочкам. При этом предсердия и желудочки сокращаются неза­
висимо друг от друга (АВ-диссоциация) <=> полный поперечный блок.
По клиническим проявлениям различают три степени АВ-блокады. • I сте­
пень — удлинение интервала Р—К (Р—О) более 200 мс из-за замедленного
проведения импульса через АВ-соединение. Причинами могут быть увели­
чение тонуса парасимпатической нервной системы, прием ЛС (сердечных
гликозидов, р-адреноблокаторов, верапамила, дилтиазема), а также пораже­
ния проводящей системы (фиброз, миокардит). • II степень подразделяется
на два типа. 0 тип Мобитца I характеризуется периодикой Венкебаха — удли­
нением интервала Р—К от сердечного цикла к циклу вплоть до прекращения
проведения импульса на желудочки и выпадения комплекса ()К5. Причины
возникновения этого типа АВ-блокады такие же, как при АВ-блокаде I сте­
пени. Дополнительно к этиологическим факторам относят инфаркт миокар­
да нижней стенки левого желудочка. 0 тип Мобитца II характеризуется
внезапным выпадением комплекса ()К8 без предшествующего удлинения
интервала Р—К (при этом интервал Р—К может быть как постоянно нормаль­
ным по продолжительности, так и постоянно удлиненным более 200 мс).
В этом случае блокада обычно возникает ниже АВ-соединения. Наиболее
частые причины этого типа АВ-блокады — инфаркт миокарда нижней стен­
ки левого желудочка, фиброз проводящей системы сердца (болезнь Лева),
хирургические вмешательства на сердце. АВ-блокада II степени 2-го типа
обычно имеет тенденцию к прогрессированию и переходу в АВ-блокаду III
степени. • III степень АВ-блокады характеризуется отсутствием проведения
импульса на желудочки. Ритм желудочкам задается из центров автоматизма
низшего порядка — желудочков. Частота сокращений желудочков обычно
составляет 35—50 в минуту. При редком ритме сокращения желудочков, неза­
висимо от степени АВ-блокады (II или III), возможны головокружение
и обмороки в результате ухудшения мозгового кровообращения (приступы
Морганьи—Адамса—Стокса). Лечение. При I степени АВ-блокады необходи­
мости в лечении нет. При II степени АВ-блокады (тип Мобитца II),
АВ-блокаде III степени при наличии симптомов (головокружение, обморо­
ки) показана установка электрокардиостимулятора. • Бифасцикулярная б. с.
Сочетание б. с. правой ножки пучка Гиса (предсердно-желудочкового пучка)
с блокадой одной из ветвей левой ножки или блокада обеих ветвей левой
ножки при сохранении проведения возбуждения по правой ножке.
• Внутрижелудочковые бб. Нарушение проведения импульса по левой
или правой ножкам пучка Гиса приводит к удлинению интервала (2К8.
Различают полную (интервал (2К.5 удлиняется более 0,12 с) и неполную
(ширина интервала ()К8 составляет 0,10—0,12 с) блокаду ножек пучка Гиса.
Блокироваться могут также ветви (передняя или задняя) левой ножки пучка
Гиса. Кроме того, блокада ножек пучка Гиса может быть постоянной или пре­
ходящей (перемежающейся). 0 Полная блокада правой ножки пучка Гиса
возникает чаще, чем блокада левой. Она может появляться и у здоровых
людей (без заболевания сердца). Причинами полной блокады правой ножки
пучка Гиса могут быть врожденные пороки сердца (например, дефект межпредсердной перегородки), приобретенные пороки сердца (например, стеноз
митрального отверстия), ИБС. Признаки. • Ш ирина комплекса ()К8 более
0,12 с. • Трехфазный (г8К ’) комплекс в отведениях V,—V-, с дискордантными
сегментом 8 Т и зубцом Т. • Ш ирокие зубцы 5 в отведении У6. 0 Неполная
блокада правой ножки пучка Гиса характеризуется комплексами типа г8К ’
в отведениях V,—У3 при нормальной длине интервала ()К8. 0 Полная блока­
да левой ножки пучка Гиса наиболее часто — признак органического пора­
жения сердца. Причинами ее могут быть ИБС, ГБ при длительном течении,
пороки аортального клапана, кардиомиопатии. Внезапное появление блока­
ды левой ножки пучка Гиса может быть одним из проявлений инфаркта
миокарда. ЭКГ-признаки. • Ш ирина ()К8 более 0,12 с. • Ш ирокие зубцы К
в отведениях У5, У6, I, аVI. с дискордантными сегментом 8 Т и зубцом Т.
• Низкоамплитудные (невысокие) зубцы К и глубокие зубцы 5 в отведениях
II, III, аУР. • Низкоамплитудные (маленькие, могут даже отсутствовать)
зубцы К и глубокие зубцы 5 в отведениях V,—У3. Сегмент 8 Т в этих отведе­
ниях может быть расположен выше изолинии. 0 Блокада ветвей левой ножки
пучка Гиса. Левая ножка пучка Гиса имеет две ветви. Передняя ветвь, более
длинная и тонкая, снабжается кровью из одного сосуда. Задняя ветвь пре­
вышает по толщине переднюю, ее кровоснабжение осуществляется двумя
сосудами. Это объясняет тот факт, что задняя ветвь левой ножки пучка Гиса
блокируется реже передней. Причиной блокады задней ветви левой ножки
пучка Гиса, как правило, выступает выраженная ИБС. Причинами блокады
передней ветви левой ножки пучка Гиса могут быть ИБС, кардиомиопатии.
кальцификация аортального клапана, преходящая гиперкалиемия. Изредка
блокаду передней ветви левой ножки пучка Гиса обнаруживают в норме.
Блокада ветвей левой ножки пучка Гиса обычно не приводит к расширению
комплекса ()К8, но выражается в резком отклонении электрической оси
сердца во фронтальной плоскости. ЭКГ-признаки блокады передней ветви
левой ножки пучка Гиса. • Отклонение электрической оси сердца влево (—45"
и менее). • Ш ирина комплексов (Ж 8 менее 0,1 с. • Маленькие зубцы К
и глубокие зубцы 5 в отведениях II, III, аУР, маленькие зубцы 0 в отведе­
ниях I, аУЬ. ЭКГ-признаки блокады задней ветви левой ножки пучка Гиса.
• Отклонение электрической оси сердца вправо (более +120°). • Ш ирина
комплексов ()К5 менее 0,10 с. • Маленькие зубцы 0 и высокие зубцы Я
в отведениях II, III, аУР, маленькие зубцы К в отведениях I, а\Ъ .
• Внутрипредсердная б. с. — нарушение проведения возбуждения в миокарде
предсердий (на ЭКГ проявляется только расширением и деформацией
зубца Р). • Входа б. с. Невозможность распространения возбуждения
на определенный участок миокарда вследствие того, что клетки этого участ­
ка временно или навсегда утрачивают способность проводить возбуждение.
• Выхода б. с. Невозможность выхода возбуждения за пределы определен­
ного участка миокарда вследствие того, что клетки этого участка временно
или навсегда утрачивают способность проводить возбуждение. • Левой ножки
пучка Гиса б. — полное прекращение проведения возбуждения по левой
ножке предсердно-желудочкового пучка или одновременно по ее передней
и задней ветви. См. «Блокада сердца внутрижелудочковая». • Монофасцикулярная б. с. Изолированная блокада правой ножки и/или одной из ветвей
левой ножки пучка Гиса (предсердно-желудочковый пучок) — см. «Блокада
сердца внутрижелудочковая». • Периинфарктная б. с. Транзиторная б. с.
в отделах проводящей системы сердца, прилежащих к некротизированному
участку при инфаркте миокарда. • Правой ножки пучка Гиса б. Полное пре­
кращ ение проведения возбуждения по правой ножке предсердно­
желудочкового пучка — см. «Блокада сердца внутрижелудочковая». • Пред­
сердно-желудочковая б. с. — см. «Блокада сердца атриовентрикулярная».
• Синоатриальная б. — замедление проведения импульсов из синусно­
предсердного узла к предсердиям или их блокирование на участке между
синусно-предсердным узлом и предсердием. Различают три степени с. б.
• I степень характеризуется задержкой проведения импульса от синусового
узла к предсердию. На ЭКГ ее не выявляют (определяют только с помощью
электрофизиологического исследования). • II степень может быть двух
типов. Тип 1-й характеризуется периодикой Венкебаха (постепенное укоро­
чение интервала Р —Р вплоть до укорочения очередного цикла). Тип 2-й
проявляется внезапным удлинением интервала Р—Р до расстояния, кратно­
го обычным интервалам Р—Р (например, 2 Р—Р, 3 Р—Р). • III степень —
остановка синусно-предсердного узла. В этом случае на ЭКГ регистрируется
изолиния, а затем активизируется нижележащий водитель ритма либо воз­
никает асистолия. С. б. возникает при электролитных нарушениях, воздей­
ствии ЛС (сердечные гликозиды, антиаритмические средства I класса)
или при изолированном поражении синусового узла. При выраженной брадикардии с соответствующими симптомами (головокружение, обмороки,
приступы Морганьи—Адамса—Стокса) рекомендуют установку электрокар­
диостимулятора. • Трифасцикулярная б. с. Постепенно развивающаяся пол­
ная АВ-б. с., при которой происходит последовательное вовлечение в про­
цесс всех разветвлений предсердно-желудочкового пучка.
Блокаторы кальциевых каналов (блокаторы медленных кальциевых каналов,
антагонисты кальция) — класс ЛС, способных ингибировать вход Са2+
в клетку. Вследствие своих фармакологических эффектов (отрицательный
инотропный, хронотропный и дромотропный) препараты имеют особое
значение в терапии сердечно-сосудистых заболеваний. Представители —
нифедипин, верапамил, дилтиазем и др. о блокаторы медленных каль­
циевых каналов <» антагонисты кальция.
Блот-гибридизация по Саузерну — идентификация участка Д Н К , содержа­
щего искомую нуклеотидную последовательность, путем гибридизации
разделенных гель-элекгрофорезом и фиксированных на твердом матриксе
фрагментов Д Н К с меченым комплементарным ДНК-зондом.
Болезни. • Аутоиммунные бб. — бб., в патогенезе которых основная роль отво­
дится аллергической реакции на аутоантигены: а1ореаа агеаш (гнездная
алопеция), витамин В 12-дефицитная анемия, синдром Ш мидта (аутоим­
мунный полигландулярный синдром II), СКВ, синдром Ш егрена, аутоим­
мунная гемолитическая анемия. • Витаминной недостаточности б. — син­
дромы, обусловленные недостаточным поступлением, а также повышением
потребности или нарушением усвоения витаминов на фоне нормального
содержания в пище. Клинические проявления развиваются медленно,
что существенно затрудняет диагностику. Чаще возникают сочетанные
формы недостаточности витаминов; изолированные формы наблюдаются
редко. Классификация. • По этиологии: экзогенные, или первичные гиповитаминозы возникают вследствие недостаточного содержания витамина
в пище; эндогенные, или вторичные гиповитаминозы связаны с наруше­
нием всасывания витаминов в Ж К Т или повышенным их потреблением.
• По степени недостаточности витамина: прегиповитаминоз, гиповитами­
ноз, авитаминоз. • По числу витаминов, недостаточность которых обусло­
вила клиническую картину, — моногиповитаминозы и моноавитаминозы,
полигиповитаминозы и полиавитаминозы (возникают гораздо чаще моногипо- и моноавитаминозов). Этиологические факторы. • Экзогенные.
О Длительное однообразное питание с недостаточным содержанием белка.
О Особое значение придают питанию консервированной пищей и сухими
продуктами, содерж ащ ими недостаточное количество витаминов.
О Тепловая обработка в процессе приготовления пищ и способна разрушить
или изменить структуру витаминов. Особенно чувствителен к высокой
температуре витамин С. • Эндогенные. О Беременность и период лактации.
О Чрезмерно низкая или высокая температура окружающей среды, регу­
лярно и /и л и в течение продолжительного времени воздействующая
на организм (в том числе в холодное время года — одна из причин сезон­
ности гиповитаминозов). Ф Длительное ф изическое или нервно­
психическое напряжение. Ф Период реконвалесценции при хронических
заболеваниях. ФДисбактериоз, в том числе вызванный приемом антибио­
тиков (например, тетрациклина). О Заболевания ЖКТ, сопровождающиеся
симптомами мальабсорбции и мальдигестии. О Нарушение проходимости
желчных путей (опухоль, закупорка камнем и др.), сопровождается пре­
кращением поступления желчи в кишечник, что обусловливает нарушение
всасывания жирорастворимых витаминов. О Тяжелые инфекционные забо­
левания. О Гельминтозы (повышают потребность в витаминах и нарушают
их всасывание). Факторы риска. • Алкоголизм. • Наркомания. • Паренте­
ральное питание. • Гемодиализ. • Другие гиповитаминозы. • Первый год
жизни. • Пожилой возраст. • Низкий социальный статус. • Длительное
применение слабительных средств. • Генетические заболевания (например,
абеталипопротеинемия). • Хирургические операции на органах ЖКТ.
Клиническая картина полиморфна, особенно в случае полигиповитаминоза.
Особенно часты слабость, ухудшение памяти, снижение трудоспособности,
нарушения сна, понижение аппетита, одышка при обычной физической
нагрузке. • Прегиповитаминоз (субнормальная обеспеченность витамина­
ми). Характерны общие проявления в виде слабости, повышенной утом­
ляемости, нарушения сна, аппетита. Наличие витаминной недостаточно­
сти в этой стадии можно обнаружить только при применении лабораторных
методов исследования. • Гиповитаминоз (относительный дефицит витами­
на или витаминов). Проявляется характерными для того или иного вида
витаминной недостаточности клиническими симптомами. Диагноз под­
тверждают лабораторными исследованиями [определение содержания
витамина в сыворотке или в цельной крови (часто более информативно),
экскреция витамина или продуктов его метаболизма с мочой]. • Авитаминоз.
К райняя степень витаминной недостаточности вследствие полного
или почти полного отсутствия поступления витамина в организм.
Проявляется характерной симптоматикой и значительным снижением
содержания витамина в организме. Лечение обычно амбулаторное.
Госпитализация показана при тяжелом течении. Тактика ведения. • Лечение
основного заболевания. • Пероральное или парентеральное возмещение
витаминов. Диета • При гиповитаминозах, связанных с недостаточным
поступлением витамина или витаминов с пищей, необходима консульта­
ция диетолога. • Воздержание от употребления алкогольных напитков.
Примечание: см. также статью «Гиповитаминоз». • Врожденные — бб.,
проявившиеся при рождении (чаще, но не обязательно наследственные).
• Идиопатические — бб., причина которых не ясна. • Коллагеновые бб. —
группа генерализованных бб., поражающих соединительную ткань и харак­
теризующихся фибриноидным некрозом или васкулитом. К к. бб. относят
СКВ, системную склеродермию, ревматоидный артрит, ревматическую
лихорадку, узелковый периартериит и дерматомиозит. Термин «коллагено­
вые бб.» не точен, так как не показано, что первично или преимуществен­
но поражаются именно коллагены (речь идет скорее об использовании
понятия «коллаген» как синонима термина «соединительная ткань») о
коллагеново-сосудистые бб. о коллагенозы. • Лизосомные (иногда «бб.
накопления») — группа наследственных бб., характеризующихся унаследо­
ванной недостаточной продукцией лизосомных ферментов. • Мультифакториальные (многофакторные) — бб., которые развиваются в результате
взаимодействия определенных комбинаций аллелей разных локусов и спе­
цифических воздействий факторов окружающей среды. • Обструктивные
бб. легких развиваются при нарушениях функции внешнего дыхания,
характеризуются ухудшением проходимости дыхательных путей и возрас­
танием сопротивления воздушному потоку. • Накопления бб. (болезни
депонирования, тезаурисмозы) — наследственные болезни, вызванные
наруш ениями обмена, проявляю щ иеся прогрессирующим отложением
веществ определенного типа в клетках различных тканей, например гликогенозы. ОЛизосомные н. бб. характеризуются неспособностью ферментов
лизосом (вследствие дефектов их синтеза или структуры) расщеплять мета­
болиты. Так, болезнь Тея—Сакса развивается вследствие недостаточности
гексозаминидазы А (в нейронах накапливается моносиалоганглиозид);
синдром Хюрлер (Гурлер): недостаточность а-/,-идуронидазы (во многих
тканях и органах возрастает количество гликозаминогликанов); гликогеноз
типа 2: недостаточность лизосомальной а-1,4-глюкозидазы (избыточное
содержание гликогена в сердце, скелетных мышцах, печени и мозге).
ОПероксисомные н. бб. — наследственные бб. обмена веществ, обусловлен­
ные нарушением биогенеза или функции пероксисом. Синтез плазмалогенов недостаточен, нарушена организация органоидов или пероксисомы
отсутствуют полностью. К этой группе болезней отнесены синдром
Цельвегера, болезнь Рефсума, адренолейкодистрофия новорожденных,
гиперпипеколическая ацидемия. При этом в крови повышено содержание
фитановой кислоты и длинноцепочечных жирных кислот. При хондродисплазии точечной ризомелической, некоторых формах ихтиоза и наследу­
емой катаракты происходит накопление фитановой кислоты, но не длин­
ноцепочечных жирных кислот. • Наследственные — бб., для которых
этиологическим фактором является генная, хромосомная или геномная
мутация. • Семейные — бб., наблюдающиеся у нескольких членов семьи
в одном или нескольких поколениях. • Соединительной ткани бб. — группа
генерализованных бб., поражающих соединительную ткань, особенно бб.,
для которых не характерно менделевское наследование. Типичны ревмати­
ческая лихорадка и ревматоидный артрит. Сюда же относят коллагеновые
бб. • Хромосомные бб. — большая группа заболеваний (более 300 синдро­
мов), вызванных аномалиями в количестве или структуре хромосом.
Патологические изменения при х. б. включают дупликации, делеции
и транслокации генетического материала. Классификация. • Геномные
аберрации [аномальное количество хромосом, кратное гаплоидному набо­
ру (23п)]. 0 Триплоидия (у живорожденных) — см. «Триплоидии».
0 Тетраплоидии (спонтанное прерывание беременности). • Числовые ано­
малии (аномальное количество хромосом, не кратное гаплоидному набору)
возникают вследствие нарушений в расхождении хромосом при мейозе
(как правило, у лиц со структурно поврежденными хромосомами). По мере
взросления женщ ины риск рождения у нее ребенка с числовыми аберра­
циями увеличивается (особенно резко он возрастает после 35 лет).
0 М оносомии аутосомные (наблюдают крайне редко). 0 Трисомии аутосомные: 0 синдром Дауна (трисомия 21), ® синдром Эдвардса (трисомия 18),
0 болезнь Патау (трисомия 13), 0 трисомия 8. 0 М оносомии по половым
хромосомам: 0 синдром Тернера (45,X), 0 моносомии по У-хромосоме
не бывает. ❖Трисомии (и полисомии) по половым хромосомам: 0 трисомия
и полисомия X (47,XXX), 0 синдром Клайнфелтера (47,ХХУ), 0 полиУ-синдром — случайная находка, не имеющая клинического значения.
• Структурные аномалии (нормальное количество хромосом со структур­
ными изменениями хотя бы в одной из них). Все виды структурных ано-
малий, проявляющихся клинически (кроме нарушений репродуктивной
функции), можно охарактеризовать как частичные моносомии, частичные
трисомии или их сочетания. Виды структурных аномалий, не ведущие
к утрате или избытку хромосомного материала, считают сбалансированны­
ми, но они могут вызывать аномальное расхождение хромосом в мейозе
и приводить к нарушению репродуктивной функции. О Делеции (потеря
хромосомного материала) приводят к частичным моносомиям: ® синдрому
кошачьего крика, ® синдрому Вольфа—Хиршхорна, ® синдрому делеции
короткого плеча хромосомы 9 (всего описано около 70 случаев. Отмечают
тригоноцефалию , остальные признаки напоминаю т синдром Дауна.
Прогноз для жизни благоприятный), ® синдрому 11р-----делеция корот­
кого плеча хромосомы 11, особенно участка 11р13; проявляется врожден­
ным отсутствием радужки (аниридия) и часто сочетается с опухолью
Вильмса, ® синдром 13я---- делеция длинного плеча хромосомы 13; поми­
мо множественных аномалий развития, сопровождается ретинобластомой,
вызванной утратой гена-супрессора, ® другим синдромам делеций. Описаны
делеции как коротких, так и длинных плеч хромосомы 18, приводящие
к различным степеням задержки умственного развития и черепно-лицевым
аномалиям. Утрата материала хромосом 19, 20, 21 и 22 обычно связана
с образованием кольцевых хромосом. Синдромы, связанные с 21д- и 22<\-,
наиболее часты и могут иметь сходство с синдромом Дауна. Ф Дупликация
(вставка) — синдромы частичных трисомий вследствие вставки нового
хромосомного материала. Ф Инверсия и транслокация. Как инверсии, так
и транслокации представляют собой перенос генетического материала
с одного места на другое; в первом случае этот перенос касается только
одной хромосомы, а во втором задействованы две или более хромосом:
® сбалансированная транслокация (полный перенос) не сопровождается
утратой или приобретением нового материала и не имеет клинического
значения, ® несбалансированная транслокация (частичный перенос),
например синдром Дауна, проявляется клинически.
Болезнь. • КЬ-ноль б., см. «Синдром ЯЬ-ноль». • Аддисона б. — эндокринная
патология, связанная с двусторонним поражением коры надпочечников
с выключением или уменьшением продукции ее гормонов; характеризует­
ся гиперпигментацией кожи и слизистых оболочек, исхуданием, артери­
альной гипотензией, нарушениями водно-солевого обмена. Частота —
приблизительно 4:100 ООО. Ж енщ ины страдают чаще мужчин. Этиология
и патогенез. • Первичная недостаточность коры надпочечников (собствен­
но болезнь Аддисона). Ф Недостаточность коры надпочечников аутоиммун­
ной природы. Ф Туберкулез. Ф Ятрогенные причины (двусторонняя адреналэктом и я, последствия длительной терапии глю кокортикоидами).
Ф Грибковые заболевания (гистоплазмоз, бластомикоз, кокцидиоидомикоз). 0 Саркоидоз. Ф К ровоизлияния в надпочечники. Ф Опухоли.
Ф Амилоидоз. Ф СПИД (за счет цитомегаловирусных и других инф екцион­
ных поражений надпочечников). Ф Сифилис. Ф Адренолейкодистрофия.
Сопровождается повы ш ением уровня АКТГ, вместе с а-м еланостимулирующим гормоном, обусловливающим гиперпигментацию кожных
покровов и слизистых оболочек, — отличительный признак болезни
Аддисона. • Вторичная недостаточность коры надпочечников, обусловлен­
ная недостаточностью гипофиза (дефицит АКТГ); в отличие от первичной,
никогда не сопровождается гиперпигментацией. Генетические аспекты.
Проявления болезни Аддисона наблюдаются при ряде наследуемых состо­
яний. • Адренолейкодистрофия. • Врожденная болезнь Аддисона (103230,
'Л): гиперпигментация кожи, гипернатриурия, гипокалиурия. • Гипоадренокортицизм семейный (*240200, р): рвота, пигментация кожи, судорожные
припадки, сосудистый коллапс, гипогликемия, гипонатриемия, гиперкалиемия. • Гипоплазия коры надпочечника семейная (*300200, Хр21.3—р 2 1.2,
дефекты гена ИАХ1, р). • Дефекты глюкокортикоидных рецепторов [* 138040,
5я31, ген С Ш , $Н; также *202200 (дефект гена МС2К, 18р11.2, р)]: нет отве­
та на АКТГ, артериальная гипертензия; тяжелая артериальная гипертензия
и гипокалиемический алкалоз у гомозигот. • Недостаточность глицеролкиназы. • Синдром Оллгрова. • Синдром полигландулярный аутоиммунный.
Факторы риска. • Недостаточность надпочечников аутоиммунной природы
у родственников (I или II степени родства). • Лечение глюкокортикоидами
в течение длительного времени, а также тяжелые инф екции, травмы
или хирургические вмешательства. Проявления. • Общие симптомы.
0 Выраженная слабость. О Утомляемость. 0 С нижение массы тела.
0 Артериальная гипотензия. О Головокружение. 0 Гиперпигментация кожи
и слизистых оболочек (при первичной недостаточности). 0 Анорексия.
0 Рвота. 0 Диарея. 0 Плохая переносимость холода. • Возможны психиче­
ские расстройства (депрессия, психоз, спутанность сознания).
• Адреналовые кризы, возникающие вследствие острого инфекционного
заболевания, травмы, оперативного вмешательства, быстрой потери соли.
Симптомы криза: выраженная слабость, интенсивные боли в животе,
рвота, сосудистый коллапс, почечная недостаточность, изменения психи­
ческого статуса. Лабораторные исследования. • Низкая концентрация натрия
в плазме крови (менее 130 мэкв/л). • Высокая концентрация калия в плаз­
ме крови (более 5 мэкв/л). • Увеличение содержания азота мочевины
крови. • Гипогликемия. • Уменьшение содержания кортизола, повышение
уровня ренина (радиоиммунологическое исследование). • Увеличение
содержания АКТГ (при вторичной недостаточности — снижение)
• Уменьшение концентрации 17-гидроксикортикостероидов в моче
• Умеренные нейтропения и эозинофилия. Лечение. • Лечение недостаточ­
ности коры надпочечников с помощью заместительной терапии глюкои минералокортикоидами. • Соответствующее лечение основного заболе­
вания (например, туберкулеза). • Аддисона—Бирмера б. — см. «Анеми:
витамин В12-дефицитная». • Альцхаймера (Альцгеймера) б. — органическое
слабоумие, возникающее после 50 лет [различают раннее и позднее (после
65 лет) начало А. б.] и сопровождающееся альцгеймеровским склерозом
нейрофибриллярной дегенерацией (при участии т-белка микротрубочек!
старческими бляшками (отложения р-амилоидного белка, образовани.
амилоидных волокон) <-> деменция пресенильная (А. б. с ранним началом
• 104760). • Бадда-Киари б. — см. «Синдром Бадда—Киари». • Барракера-
Симонса б. — см. «Липодистрофия прогрессирующ ая сегментарная».
• Бехтерева б. — см. «Спондилит деформирующий». • Брайта б. (брайто­
ва б.) — устаревшее название гломерулонефрита. • Брутона б. — см.
«Агаммаглобулинемия». Вакеза б. (ро1усу(кет1а чет) — см. «Полицитемия».
• Вальденстрема б. — см. «Макроглобулинемия Вальденстрема». • Вестфаля—
Вильсона—Коновалова б. — см. «Дегенерация гепатолентикулярная». • Виб­
рационная б. — профессиональная б., которая вызывается воздействием
вибрации, передающейся через руки или поверхность опоры тела, и харак­
теризуется развитием ангиотрофоневроза, сопровождающегося другими
нарушениями функции ряда органов и систем о ангионевроз вибрацион­
ный о псевдо-Рейно болезнь <=> синдром «белых пальцев» <=> синдром
вибрационный. • Виллебранда б. — см. «Болезнь фон Виллебранда».
• Вильсона—Коновалова б. (рекомендуется Уилсона—Коновалова б.) — см.
«Дегенерация гепатолентикулярная». • Геморрагическая новорожденных б. —
патологическое состояние периода новорожденности, связанное с недо­
статочностью зависимых от витамина К факторов свертываемости крови.
• Гипертоническая б. — гипертензия артериальная эссенциальная. • Гоше б.
(230800, Ц21, ген СВА, р) — наследственное заболевание [в том числе
недостаточность глюкоцереброзидазы (кислая р-галактозидаза)], сопрово­
ждающееся накоплением глюкоцереброзидов в макрофагах с развитием
гепатоспленомегалии, лимфаденопатии, разруш ением костной ткани,
поражением ЦНС, отставанием в умственном развитии. Лечение: спленэктомия при кровотечениях, ортопедическая иммобилизация, пересадка
костного мозга, анальгетики о цереброзидоз, цереброзидный липидоз.
• Грейвса б. — зоб диффузны й токсический — см. «Гипертиреоз».
• Дауна б. — хромосомная болезнь, обусловленная трисомией по хромосо­
ме 21. Проявляется олигофренией, замедленным ростом, мышечной гипо­
тонией, недостаточностью эндокринных желез (особенно щитовидной)
и своеобразным внешним видом: косо расположенными глазными щ еля­
ми, широкой уплощенной переносицей, эпикантусом, полуоткрытым ртом,
короткими пальцами, маленькими круглыми ушами с выдающимся противозавитком, ш ирокими руками и ногами и поперечными складками
на ладонях. С возрастом увеличивается вероятность развития лейкоза
и болезни Альцгеймера <=> трисомия по хромосоме 2 1 <=> синдром Дауна.
• Дента б. — форма нефролитиаза вследствие мутации гена С1ХЛЧ5 (хлор­
ный канал, 300008), дефекты которого также ведут к развитию гипофосфатемического рахита (К, рецессив). • Деркума б. В подкожной клетчатке
формируются множественные болезненные липомы и/и ли очаги диффуз­
ного отложение жира, встречается преимущественно у ж енщ ин (5:1)
в менопаузе, сопровождается адинамией, астенией, депрессией. <=> АШрош
йоЪгоза
адипозалгия о Деркума синдром «• липалгия <=> липоматоз
болезненный о ожирение болезненное. • Желчнокаменная б. — б., харак­
теризующаяся образованием конкрементов в желчном пузыре или желч­
ных протоках. • Жильбера б. — см. «Желтуха семейная негемолитическая».
• Иценко-Кушинга б. — гиперкортицизм в связи с избыточной секрецией
АКТГ передней долей гипофиза (70% всех случаев гиперкортицизма).
Наиболее часто возникает в возрасте 20—40 лет. У женщ ин наблюдают
в 5 раз чаще, чем у мужчин. Этиология и патогенез обусловлены изменени­
ем контроля над секрецией АКТГ (что вызывается уменьшающимся инги­
бирующим воздействием гипоталамического дофамина на секрецию кортиколиберина и АКТГ), в дальнейшим — повышением содержания АКТГ
и глюкокортикоидов. • Секретирующая АКТГ базофильная или хромофоб­
ная аденома гипофиза. • Усиление синтеза АКТГ вызывает надпочечнико­
вые и вненадпочечниковые эффекты. Ф Надпочечниковые — повышение
содержания глюкокортикоидов и андрогенов сетчатого слоя коры надпо­
чечников. В меньшей степени АКТГ влияет на повышение концентрации
минералокортикоидов. Ф Вненадпочечниковые — гиперпигментация кожи
и слизистых оболочек (в 10% случаев) и психические расстройства.
• Повышение продукции глюкокортикоидов — ключевой момент патоге­
неза болезни И ценко—Кушинга. Эффекты. Ф Катаболическое действие
на белковый и углеводный обмен приводит к атрофии мышечной (в том
числе сердечной) ткани и кожи, а также к гипергликемии с развитием
стероидного диабета (20% случаев). В патогенезе стероидного диабета
также играют роль относительная недостаточность инсулина (с усилением
глюконеогенеза в печени) и инсулинрезистентность. Ф Анаболическое дей­
ствие на жировой обмен приводит к ожирению (более 90% случаев).
Ф М инералокортикоидная активность способствует активации системы
ренин—ангиотензин—альдостерон с развитием артериальной гипертензии
и гипокалиемии. Определенное значение в патогенезе артериальной гипер­
тензии имеет потенцирование кортикостероидами эффекта катехоламинов
и биогенных аминов (в частности, серотонина). Ф Вследствие катаболического воздействия на костную ткань снижается способность костной ткани
фиксировать кальций и развивается остеопороз (более 80% случаев).
Определенную роль в патогенезе остеопороза играет уменьшение кортико­
стероидами реабсорбции кальция в ЖКТ, что связано с торможением про­
цессов гидроксилирования кальциферола. Ф Подавление специфического
иммунитета влечет за собой вторичный иммунодефицит. • Повышение
секреции андрогенов (тестостерона) надпочечниками приводит к сниже­
нию гонадотропной функции гипофиза и развитию половых расстройств.
• Изменение секреции других тропных гормонов — снижение уровня СТГ
и ТТГ, увеличение содержания пролактина. Проявления. • Симптомы нару­
шения жирового обмена определяют кушингоидную внешность: Ф луноо­
бразное лицо (отложение жира на лице), Ф отложение жировой ткани
на шее (бизоний горб) и в верхней части туловища, но не на конечностях.
• Симптомы нарушения белкового обмена: Ф мышечная слабость — стеро­
идная миопатия. Ф Стероидная кардиопатия. Ф Полосы растяжения (стрии)
красно-фиолетового цвета на коже живота, груди и бедер, Ф предрасполо­
женность к возникновению синяков (в связи с повышенной ломкостью
капилляров). • Симптомы нарушения углеводного обмена — стероидный
диабет или снижение толерантности к глюкозе с быстро нарастающей
гипергликемией и/или глюкозурией. • Артериальная гипертензия. • Остео­
пороз. • Расстройства гормональной регуляции (аменорея, бесплодие,
гирсутизм, акне). • Образование трофических язв и гнойничковых пора­
ж ений кожи (вследствие развития вторичного иммунодефицита).
• Гиперпигментация кожи и слизистых оболочек (эффект АКТГ), обычно
в местах трения кожи — важный дифференциально-диагностический при­
знак. • Эмоциональная лабильность, депрессия, эйфория. • У детей —
задержка роста о гипофизарный базофилизм. Примечание. От болезни
И ценко-К уш инга следует отличать синдром И ценко-К уш инга. • Ишеми­
ческая б. сердца (ИБС) — заболевание, обусловленное несоответствием
между потребностью миокарда в кислороде и его доставкой, что приводит
к нарушениям функций сердца. Главная причина развития И БС (96% всех
случаев) — атеросклероз. Факторы риска ИБС подразделяются на изме­
няемые (модифицируемые) и неизменяемые (константные, немодифицируемые). • Константные: Ф возраст (чем он больше, тем сильнее выражен
атеросклероз и выше заболеваемость И БС ), 0 пол [до 55 лет заболеваемость
И БС среди мужчин в 3—4 раза больше, чем у женщ ин (исключение состав­
ляют женщ ины, страдающие артериальной гипертензией, гиперлипидемией, СД, при ранней менопаузе); после 75 лет заболеваемость И БС среди
мужчин и женщ ин одинакова], 0 семейный анамнез ИБС. Риск развития
И БС повышен: ® у близких родственников больного И БС (у родственни­
ков I степени родства: родителей, братьев, сестер, сыновей, дочерей — он
серьезнее, чем для родственников II степени родства: дядьев, теть, бабу­
шек, дедушек); ® при большом количестве больных И БС в семье;
® при возникновении И БС у родственников в сравнительно молодом воз­
расте. • М одифицируемые: 0 курение, Ф артериальная гипертензия,
Флипидный профиль: высокая концентрация общего холестерина, высокая
концентрация холестерина Л П Н П , низкая концентрация холестерина
ЛП ВП , высокое содержание триглицеридов, Ф гипергликемия и СД,
О малоподвижный образ жизни, Ф ожирение, Ф гипергомоцистеинемия
О дефицит эстрогенов (отсутствие заместительной терапии в менопаузе),
Ф применение гормональных противозачаточных средств. Классификация
ИБС. В России в практической деятельности продолжают использовать
классификацию И БС , разработанную сотрудниками ВКН Ц АМ Н СССР
в 1983 г. на основе предложений экспертов ВОЗ (1979). • Внезапная сердеч­
ная смерть (первичная остановка сердца). • Стенокардия: Ф напряжения,
в том числе стабильная и впервые возникшая; Ф прогрессирующая стено­
кардия напряжения — нестабильная; Ф спонтанная: покоя, вариантная,
Принцметала. • Инфаркт миокарда: Ф крупноочаговый (трансмуральный),
Ф мелкоочаговый (нетрансмуральный). • Постинфарктный кардиосклероз.
• Нарушения сердечного ритма. • Сердечная недостаточность. • «Немая»
форма ИБС. • Киари б. — тромбоз печеночной вены с мощным развитием
коллатерального кровотока, гепатомегалией, асцитом, портальной гипер­
тензией <=> Рокитанского б. <=> Бадда-К иари б. • Кистозная мозгового
вещества б. обусловлена мутацией гена ИРН1. Характеризуется наличием
большого количества кист на границе коркового и мозгового вещества,
атрофией канальцев и развитием склероза почек с последующей тяжелой
почечной недостаточностью. • «Кленового сиропа» б. (лейциноз, 248600) —
тяжелое наследственное заболевание (ацидоз, рвота, гипогликемия, судо­
рожные припадки, отставание умственного и физического развития, спец­
ифический запах мочи). Частота заболевания примерно 1:300 ООО. Биохимия:
известно несколько приводящих к развитию заболевания дефектов СЕ
митохондриального ферментного комплекса (см. «Дефекты катаболизма
лейцина»). При некоторых формах эффективен тиамин. • КрейтцфельдаЯкоба б. Спастический псевдосклероз с кортико-стриоспинальной дегене­
рацией; форма спонгиозной энцефалопатии, вызываемая медленными
вирусными инфекциями [обсуждается вопрос о передаче инфекционного
начала (прион) от крупного рогатого скота, страдающего губчатой энцеф а­
лопатией] и характеризующаяся слабоумием, миоклониями, атаксией
и другими неврологическими проявлениями (быстро приводит к коме
и смерти) <=> дегенерация кортико-стриоспинальная. • Крона б. — грану­
лематозная патология с вовлечением терминальных отделов подвздошной
кишки, реже — других отделов ЖКТ. Предрасположенность к развитию б.
связана с дефектами гена N 0 0 2 хромосомы 16. Характерны отдельные
глубокие язвы с возможным образованием фистул, сужение просвета
и утолщение стенок киш ечника за счет фиброза и лимфоцитарной инфиль­
трации, а также неказеозные туберкулоидные гранулемы в регионарных
лимфатических узлах <=> регионарный энтерит <=> регионарный илеит.
• «Курчавых волос» б. — см. «Синдром Менкеса». • Лаймская б. [по назва­
нию города Лайм (Ьуте , штат Коннектикут, США), в окрестностях кото­
рого б. была впервые обнаружена в 1980 г.] — трансмиссивное природно­
очаговое заболевание, передающ ееся клещ ами, с поражением кожи,
сердца, нервной системы и суставов <=> лаймский артрит о лаймоборрелиоз <=> Лайма б. • Лева б. — блокада ножки пучка Гиса у больных со здоро­
выми миокардом и коронарными артериями. Результат фиброза или каль­
цификации проводящей системы и вовлечения мембранозной перегородки,
верхушки мышечной перегородки, колец митральных и аортальных клапа­
нов. Проявляется в виде аритмий, полиморфных и политопных экстраси­
стол, обмороков. Описано несколько форм: Л, 115000, 108770 — предсерд­
ная кардиомиопатия с блокадой, *115080. • Легионеров б. — наиболее
распространенная клиническая форма легионеллеза, которая протекает
в виде тяжелой токсической пневмонии, вызываемой Ь. рпеиторкПа.
• Легких обструктивная хроническая б. — хроническое, медленно прогрес­
сирующее заболевание, характеризующееся необратимой или частично
обратимой (например, при применении бронходилататоров) обструкцией
бронхиального дерева. • Леффлера б. — утолщение эндокарда или инфиль­
трация миокарда (амилоидоз, гемосидероз и др.), сопровождаемые гибелью
кардиомиоцитов, компенсаторной гипертрофией и фиброзом, что приво­
дит к нарушению функций предсердно-желудочковых клапанов. Стенки
желудочков становятся нерастяжимыми, повышается давление наполнения
желудочков. Распространение процесса на проводящую систему сердца
ведет к возникновению аритмий <=> эндокардит Леффлера <=> эндокардит
фиброзный <=> эндокардит фибропластический <=> эндомиокардиальная
болезнь. • Миеломная б. — см. «Миелома множественная». • Олбрайта б. —
наследуемое заболевание, обусловленное нарушением регистрации тканя­
ми гормонов паращитовидных желез (резистентность к ПТГ), часто соче­
тающееся с СД, артериальной гипертензией, артериитом, полиартрозом <=>
Олбрайта наследственная остеодистрофия <=> М артина—Олбрайта псевдогипопаратиреоидный синдром. • Нестабильного НЬ б. — гемоглобинопатия,
обусловленная точечны ми мутациями генов глобинов. Развивается
при дефектах, относящихся к гидрофобным областям молекулы НЬ (напри­
мер, НЬ Цюрих). • Нимана—Пика б. — липоидный гистиоцитоз с накопле­
нием фосфолипидов (главным образом, сфингомиелина) в макрофагах
печени, селезенки, лимфатических узлах и костном мозге; вовлечение
головного мозга (клетки базальных ганглиев) возможно в поздних стадиях.
Известно множество клинических форм, в том числе развивающиеся
при недостаточности сфингомиелиназы (257200, сфингомиелин фосфодиэстераза, КФ 3.1.4.12, 11р15.4-р15.1, ген 5М Р 01). Лечение: спленэктомия, пересадка костного мозга <=> сфингомиелиновый липидоз о фосфатидоз <=> гистиоцитоз липоидный о липоидоз фосфатидный <=> ретикулез
нелейкемический <=> ретикулоэндотелиоз метаболический о спленогепатомегалия липоидно-клеточная о сфингомиелиноз. • Олпорта б. (наслед­
ственный нефрит) — прогрессирующая нефропатия с развитием почечной
недостаточности; глухота, связанная с поражением слухового нерва, иногда
присоединяется нарушение зрения. Обычно постепенно развивается почеч­
ная недостаточность и смерть в подростковом возрасте. Этиология: крайняя
гетерогенность мутаций, в том числе СЕ коллагенов (р) типа 4 (а 3), а 3;
также $К, К. • Паркинсона б. — см. «Паркинсонизм». • Педжета б. (остоз
деформирующий, остеодистрофия деформирующая, остит деформиру­
ющий) — болезнь, характеризующаяся деформацией бедренных и больше­
берцовых костей, позвоночника и черепа с выраженным гиперостозом,
утолщением и искривлением костей. • Поликистозная болезнь почек —
врожденное заболевание, характеризующееся образованием и ростом кист
в обеих почках (обычно не менее 4—5 кист). Кисты могут располагаться
в кортикальном или мозговом слое почек. В последнем случае говорят
о нефроноф тизе. • Реклингхаузена б. — см. «Нейрофиброматоз».
• Рефсума б. — пероксисомная болезнь (р) накопления (б. накопления
фитановой кислоты, дефекты генов пероксисом РЕХ1, РНУН, РАЯХ).
Проявления — пигментный ретинит, миоз, птоз, атаксия, аносмия, глухота,
демиелинизирую щ ая полиневропатия, увеличенное содержание белка
в СМ Ж, изменения ЭКГ, ихтиоз; при ювенильной форме выражено отста­
вание в умственном развитии. Характерны лицевой дисморфизм (плоское
лицо), гепатомегалия, стеаторея, остеопороз. Диета с исключением фита­
новой кислоты и плазмаферез приостанавливают прогрессирование невро­
логических нарушений. • Симмондса б. — первичная гипофизарная недо­
статочность, при которой снижена секреция большинства гипофизарных
гормонов, с вторичной гипофункцией периферических эндокринных
желез. Причины — гипо- или атрофия передней доли гипофиза, недоста­
точность гипоталамических либеринов, травмы, инфекция (сепсис, энце­
фалиты, туберкулез, сифилис), сосудистые нарушения (спазм, коллапс).
Клиническая картина зависит от степени снижения функций гипофиза.
Резкое снижение массы тела — доминирующий признак заболевания <=>
кахексия гипофизарная <=>пангипопитуитаризм о кахексия диэнцефальногипофизарная <=> болезнь Симмондса—Глинского. • Сывороточная б. —
аллергическая б., вызываемая парентеральным повторным введением
чужеродной сыворотки крови или препаратов, содержащих ее белковые
компоненты (антитоксические сыворотки, вакцины и др.). Основу патоге­
неза составляют аллергические реакции типа I и III. Проявляется лихорад­
кой, болью в суставах, эритемой и увеличением лимфатических узлов.
• Такаясу б. — прогрессирующий облитерирующий артериит сосудов, отхо­
дящих от дуги аорты <=> Такаясу синдром <=> коарктация аорты инвертиро­
ванная <=> панартериит множественный облитерирующий <=> синдром дуги
аорты о отсутствия пульса б. • Танжье б. (*205400, дефект посттрансляционной модификации, р) — наследуемая недостаточность ЛПВП в сочета­
нии с низким содержанием апоЛП А, (синтез последнего не нарушен),
накоплением пенистых клеток, содержащих холестериновые эфиры, уве­
личением и яркой гиперемией миндалин, гепатоспленомегалией, лимфаденопатией, гиперхолестеринемией о анальфалипопротеинемия. Приме­
чание. Танжье (Тап^ег) — остров в Чесапикском заливе (Вирджиния,
СШ А), где впервые зафиксирована болезнь. • Тея—Сакса б. — см.
«Ганглиозидоз». • Тяжелых цепей 1§ б. относят к группе парапротеинемических гемобластозов, вызванных нарушением синтеза 1§ с появлением
в крови и моче атипичного белка (парапротеин), представленного фраг­
ментами тяжелых цепей (тц) 1§ одного из классов — а , у или ц. Класси­
фикация. • Болезнь а-тц — иммунодефицит в сочетании с выраженной
мальабсорбцией и выявляемым в сыворотке крови белком, реагирующим
с антисывороткой к а-тц. • Болезнь у-тц — иммунодефицит в сочетании
с лимфаденопатией, гепатоспленомегалией, фиброзом легких, ревматоид­
ным артритом и наличием в крови и моче димера патологически изменен­
ных у-тц <=> моноклональная гаммапатия <=> болезнь Франклина. • Болезнь
ц-тц — редчайшая форма болезней тц, протекающая с гепатоспленомега­
лией, впервые обнаруженная у пациентов с длительно текущим ХЛЛ; диа­
гноз установлен с помощью иммуноэлектрофореза при обнаружении ком­
понента, реагирующего с антисывороткой к ц-тц. • Вильсона б. (Виль­
сона-Коновалова б.) — см. «Дегенерация гепатолентикулярная». • Уипла
б. — редкая б. системного характера, которой свойственны стеаторея, часто
генерализованная лимфаденопатия, артрит, лихорадка и кашель. Возможный
инфекционный агент — Тгоркегута м/ЫрреШ о интестинальная липодистрофия. • Фабри б. — наследственная недостаточность а-галактозидазы
(*301500, КФ 3.2.1.22, X), при которой в стенке кровеносных сосудов про­
исходит накопление перегруженных нейтральными гликолипидами клеток,
а в коже — формирование образований типа ангиом. Смерть наступает
в результате осложнений со стороны сердца, почек, мозга <=> ангиокератома диффузная туловища о липоидоз дистопический наследственный <=>
Фабри ангиокератома о Фабри синдром. • Фон Виллебранда б. [* 193400,
12рГег—р12, дефекты генов УТУР, Р8У]УР, 9?; *277480, р; * 177820, дефект
рецептора фактора ф. В. (ффВ)] — врожденное отсутствие мультимерных
форм ффВ (VIII: К, ффВ), необходимых для агрегации тромбоцитов.
Известно несколько клинических форм. Проявления. Геморрагический
диатез при нормальном количестве тромбоцитов и нормальной ретракции
сгустка, частичная недостаточность фактора VIII: К. Тип III болезни имеет
более тяжелое течение, содержание фактора VIII: К резко снижено, воз­
можны аортальный стеноз и пролапс митрального клапана. Лечение: заме­
щ ение фактора VIII, кислота аминокапроновая <=> ангиогемофилия <=>
наследственная псевдогемофилия <=> сосудистая гемофилия <=> конститу­
циональная тромбопатия <=> капилляропатия геморрагическая <=> псевдоге­
мофилия сосудистая <=> пурпура атромбопеническая <=> пурпура атромбоцитопеническая <=> Ю ргенса синдром <=> фон Виллебранда—Ю ргенса
конституциональная тромбопатия. • Франклина б. — см. «Болезнь тяжелых
цепей 1§». • Хантера б. — см. «Мукополисахаридозы». • Хиппеля—Линдау б. — наследственная ('Л) болезнь, обусловленная аномалией развития
кровеносных капилляров, проявляющаяся ангиоматозом сетчатки в соче­
тании с ангиоретикулемами мозжечка, спинного мозга и других органов <=>
ангиоматоз цереброретинальный. • Хиршспунга б. — см. «Мегаколон врож­
денный». • Ходжкина б. — см. «Лимфогранулематоз». • Хашимото б. — см.
«Тиреоидит хронический аутоиммунный». • Хроническая гранулематозная б.
(р, К, *233700, *233710, *233690, *306400, Хр21.1; дефекты цитохрома Ь,
недостаточность нейтрофильного цитозольного фактора 1 и др.) — врож­
денное наруш ение п еревари ван ия ф агоцитированны х бактерий
полиморфно-ядерными лейкоцитами, приводящее к снижению резистент­
ности против инфекций о врожденный дисфагоцитоз. • Хенда (Хэнда)—
Шюллера—Крисчена б. [впервые болезнь Хенда—Ш юллера—Крисчена опи­
сал в 1865 и 1876 гг. английский патолог Томас Смит (8тШ Ткотаа,
1833—1909)] — генерализованный жировой гистиоцитоз костной ткани,
в особенности черепа. Сопровождается разрушением костей, эозинофиль­
ным лейкоцитозом, а также накоплением клеток, содержащих эфиры
холестерина. Клинически проявляется полиурией, экзофтальмом, очагами
деструкции костей (особенно черепа) о Ш юллера б. <=> Шюллера синдром
<=> гистиоцитоз X <=> гранулематоз липоидный <=> К ри счен а- Шюллера б.
<=> липогранулематоз. • Гурлер (Хюрлер) б. — см. «Мукополисахаридозы».
• Центрального стержня б. — спорадическая миопатия, возникаю щ ая
в результате мутации гена рецептора рианодина. • Шарко—Мари—Тута б.
Группа семейных ('Л — тип 1, р — тип 2, имеются типы с К) нервно-мыш еч­
ных расстройств в виде прогрессирующей дегенерации мышечных волокон
в дистальных мышцах конечностей, начинающейся с мышц ног; слабые
или отсутствующие глубокие сухожильные рефлексы, сенсорная полинев­
ропатия, гипергидроз, сердечная блокада, диарея, тошнота; дегенерация
мотонейронов спинного мозга, уменьшение скорости проведения по
нервам, сегментарная демиелинизация. Тяжесть течения зависит от типа
наследования. Классификация: тип 1В (мутации белка миелина Р0), тип 1А
(мутации белка 22 миелина), типы 2 и X (мутация белка щелевых контак­
тов — коннексина-32, тип 4 (мутация белка 2 миелина), тип 5 (характерны
пирамидные знаки), также другие формы <=> Ш арко—М ари мышечная
атрофия <=> мышечная перонеального типа атрофия <— амиотрофия наслед­
ственная невральная.
Боррелиоз — заболевание, вызываемое спирохетами рода ВоггеЫа (например,
лаймоборрелиоз — лаймская болезнь).
Брадикардия — пониженная ЧСС. • Синусовая б. (СБ) обусловлена наруше­
нием способности синусно-предсердного узла генерировать электрические
импульсы с частотой более 60 в минуту. У 25% здоровых молодых мужчин
ЧСС составляет от 60 до 50 в минуту, во время сна происходит снижение ЧСС
на 30%. Классификация. • Экстракардиальная СБ (нейрогенная). Причины —
массаж каротидного синуса, давление на глазные яблоки (рефлекс Ашнера),
повышение внутричерепного давления (например, менингит, ушиб мозга,
субарахноидальное кровоизлияние, отек мозга), синдром Меньера, интуба­
ция, язвенная болезнь желудка и двенадцатиперстной кишки, микседема.
• Органическая СБ: атеросклероз коронарных артерий, инфаркт миокарда,
миокардиты, дегенеративные и фиброзные изменения в синусовом узле
(см. «Синдром слабости синусно-предсердного узла»). • Лекарственная СБ:
хинидин, (3-адреноблокаторы, симпатолитические препараты (например,
резерпин), блокаторы кальциевых каналов (например, верапамил, нифедипин), сердечные гликозиды, морфин. • Токсическая СБ: сепсис, желтуха,
уремия, брюшной тиф, отравление фосфорорганическими соединениями.
• СБ спортсменов: ЧСС в покое 40-35 в минуту даже в дневное время.
Причина — особенности нейровегетативной регуляции сердечного выброса
у людей, занятых тяжелой физической работой или профессиональным
спортом. Проявления зависят от выраженности СБ, величины ударного
объема, состояния вегетативной нервной системы и/или характера основ­
ного заболевания. • ЧСС менее 40 в минуту характернее для АВ-блокады,
чем для СБ. • Активация эктопических центров автоматизма — предсердные
и желудочковые аритмии. • Приступы Морганьи—Адамса—Стокса при паузе
перед началом функционирования нижележащего центра автоматизма дли­
тельностью более 10—20 с. ЭКГ-идентификация — ЧСС менее 60 в минуту,
каждому зубцу Р соответствует комплекс О ЯЗ. Характерно частое сочетание
СБ с дыхательной аритмией. Лечение. При сочетании умеренной СБ с арте­
риальной гипотензией — препараты красавки, например капли Зеленина,
беллатаминал* (противопоказаны при глаукоме).
Брадикинин — нонапептид А р г -П р о -П р о -Г л и -Ф е н -С е р -П р о -Ф е н -А р г ,
получаемый из декапептида (каллидина II, брадикининогена), который,
в свою очередь, синтезируется из а 2-глобулина под действием калликреинов. Б. — один из кининов плазмы, потенциальный вазодилататор, один
из физиологических медиаторов анафилаксии; высвобождается из туч­
ных клеток, покрытых цитофильными АТ при взаимодействии последних
со специфичным Аг (аллергеном).
Брахидактилия — укорочение пальцев рук или ног.
Бронхоэктазы — необратимое патологическое расширение бронхов в результате
гнойно-воспалительной деструкции бронхиальной стенки. • Ателектатический б. — б., развивающийся в зоне обширных ателектазов легких
и характеризующийся равномерным расш ирением многих бронхиаль­
ных ветвей из-за «эффекта клапана» в период дистелектаза (неполного
ателектаза). При этом паренхима легких приобретает вид пчелиных сот.
• Веретенообразный б. — расширенная часть просвета бронха постепенно
переходит в бронх обычного калибра.
Бруцеллез — инф екционная болезнь из группы бактериальных зоонозов,
вызываемая микроорганизмами рода ВгисеИа, передающаяся от больных
животных человеку алиментарным или контактным путем. Обычно про­
текает по типу хрониосепсиса с полиморфной клинической картиной,
рецидивами и обострениями.
Бужирование — введение бужей в некоторые органы трубчатой формы (моче­
испускательный канал, пищевод и др.) с диагностической или лечебной
целью.
Булимия (от греч. Ьиз — бык, Птоз — голод; голод «волчий», кинорексия) —
расстройство в виде повторных и неконтролируемых приступов погло­
щения большого количества пищ и в течение короткого периода времени
с последующими вызыванием рвоты, очищением кишечника и анорекси­
ей. Сопровождается чувством вины, депрессией, заниженной самооцен­
кой. Стертая б. возможна как симптом некоторых заболеваний (преиму­
щественно психических), состояния гипогликемии. Б. наблюдается у 4%
женщ ин и 0,5% мужчин, преимущественно в молодом возрасте.
Буллэктомия — удаление булл. Применяют при лечении некоторых форм
буллезной эмфиземы, при которых гигантские буллы сдавливают здоровую
легочную ткань.
Бурсит — воспаление синовиальной сумки, сопровождающееся скоплением
в ее полости экссудата.
Вазодилататор — средство (в том числе ЛС), расширяющее просвет крове­
носного сосуда и снижающее АД. Атриопептин, оксид азота (N 0 ), У1Р,
брадикинин и некоторые ПГ — естественные вазодилататоры.
Вазопрессин (аргинин-вазопрессин, антидиуретический гормон — АДГ,
С46Н65N 1501282.) — нанопептид, оказываю щ ий антидиуретический
(регулятор реабсорбции в канальцах почки) и сосудосуживающий (вазокон­
стриктор) эффекты. Ген АВР кодирует АДГ и нейрофизин II. Экспрессия
АДГ происходит в части нейросекреторных нейронов околожелудочкового и надзрительного ядер гипоталамуса. Внегипоталамическая секреция
АДГ возможна в клетках злокачественных опухолей (например, овсяно­
клеточная карцинома легкого, рак поджелудочной железы). Секрецию
АДГ стимулируют гиповолемия через барорецепторы каротидной области,
гиперосмолярность через осморецепторы гипоталамуса, переход в верти­
кальное положение, стресс, состояние тревоги, а ингибируют алкоголь,
а-адренергические агонисты, глюкокортикоиды. Главная функция АДГ —
регуляция обмена воды (поддержание постоянного осмотического давле­
ния жидких сред организма), что происходит в тесной связи с обменом
натрия; другие функции — стимуляция гликогенолиза, сосудосуживающий
эффект, агрегация тромбоцитов. Рецепторы АДГ относятся к связанным
с О-белком трансмембранным гликопротеинам. Взаимодействие аргинин-
вазопрессина с его рецепторами обусловливает стимуляцию фосфолипазы
С, образование фосфатидилинозитола и увеличение содержания вну­
триклеточного Са2+. Выделено три подтипа рецепторов. • У1а-подтип
экспрессируется в разных клетках, реализуя многочисленные эффекты
аргинин-вазопрессина: гепатоциты (стимуляция гликогенолиза), сосуди­
стые ГМ К (сосудосуживающий эффект), тромбоциты (агрегация кровяных
пластинок). • У1Ь. Эти рецепторы обнаружены в аденогипофизе (модуляция
секреции АКТГ, р-эндорфина и пролактина). • У2. Подтип У2 экспрес­
сируется только в почке. Нефрогенный несахарный диабет (тип I, 304800,
Хд28, X, имеются и 'Л-формы) — следствие мутаций гена для рецептора
подтипа У2.
Вакцина БЦЖ — вакцинный штамм МусоЬас1епит Ъоухз пониженной вирулент­
ности. Используют для профилактики туберкулеза <-> вакцина К альметтаГерена о ВСС уассте, от ЬасШш Са1теПе—Оиепп.
Вальвулопластика — хирургическая реконструкция деформированного сер­
дечного клапана. • Катетерная чрескожная баллонная в. — расширение
суженного предсердно-желудочкового отверстия специальным баллоном
под высоким давлением.
Вальвулотомия — рассечение стенозированного сердечного клапана.
Вегетация. 1. Рост или разрастание тканей любого типа. 2. Патологические
наложения на эндокарде при инфекционном эндокардите, состоящие
из фибрина, тромбоцитов и бактерий.
Везикула — первичный морфологический элемент сыпи в виде пузырька (диа­
метром до 5 мм), наполненного серозным экссудатом.
Вектор — материал, применяемый для переноса в клетку генетической инфор­
мации, наиболее часто вирусы (ретровирусы, аденовирусы и др.); возмож­
ны и синтетические конструкты • для клонирования в. — любая небольшая
плазмида, фаг или ДНК-содержащий вирус, в которые может быть встрое­
на чужеродная ДНК.
Велоэргометрия — метод функционального исследования с применением
дозированной физической нагрузки, заключающейся во вращении ногами
или руками педалей велоэргометра. Применяется для выявления латентной
коронарной недостаточности, определения показателей функции внеш не­
го дыхания и т.п.
Вентиляция — движение газа в легкое и обратно. • Альвеолярная в. — объем
газа, выдыхаемый из альвеол за 1 мин. Расчет: частота дыхания, умножен­
ная на различие между дыхательным объемом и «мертвым» пространством
(Ут—Ус ); выражается в мл/мин. • Вспомогательная искусственная в. легких —
в. при сохраненном ритме, но недостаточном объеме естественного дыхания,
когда в легкие при вдохе нагнетают дополнительный объем газовой смеси о
вспомогательное дыхание. • Искусственная в. легких (ИВЛ) — периодическое
приложение механического или вручную создаваемого положительного давле­
ния к (газу) газам, находящимся в пределах дыхательных путей, чтобы запол­
нить газом легкие при отсутствии спонтанного дыхания <=> искусственное
дыхание. • Легочная в. — общий объем газа, вдыхаемый (V,) или выдыхаемый
(УЕ) за 1 мин; выражается в л/мин <=> минутный объем дыхания (МОД).
Вещество. • В. Р (от англ. ра'т — боль) — нейропептид из семейства тахикининов, продуцируемый как нейронами, так и ненервными клетками
и функционирующий как нейромедиатор (базальные ганглии, гипотала­
мус, спинной мозг, где в. Р передает возбуждение с центрального отростка
чувствительного нейрона на нейрон спиноталамического тракта; через
опиоидные рецепторы энкефалин из вставочного нейрона тормозит секре­
цию в. Р из чувствительного нейрона и проведение болевых сигналов).
В. Р. также усиливает проницаемость стенки сосудов кожи, осуществляет
вазодилатацию или вазоконстрикцию ГМ К артериол мозга, стимулирует
секрецию слюнных желез и сокращение ГМ К воздухоносных путей и ЖКТ.
В. Р. функционирует также как медиатор воспаления. • Межклеточное в. —
см. «Матрикс тканевый».
Взрыв респираторный (на примере активированного нейтрофила). В течение
первых секунд после стимуляции резко увеличивается поглощение кис­
лорода и быстро расходуется значительное его количество. Это явление
известно как респираторный (кислородный) взрыв. При этом образуются
токсичные для микроорганизмов Н20 2, супероксид О2* и гидроксильный
радикал ОН*.
Вирилизм — развитие у женщ ин мужских вторичных половых признаков.
Вирус. • Аденовирус — в. семейства Адепочтдае. Более 80 типов инфицируют
человека, вызывая инфекции верхних дыхательных путей, конъюнктивит,
гастроэнтерит, геморрагический цистит, серозные воспаления новорожден­
ных. • Ветряной оспы в. — герпесвирус, морфологически идентичный виру­
су простого герпеса. Вызывает ветряную оспу и опоясывающий лишай,
отнесен к подсемейству А1ркакегре^1пс1ае <=> опоясывающего лиш ая в.
• Гепатита вв., см. «Гепатит». • Герпеса в. — в. рода Негреш гш . Выделяют
8 типов; основное значение имеют: V в. простого герпеса типа 1 — воз­
будитель стоматита, герпетической лихорадки, кератоконъю нктивита,
менингоэнцефалита; V в. простого герпеса типа 2 — возбудитель герпеса
половых органов и герпеса новорожденных. • Гриппа вв. — вирусы семей­
ства Опкотухо\'1гШае, возбудители гриппа и гриппоподобных заболеваний.
Существуют типы А и В (род 1пЦиещ,ау1гш), вызывающие грипп типа А
и В, а также тип С, принадлежащий к отдельному роду и вызывающий
грипп типа С. • ЕСНО-вв. [от Еп(его Су1ора(ко§емс Нитап Огркап — энтероцитопатогенные сиротские вирусы человека; термин «сиротский» под­
разумевает отсутствие (при выделении) прямой связи с болезнью] — вв.
семейства Р кот аут дае, возбудители лихорадок и асептического менин­
гита. Некоторые серотипы, по-видимому, вызывают слабовыраженные
респираторные заболевания. • Иммунодефицита человека в. (ВИЧ) — воз­
будитель инфекции, проявляющейся развитием прогрессирующих нару­
ш ений иммунного реагирования в результате длительного циркулирования
вируса в лимфоцитах, макрофагах и клетках нервной ткани; следствие
ВИЧ-инфекции — СПИД; ВИЧ входит в состав подсемейства ЬеШтппае
семейства Ке1гоутйае. • Коксаки в. [Сохзаск1е (Коксаки), штат Нью -Й орк
(США); город, где впервые выделен представитель группы вирусов] —
группа пикорнавирусов сферической формы рода ЕМегоу1ги$. Вызывают
миозиты, параличи, отвечают за развитие инфекций с разнообразной
симптоматикой. Вв. типа А вызывают герпетическую ангину; вв. типа
В — возбудители эпидемической плевродинии; вв. обеих групп могут
вызывать асептический менингит, мио- и перикардит, ювенильный СД.
• Кори в. — РНКовый в. рода МогЬШтгиз (семейство Рагатухоутйаё),
возбудитель кори (аэрогенная передача). В. обладает гемагглютинирующими и гемолизирующими свойствами. • Краснухи в. — РНКовый в.
семейства тогавирусов рода КиЬтгив, возбудитель краснухи. • Парагриппа
вв. — вв. рода Рагатухоуггш, пяти типов. V Типа 1 — в. гемадсорбции
типа 2, возбудитель острого ларинготрахеита. V Типа 2 — в. псевдокрупа,
возбудитель острого ларинготрахеита или крупа, а также малых респира­
торных инфекций. V Типа 3 — гемадсорбции типа 1, кроме заболеваний
фарингитом, капиллярным бронхитом и пневмонией, вызывает спора­
дические респираторные инфекции. • Парвовирусы — род в. с мелкими
вирионами (18—26 нм), не имеющими наружной оболочки. Геном пред­
ставлен однонитчатой молекулой ДНК. В19-парвовирус (РУ В19) патоге­
нен для человека; вызывает артриты и артралгии, а также инфекционные
эритемы и апластические кризы при наличии у больного гемолитической
анемии. • Пикорнавирусы — семейство в., содержащих одноцепочечную
Р Н К и характеризующихся кубической симметрией капсида. Поражают
позвоночных, распространяются без переносчиков. Многие представители
п., относящиеся к энтеровирусам и риновирусам, патогенны для человека.
• Респираторно-синцитиальный в. — РНК-содержащий в. рода Рпеитотгиз,
возбудитель так называемых малых респираторных инфекций у взрослых,
бронхита и бронхопневмонии у детей. • Риновирусы — род кислотолабильных
в. семейства Ргсот т т йае, включает возбудителей острых респираторных
инфекций человека и ящура крупного рогатого скота. • Цитомегаловирус —
группа герпетических вв., поражающая людей. Многие из этих в. обладают
специфическим сродством к слюнным железам, образуют характерные
включения в цитоплазме или ядре; видоспецифичны <=> вирус цитомегалии
<=> герпесвирус 5 человека. • Эпштейна-Барр в. — в. герпеса, обнаруженный
в клеточных культурах лимфомы Беркитта. Возбудитель инфекционного
мононуклеоза (семейство Н егрет пйае, подсемейство Саттакегрезутпае)
<=> Беркитта лимфомы в.
Витамин (от лат. уИа — жизнь + атт — аминосодержащее вещество) —
см. также «Болезни витаминной недостаточности», «Гиповитаминозы»,
«Гипервитаминозы». • В2 — термонеустойчивый в., содержащийся в моло­
ке, дрожжах, зерне злаков. Получают также синтетическим путем; ростовое
вещество о тиамин. • В6 — пиридоксин и родственные соединения. • В12 —
цианокобаламин, антианемический в. — фактор кроветворения, поступает
с пищей и всасывается в тонкой кишке. В12 деметилирует фолаты, предот­
вращая их выход из клеток; участвует в синтезе ДНК. Для всасывания в. Вг
в кишечнике необходим (внутренний) фактор Касла, синтезируемый парие­
тальными клетками желудка. Фактор связывает в. В12 и защищает его от раз­
рушения ферментами. Комплекс внутреннего фактора с в. В12 в присутствии
ионов Са2+ взаимодействует с рецепторами энтероцитов дистального отдела
подвздошной кишки. При этом в. В |2 поступает в клетку, а внутренний
фактор высвобождается. Из эпителия кишечника в. В12 с помощью транскобаламина II транспортируется в костный мозг и печень (для запасания).
Транскобаламин II вырабатывается эпителиальными клетками кишечни­
ка. Отсутствие или недостаток внутреннего фактора приводит к развитию
витамин В12-дефицитной анемии. Алиментарный дефицит в. В12 в развитых
странах встречается редко; исключение составляют грудные дети матерей —
строгих вегетарианок. Обычная причина дефицита — нарушение процессов
всасывания; одна из причин — дифиллоботриоз (гельминтоз, вызванный
БгркуНоЬоГкпит Шит — лентецом широким). « Б — жирорастворимые сте­
роиды, необходимые для нормального развития костей и зубов, всасывания
кальция и фосфатов в кишечнике. Существуют ядерные рецепторы, связыва­
ющие активную форму витамина Э3 — кальцитриол. Все остальные соедине­
ния этой группы подвергаются различным модификациям для превращения
в активную форму: V провитамин Б3 — (Зр)-7-дегидрохолестерин, С27Н440 ,
мол. масса 384,65, в эпидермисе под влиянием УФ превращается в витамин
Б 3; V витамин Б3, 9,10-секохолестатриен-5,7,10 (19) -ол-Зр (активирован­
ный 7-дегидрохолестерин, холекальциферол), антирахитическое средство
животного происхождения (печень рыб и млекопитающих, мозг, яичный
желток), образуется в коже в результате фотолиза из провитамина Б 3; V кальшииол, 25-гидроксихолекальциферол, 25-гидроксивитамин Э 3, С ^ Н ^ О ^
мол. масса 400,65, промежуточный продукт биологического превращения
витамина 0 3 в кальцитриол, образуется в печени при гидроксилировании
по 25С (1а-гидроксилаза кальцидиола — монооксигеназа, превращающая
при участии 0 2 и НАДФН кальцидиол в кальцитриол. Недостаточность фер­
мента приводит к дефициту витамина Э и витамин 0-зависимому рахиту);
V провитамин Б2 — эргостерол, витамин О растительного происхождения;
V витамин Б2 (эргокальциферол, кальциферол), С28Н440 , мол. масса 396,66,
активированный эргостерол (образуется при облучении эргостерола УФ),
антирахитическое средство. В течение многих лет эргокальциферол был
стандартным препаратом витамина Б. Для полного эффекта необходимо
превращение в кальцитриол (стимулятор — ПТГ). Суточная потребность:
дети — 10 мкг холекальциферола (400 МЕ), взрослые после 25 лет — вдвое
меньше. Недостаточность витамина Б приводит к развитию рахита (у детей),
остеомаляции, остеопорозу, остеодистрофии. Гипервитаминоз вызывает раз­
витие токсического синдрома (анорексия, рвота, диарея), кальцификацию
мягких тканей (сердце, сосуды, почка, легкие). « К — общее название жиро­
растворимых термостабильных соединений, обладающих биологической
активностью филлохинона; содержатся в люцерне, свиной печени, рыбной
муке и растительном масле; важны для образования протромбина о антигеморрагический фактор.
Витилиго — очаговая депигментация кожи.
Волокно нервное. В зависимости от того, формируют ли шванновские клетки
вокруг осевого цилиндра миелин, выделяют безмиелиновые и миелинизированные нервные волокна. Скорость проведения возбуждения существен­
но зависит от диаметра и миелинизации нервного волокна (табл. п. 1).
Таблица п. 1. Классификация нервных волокон по диаметру и скорости проведения
Тип
А
а-мотонейроны
А
у-мотонейроны
В
Диаметр,
мкм
Скорость
проведе­
ния, м/с
Структуры
Соматические и висцеральные эфференты
12-20
70-120
Экстрафузальные мышечные волокна
2 -8
10-50
Интрафузальные мышечные волокна
<3
3-15
С
0,2-1,2
0,7-2,3
Преганглионарные аксоны к нейронам
вегетативных ганглиев
Постганглионарные аксоны для ГМК
и желез
Аа
Ар
12-20
А6
2 -6
4 -3 0
С
<2
0 ,5 -2
А
С
2 -1 2
<2
1а
12-20
1Р
II
12-20
6 -1 2
III
2 -6
4 -3 0
IV
<2
0 ,5 -2
6 -1 2
Кожные афференты
70-120
Рецепторы суставов
30-70
Тельца Пачини и осязательные рецепто­
ры
Осязательные, температурные и болевые
рецепторы
Болевые, температурные, некоторые
механорецепторы
Висцеральные эфференты
4 -7 0
Рецепторы внутренних органов
0 ,2 -2
Рецепторы внутренних органов
Мышечные афференты
70-120
Аннулоспиральные окончания мышеч­
ных веретен
70-120
Сухожильные органы Гольджи
30-70
Вторичные окончания мышечных вере­
тен
Окончания, отвечающие за болевое дав­
ление
Болевые рецепторы
По: БеМуег\У. ИеигоапаШту. — ВаШтоге: ХУПНатз а. \УПк1пз, 1988. — Р. 63.
Волчанка — термин, ранее прим енявш ийся для описания эрозий кожи
(как после волчьих укусов), сейчас (с определяющим словом) — для обо­
значения различных болезней. Согласно отечественной литературе, пред­
лагают такую классификацию: в. — в. обыкновенная (1ири.ч уи1%ат, она же
в. туберкулезная), в. красная (1ириз егу1кета1о$и$), в. ознобленная (1ирш
регто). • Обыкновенная в. — кожный туберкулез с характерными узелковы­
ми поражениями лица, особенно вокруг носа и ушей <=> в. туберкулезная
• Системная красная в. (СКВ) — воспалительное заболевание соединитель­
ной ткани с разнообразными проявлениями, часто сопровождающееся
лихорадкой, слабостью и утомляемостью, болями в суставах или артрита­
ми, напоминающими ревматоидный артрит; диффузными эритематозны-
ми поражениями кожи лица, шеи, верхних конечностей с дегенерацией
по типу разжижения базального слоя эпидермиса и его атрофией, с лимфоаденопатией, плевритом, перикардитом, поражением почечных клу­
бочков, анемией, гиперглобулинемией, положительным тестом на клетки
СКВ (ЬЕ-клетки), а также другими признаками аутоиммунного процесса
о диссеминированная красная в.
Время. • Активированное парциальное тромбопластиновое в. — в., необходимое
для получения фибринового сгустка после добавления к плазме кальция
и фосфолипидного реагента; используется для оценки внутренней сверты­
вающей системы. • Протромбиновое в. (ПВ) — в., необходимое для свер­
тывания после добавления тромбопластина и Са2+ в достаточном количе­
стве в кровь с нормальным содержанием фибриногена. При уменьшении
содержания протромбина п. в. увеличивается, о Квика в. • Свертывания
в. — в., необходимое для свертывания крови. • Тромбиновое в. — в., необ­
ходимое для образования фибринового сгустка после добавления тромбина
к цитратной плазме; увеличенное т. в. наблюдается у больных, получающих
гепарин натрия.
Галактоземия — врожденное нарушение метаболизма в виде галактоземии
и галактозурии, развития катаракты, гепатомегалии, отставания в умствен­
ном развитии. Характерны рвота, желтуха. Возможна нейросенсорная
тугоухость, гипогонадотрофный гипогонадизм, гемолитическая анемия.
П ричины — врожденная недостаточность галактокиназы (230200, КФ
2.7.1.6), галактозоэпимеразы (*230350, КФ 5.1.3.2) или галактозо-1-фосфат
уридилтрансферазы (*230400, КФ 2.7.7.10).
Гамартома (прогонобластома) — узловое опухолевидное образование, возника­
ющее в результате нарушения эмбрионального развития. Состоит из тех же
компонентов, что и орган, где г. находится, но отличается неправильным
расположением и степенью дифференцировки.
Гаммапатия моноклональная — см. «Болезнь тяжелых цепей 1§».
Ганглиозидоз — лю бая наследственная (р) болезнь, характеризую щ ая­
ся патологическим накоплением (особенно в нервной системе) ганглиозидов. • Г. СМ1 [230500, недостаточность С М]-р-галактозидазы
(КФ 3.2.1.23), характеризующаяся накоплением моносиалоганглиозида
С М|]. Заболевание клинически похоже на болезнь Тея—Сакса, но имеет
генерализованный характер; различают три типа генерализованного г.
(г. С М1). Ф Тип I (детский, *230500). К нему также относят мукополисахаридоз 1УВ (253010, 230500, синдром М оркио). Клинические прояв­
ления — карликовость, гипертрихоз, характерные черты лица, короткая
шея, гепато- и спленомегалия, грыжи, умственная отсталость; вакуолиза­
ция эпителия почечных канальцев, диффузная ангиокератома, гипопла­
зия тел позвонков, кифоз, сколиоз. Смерть в раннем детстве. 0 Тип II
(ю венильный, #230600). Симптоматика проявляется со 2-го года жизни,
смерть к 10 годам. 0 Тип III (взрослый). • Г. СМ2 ранее именовали болез­
нью Т ея-С акса. Г. С М2 — прогрессирующее нейродегенеративное забо­
левание, которое в классической инфантильной форме обычно приводит
к смерти в возрасте 2—3 года; причина — недостаточность гексоаминида-
зы А и/или В, а также фактора активации гексоаминидаз. Частота забо­
левания намного выше среди евреев аш кенази (восточно-европейские).
О Вариант 0 (268800, р) — болезнь Сендхоффа. Клинические проявле­
ния — слабость, атрофия мышц, реакция вздрагивания на звук, прогрес­
сирующее психомоторное ухудшение, мозжечковая атаксия, дизартрия,
ф асцикуляции, пирамидная недостаточность, повы ш ение рефлексов,
ортостатическая гипотензия, макроцефалия, «кукольное» лицо, ранняя
слепота, виш нево-красное пятно на глазном дне, макроглоссия, кардиомегалия, высокий поясничный горб, хроническая диарея, эпизоди­
ческая боль в животе, гепатоспленомегалия, нарушенное потоотделение,
недержание мочи. 0 Вариант В (*272800, р) — болезнь Т ея-С акса типа 1.
Клинические проявления — реакция на звук в виде вздрагиваний; мышеч­
ная гипотония, позже переходящая в гипертонус, апатия, психомоторная
задержка, судорожные приступы, паралич, деменция, вишнево-красное
пятно на глазном дне, окруженное светло-серой областью; слепота, начало
в раннем детстве, смерть до 5-летнего возраста. Ф Вариант АВ (*272750,
р) — болезнь Т ея-С акса типа 2. 0 Вариант А (М) В (230710, р) — болезнь
Тея—Сакса ювенильная. Начало в возрасте 2 лет, смерть к 8 годам, задержка
моторных функций, психического развития, атрофия мышц, спастичность,
задержка речевого развития. • Г. 6 МЗ (305650, От(3)-УДФ-1ЧГ-ацетилгалактозаминилтрансферазы недостаточность, КФ 2.4.1.88).
Гаптен (от греч. Нар1о — прикрепляться) — неполный или частичный Аг,
который не способен сам по себе индуцировать синтез АТ, но может взаи­
модействовать со специфическими АТ. Гг. обладают антигенностью (т.е.
взаимодействуют со специфическими АТ), но гг. не иммуногенны (т.е.
не способны запускать иммунные реакции). Гг. известны как неполные Аг.
Как правило, гг. имеют небольшую молекулярную массу и не распознают­
ся иммунокомпетентными клетками. Гг. могут быть простыми и сложными:
простые гг. взаимодействуют с АТ в организме, но не способны реагировать
с ними т т 1 г о \ сложные гг. взаимодействуют с АТ т у 1у о и т у И г о . Гг. могут
стать иммуногенными при связывании с высокомолекулярным носителем,
обладающим собственной иммуногенностью. Например, хром и никель,
связываясь с белками кожи, способны вызвать аллергический контактный
дерматит, развивающийся при повторных соприкосновениях кожи с хро­
мированными или никелированными предметами.
Гаптоглобин — гликопротеид сыворотки крови, который взаимодействует с НЬ
(например, при гемолизе) с образованием комплексного соединения, обла­
дающего пероксидазной активностью и разрушаемого фагоцитирующими
клетками с высвобождением молекулярного железа.
Гаргоилизм (от фр. §аг§оиП1е — горгулья — скульптурные изображения демони­
ческих созданий и химер, украшавшие фасады средневековых готических
построек (обычно соборов) и рыльца водосточных труб) — общее название
мукополисахаридозов типа I и II. Причудливой формы лицо и другие сим­
птомы синдрома Гурлер.
Гастрин — полипептидный гормон, который секретируется О-клетками сли­
зистой оболочки, выстилающей привратниковый отдел желудка (О-клетки
островков поджелудочной железы также секретируют г. в ранних воз­
растных группах). Г. стимулирует секрецию соляной кислоты в желудке.
Рецептор г. / холецистокинина обнаружен в Ц Н С и слизистой оболоч­
ке желудка. Стимулятор секреции г. — освобождающий гормон (ОКР).
Большое количество г. вырабатывают опухоли островковых клеток под­
желудочной железы (синдром Золлингера—Эллисона).
Гемангиома — доброкачественная опухоль, развивающаяся из кровеносных
сосудов <=> невус сосудистый.
Гемералопия — снижение четкости зрения как при ярком, так и при понижен­
ном освещении вследствие дистрофических изменений палочек сетчатки,
отвечающих за сумеречное зрение. Причина — мутации генов, кодиру­
ющих синтез опсинов (описано около 40 мутаций) <=> никталопия <=> сле­
пота куриная <=> слепота ночная. См. также «Пигменты зрительные».
Гемоглобин (НЬ) — дыхательный белок эритроцитов, состоящий из гема (около
3,8%) и глобина (96,2%); транспортирует кислород (в виде оксигемоглобина — Н Ь 0 2) от легких к тканям, где кислород легко освобождается
и НЪ02 восстанавливается до НЬ; имеется пять типов нормального г.:
эмбриональные НЬ [НЬ Сом/ег- 1 (Гоуэр), НЬ Сете г-2], фетальный (НЬР)
и два типа дефинитивных НЬ взрослого человека (НЬА, НЬА2). НЬ —
тетрамер, который состоит из двух а-глобиновых цепей, содержащих 141
аминокислотный остаток (в клетках кровяных островков эмбриона экс­
прессируются цепи глобинов другого типа (р, у, 5, г или О, включающих
146 аминокислотных остатков (НЬ С ош г-1 содержит по две С,- и е-цепи).
В ходе внутриутробного развития происходит два переключения генов
глобина: с эмбрионального на фетальный, что совпадает с переходом гемопоэза из желточного мешка в фетальную печень, и переключение феталь­
ного на дефинитивный глобин, что происходит к началу перинатального
периода. Переключение с фетального на дефинитивный НЬ начинается
в III триместре беременности и заканчивается на 6-м месяце постнатальной жизни. Однако следы НЬР присутствуют и в эритроцитах у взрослых.
Усиление эритропоэза у взрослых приводит к росту числа эритроцитов
с увеличенным содержанием НЬР. • НЬА (*141800) — г., составляющий
основную часть нормального НЬ у взрослого человека; белковая часть
НЬА содержит две пары различающихся по строению полипептидных
цепей, которые обозначаются как а- и р-цепи. • НЬА2 (* 141850) — г., вхо­
дящий в небольшом количестве (до 2,5%) в состав нормального г. взрос­
лого человека и в более значительном количестве — при р-талассемии.
Отличается от НЬА строением одной пары полипептидных цепей (5-цепи
вместо р-цепей). • НЪС (* 141900—0038) — аномальный г. с заменой лизина
на глутаминовую кислоту в 6-м положении р-цепи, что понижает функ­
циональные возможности и пластичность эритроцитов. У гомозигот фор­
мируются серповидно-клеточные эритроциты. • НЬР (* 142200) — основной
НЬ эритроцитов плода, отличающийся от НЬА строением одной пары
полипептидных цепей, большим сродством к кислороду и большей ста­
бильностью. После рождения содержание НЬР резко уменьшается (<0,5%
у детей и взрослых). Повышенное содержание НЬР наблюдают при неко­
торых гемоглобинопатиях, гипопластических и витамин В12-дефицитной
анемии, остром лейкозе о фетальный НЬ. • НЪН (* 142309) — гомотетра­
мер, образующийся при ингибировании синтеза а-цепи. Транспорт кисло­
рода нарушен, развивается синдром, схожий с талассемией (у гетерозигот
по двум генам а-талассемии). • НЪМ (* 142300 и др.). Группа аномальных
гг., у которых замещение одной аминокислоты способствует образованию
МеШЬ (хотя активность метгемоглобинредуктазы в норме), гетерозиготы
имеют врожденную метгемоглобинемию, гомозиготы — погибают. • НЬ8
(* 141900) — аномальный НЬ (мутация в 6-м положении (3-цепи), у гете­
розигот имеются серповидно-клеточные эритроциты (НЬ5 от 20 до 45%,
остальное — НЬА, анемии нет), у гомозигот развивается серповидно­
клеточная анемия (НЬ5 — 75—100%, остальное — НЬГ или НЬА2) о
серповидно-клеточный НЬ. • Барта г. (* 142309, Ваг( — фамилия пациента,
у которого впервые обнаружен). Гомотетрамер, встречающийся у раннего
эмбриона и при а-талассемии, не эффективен как переносчик кислорода.
• Карбоксигемоглобин (НЬСО) — НЬ, в котором кислород ( 0 2) замещен
окисью углерода (СО); не переносит 0 2 • Метгемоглобин (МеШЬ) — НЬ,
содержащий Ре гема в трехвалентной форме; не переносит 0 2; образует­
ся в повышенном количестве при некоторых наследственных болезнях
и отравлениях.
Гемоглобинопатии — общее название заболеваний, вызываемых дефектами
разных глобинов (например, серповидно-клеточная анемия, р-талассемия)
в составе молекулы НЬ (для примера см. «Гемоглобинопатия С» в сравне­
нии с подстатьей в статье «Гемоглобин»; другие гемоглобинопатии — см.
конкретные НЬ в статье «Гемоглобин»). Гемоглобинопатии раньше называ­
лись гемоглобинозами. • Гемоглобинопатия С — наследственное заболева­
ние, характеризующееся нарушением синтеза нормального НЬ с образова­
нием аномального глобина (НЬС); наследуется по аутосомно-рецессивному
типу; НЬС содержит аномальный глобин — замена лизина на глутами­
новую кислоту в 6-м положении (3-цепи, что понижает кислородсвязывающую функцию НЬ и нарушает пластичность эритроцитов; у гомозигот
формируются серповидно-клеточные эритроциты; патогенез — гемолиз
деформированных эритроцитов. Клиническая картина: гемолитические
кризы, боли в животе, метеоризм, увеличение селезенки, желтуха, легкая
анемия, пойкилоцитоз, повышение содержания непрямого билирубина.
Лечение: заместительная терапия (гемотрансфузии), железосодержащие
препараты для коррекции анемии.
Гемоглобинурия — наличие в моче свободного НЬ; обусловлено внутрисосудистым гемолизом и последующим выделением НЬ почками. • Маршевая
г. — доброкачественное расстройство, характеризующееся покраснением
мочи, гемоглобинурией, миоглобинурией и болью в животе; протека­
ет обычно остро; встречается после тяжелых и длительных физических
нагрузок; лечения не требуется. • Пароксизмальная ночная г. — хрониче­
ская болезнь с эпизодами гемолитической анемии и г. (преимущественно
по ночам) в результате внутрисосудистого гемолиза. Характерна желтушность или «бронзовая» кожа, умеренная спленомегалия, иногда увели­
чение печени; макроцитоз, анизоцитоз. Болезнь развивается вследствие
соматической мутации стволовых клеток, ведущей к дефекту мембраны
эритроцитов <=> М аркиафавы—М икели болезнь.
Гемодиализ — метод коррекции водно-электролитного и кислотно-щелочного
равновесия (И Д Р ) и выведения различных вредных веществ из организма,
основанный на диализе и ультрафильтрации крови аппаратом «искусствен­
ная почка». Применяют при лечении больных почечной недостаточностью
и при некоторых острых отравлениях. • Хронический г. — г., проводимый
повторно через определенные промежутки времени. Один из основных
методов лечения ХПН.
Гемопоэз (кроветворение) — процесс образования, дифференцировки и созре­
вания форменных элементов крови из клеток-предшественниц в условиях
специфического микроокружения. Г. осуществляется в костном мозге
плоских костей (череп, ребра, грудина, позвонки, кости таза) и эпифизов
трубчатых костей. Другими кроветворными органами являются селезенка,
тимус и лимфатические узлы. • Пренатальный г. В пренатальном периоде
клетки крови образуются в нескольких развивающихся органах. • Клетки
кровяных островков желточного мешка до 12 нед внутриутробного разви­
тия образуют первые клетки крови — первичные эритробласты — крупные
клетки, содержащие ядро и эмбриональные типы НЬ. • В течение второ­
го месяца развития стволовые клетки крови заселяют печень, селезенку
и тимус. Образуются все виды клеток крови. • Костный мозг у эмбрио­
на закладывается к концу третьего месяца внутриутробного периода.
К четвертому месяцу в костном мозге появляются лимфоидные элементы
и родоначальные клетки крови, а с пятого месяца возникает дифферен­
цированное костномозговое кроветворение. Помимо этого созревание
лимфоцитов происходит и в других органах: в печени, тимусе, селезенке,
лимфатических узлах. Последние в антенатальном периоде также являются
органом эритропоэза. К моменту рождения, после рождения и у взрослого
кроветворение ограничивается костным мозгом и лимфоидной тканью.
При недостаточности костного мозга восстанавливается экстрамедуллярный гемопоэз (гемопоэз в печени, селезенке и лимфатических узлах).
• Постнатальный г. Зрелые клетки периферической крови развиваются
из предшественников, созревающих в костном мозге. Стволовая кровет­
ворная клетка — СР11-Ыа§1 — родоначальница всех форменных элементов
крови. Потомки стволовой кроветворной клетки — полипотентные клеткипредшественницы лимфопоэза (С Ш -Ь у ) и миелопоэза (С Ш -С Е М М ).
В результате деления СРЕГ-Ьу и СР11-ОЕМ М их потомки остаются полипотентными или дифференцируются в один из нескольких типов унипотентных стволовых клеток, которые также способны делиться, но дифф е­
ренцируются только в одном направлении (образуя один клеточный тип).
Они пролиферируют и в присутствии факторов роста дифференцируются
в клетки-предшественницы. Терминология. СРЕ1-Ыа§1: — стволовая кровет­
ворная клетка; СРЕГ-ОЕММ — полипотентная клетка-предшественница
миелопоэза; СРЕ1-Ьу — полипотентная клетка-предшественница лим ф о­
поэза; СРЕ1-СМ — полипотентная клетка-предшественница гранулоцитов
и моноцитов; С РИ -О — полипотентная клетка-предшественница нейтро­
филов и базофилов. Унипотентные предшественники: ВРЕ1-Е и СРЕ1-Е —
эритроцитов; СР11-Ео — эозинофилов; С РИ -М — моноцитов; СР11Ме§ — мегакариоцитов. • Эритропоэз. Из стволовой клетки эритропоэза
(ВРЕГ-Е) дифференцируется унипотентный предшественник эритроцитов
СР11-Е. Последний дает начало проэритробласту. Дальнейшая дифференцировка приводит к увеличению содержания НЬ и потере ядра. При этом
из проэритробласта последовательно дифференцируются базофильный,
полихроматофильный, оксифильный эритробласт (нормобласт), ретикулоцит, эритроцит. • Гранулоцитопоэз. В ходе дифференцировки предше­
ственников гранулоцитов выделяют миелобласт, промиелоцит, миелоцит,
метамиелоцит, палочкоядерный и сегментоядерный гранулоцит. По мере
дифференцировки в цитоплазме появляются гранулы, уплотняется ядро
и изменяется его форма (от округлой к сегментированной). • Моноцитопоэз.
М оноциты и гранулоциты имеют общую клетку-предшественницу — коло­
ниеобразующую единицу гранулоцитов и моноцитов (СР11-ОМ), обра­
зующуюся из полипотентной клетки — предшественницы миелопоэза
(С Р У -С Е М М ). В развитии моноцитов выделяют две стадии — монобласт и промоноцит. • Тромбоцитопоэз. Из мегакариобластов развиваются
самые крупные (30—100 мкм) клетки костного мозга — мегакариоциты.
При дифференцировке мегакариоцит увеличивается в размерах, его ядро
становится дольчатым. Образуется развитая система демаркационных мем­
бран, по которым происходит отделение («отшнуровка») тромбоцитов.
• Лимфопоэз. Из стволовой кроветворной клетки (СР1_1-Ыай) происходит
полипотентная клетка-предшественница лимфопоэза (СРЕГ-Ьу), которая
впоследствии дает начало клеткам-предш ественницам В-лимфопоэза,
Т-лимфопоэза и (частично) предшественницам N К-клеток. Дальнейшая
дифференцировка включает уровни про-В(Т)-, пре-В(Т)-, незрелых В(Т)-,
зрелых наивных В(Т)- и (после контакта с Аг) зрелых В(Т)-клеток окон­
чательных стадий дифференцировки. • Факторы гемопоэза. Образование
клеточных элементов крови активируется и регулируется факторами гемо­
поэза: гемопоэтическими факторами роста, факторами транскрипции,
фолиевой кислотой и витамином В12. • Гемопоэтические факторы роста —
фактор стволовых клеток (5СР), колониестимулирующие факторы (С8Р),
ИЛ, эритропоэтин, тромбопоэтин. • Факторы транскрипции — белки,
связывающиеся с Д Н К и регулирующие экспрессию генов кроветворных
клеток. • Фолиевая кислота и витамин В12 необходимы для синтеза ДНК.
Фолаты и витамин В12 поступают с пищей и всасываются в тонкой кишке.
Для всасывания витамина В12 в кишечнике необходим внутренний фактор
Касла, синтезируемый париетальными клетками желудка. Фактор связы­
вает витамин В ,2 и защищает его от разрушения ферментами. Комплекс
внутреннего фактора с витамином В12 в присутствии ионов кальция взаи­
модействует с рецепторами эпителиальных клеток, образующих дисталь­
ный отдел подвздошной кишки. При этом витамин В12 поступает в клетку,
а внутренний фактор высвобождается. Отсутствие внутреннего фактора
Касла приводит к развитию анемии.
Гемосидерин — темно-желтый железосодержащий пигмент, образующийся
внутриклеточно при распаде НЬ или интенсивном всасывании железа
в кишечнике.
Гемосорбция — метод выведения токсинов из организма путем экстракорпо­
ральной перфузии крови через гранулированные или пластинчатые сор­
бенты.
Гемоторакс — скопление крови в плевральной полости.
Гемофилия — геморрагическое заболевание, вызванное наследуемым деф ек­
том плазменных факторов свертывания. Различают г. А (недостаточность
фактора свертывания VIII, классическая г.), г. В (недостаточность фактора
IX, болезнь Кристмаса), г. сосудистую (см. «Болезнь фон Виллебранда»),
Дебют г. наблюдают в раннем детском возрасте. Генетические аспекты:
• г. А (306700, X^28, дефекты генов Р8С)\ • г. А с сосудистыми анома­
лиями (306800, К) сочетается с ломкостью капилляров; • аутосомная г.
А (134500,5К), вариант болезни фон Виллебранда с нормальным временем
кровотечения; • г. В (болезнь Кристмаса, 306900, X ^27.1-^27.2, дефекты
генов Р9, НЕМВ). Частота. • г. А — 10:100 000 мужчин. • г. В — 2:100 000
мужчин. • Рождение больных девочек теоретически возможно при браке
мужчины, больного г., с женщ иной — носительницей патологического
гена. Тяжесть заболевания зависит от активности факторов свертыва­
н ия в крови. • Активность менее 2% — тяжелая форма г. • Активность
2 -5 % — среднетяжелое течение. • Активность 6—25% — легкая степень г.
• Активность более 25% клинически редко себя проявляет; возможны кро­
вотечения при обширных травмах и операциях. Проявления. • Характерны
спонтанные или посттравматические кровотечения (наружные или вну­
тренние, например почечные). 0 У грудных детей кровотечение воз­
никает при обрезании, прорезывании зубов. 0 Когда ребенок начинает
ходить, возможны кровоизлияния в мягкие ткани (подкожные гемато­
мы), крупные суставы. 0 Длительность кровотечений зависит от тяжести
заболевания. • Смеш анный тип кровоточивости (образование гематом
и кровотечения из слизистых оболочек). 0 Рецидивы гемартроза приводят
к образованию хронических артритов, сопровождающихся контрактурами
и приводящих к анкилозам. Лабораторные исследования. • Содержание
факторов (VIII или IX) свертываемости крови ниже 50%. • ЧТВ повышено
при нормальном количестве тромбоцитов и ПВ. • Время кровотечения
удлинено — более 10 мин • Н ормализация ЧТВ при добавлении нормаль­
ной плазмы к сыворотке больного. Лечение. Тактика ведения. • Ж енщ инносительниц патологического гена до беременности следует предупредить
о возможном рождении мальчика, больного г. • П ри рождении мальчика
в семье с отягощ енной наследственностью по г. со стороны матери —
исследование гемостаза в первом полугодии жизни. • При установлении
диагноза г. больному ребенку проводят вакцинацию против сывороточ­
ного гепатита. • Все ЛС, в том числе профилактические прививки, при­
нимают внутрь или вводят в/в. • Хирургические вмешательства проводят
по строгим показаниям на фоне заместительной терапии (свежезаморо­
женная плазма, криопреципитат, препараты факторов). • Экстракцию
зуба проводят в условиях стационара. • Противопоказано применение
ацетилсалициловой кислоты. • Родителям больного ребенка необходи­
мо всегда иметь в запасе препарат дефицитного фактора. • Исключают
занятия физкультурой. • Д инамическое наблюдение гематолога. • Детям,
больным г., необходимо всегда иметь при себе паспорт гемофилика, где
указан тип г., группа, КЬ-принадлежность крови больного и меры первой
неотложной помощи. Лечение при кровотечениях. • Режим постельный.
• Лекарственная терапия: 0 препараты факторов (VIII или IX) в/в; рас­
чет необходимой дозы — 1 ЕД фактора (количество фактора VIII в 1 мл
плазмы) повышает его содержание в плазме пациента на 2% на I кг массы
тела. Период полувыведения фактора VIII — 8—12 ч, поэтому введение
фактора следует проводить 2 -3 раза в сутки. Ф Свежезамороженная
плазма в/в из расчета 10 м л/кг каждые 6 ч до остановки кровотечения.
0 Криопреципитат в/в при г. А. 0 Ангиопротекторы, гемостатики: этамзилат, кислота аминокапроновая внутрь или в/в. Ф Глюкокортикоиды (преднизолон) при гемартрозах, почечных кровотечениях. О При кровотечени­
ях из слизистых оболочек — гемостатическая губка (местно). Осложнения
терапии. • Инфицирование вирусом гепатита и ВИЧ при применении
компонентов крови. • Формирование ингибиторной формы г. — образо­
вание в крови больного ингибиторов к переливаемым факторам — делает
заместительную терапию неэффективной. Профилактика. Генетическое
консультирование. Примечание. • Рекомбинантный фактор VIII — дорогой
препарат, биологически активный и высокоэффективный. При введении
препарата риск инфицирования вирусами гепатита и иммунодефицита
человека отсутствует.
Гемофтиз — кровохарканье.
Гемохроматоз — наследственная болезнь, характеризующаяся нарушением
обмена железосодержащих пигментов, повышенным всасыванием в кишеч­
нике железа и накоплением его в тканях и органах; проявляется призна­
ками цирроза печени, СД, пигментацией кожи; выделены доминантно
и рецессивно наследуемые формы г.
Гепарин (гепаринсульфат) — естественный антикоагулянт, который содер­
жится во многих тканях (особенно в печени, легких) и тучных клетках;
гетерополисахарид (Мг от 16 до 17 кД) с повторяющимися остатками
сульфатированных ^/-глюкуроновой кислоты и ^/-глюкозам и на [4 -0 -(аД-глюкуронидо)-/)-глюкозамин-Л'-сульфаты]; в сочетании с кофактором
белка сыворотки крови действует как антитромбин и антипротромбин,
предупреждая агглютинацию тромбоцитов и тем самым образование
тромбов; усиливает активность осветляющих факторов (ЛП липазы —
ЛПЛазы), подавляет сокращение мышечных клеток (блокатор рецепторов
инозитол-1,4,5-трифосфата), ингибирует РНКазы, казеинкиназу. Г. синте­
зируется и запасается в секреторных гранулах тучных клеток в комплексе
с гистамином и различными протеазами.
Гепатит. • Г. А (болезнь Боткина) — инфекционное заболевание с фекально­
оральным путем передачи, характеризующееся поражением печени с раз­
витием симптомокомплекса острого г. Заболевание известно с глубокой
древности, его описание содержится в трудах Гиппократа. Вирус г. А (ВГА)
включен в род Н ераиш гт семейства Ркот ачт йае, представлен «голыми»
сферическими вирионами диаметром 25—21 нм. Геном образуется несегментированной молекулой + РНК. Эпидемиология. Резервуар возбудите­
ля — больной человек. Больной выделяет возбудитель в течение 2 -3 нед
до начала и в течение первых 3—5 сут желтушного периода. Передача воз­
будителя происходит фекально-оральным путем (через воду, пищевые про­
дукты, грязные руки, различные предметы). ВГА устойчив в окружающей
среде, при 21 °С сохраняется несколько недель; полностью инактивирует­
ся при температуре 85 °С. ВГА хорошо переносит низкие температуры,
устойчив к хлору, благодаря чему сохраняется в очищенной питьевой воде.
Фекальное загрязнение источников водопользования может вызвать фор­
мирование эпидемических вспышек. П ик заболеваемости приходится
на холодный сезон (поздняя осень или зима). После перенесенного забо­
левания формируется стойкая невосприимчивость к повторным заражени­
ям. Антигенная структура. Возбудитель представлен одним антигенным
типом и содержит главный Аг (НА-Аг), по которому его идентифицируют.
Патогенез поражений. Попадая в организм человека с водой или пищей,
ВГА размножается в эпителии слизистой оболочки тонкой кишки и реги­
онарных лимфоидных тканях. Затем наступает фаза кратковременной
вирусемии. Максимальная концентрация вируса в крови возникает в конце
инкубационного периода и в преджелтушном периоде. В это время воз­
будитель выделяется с фекалиями. Основная мишень цитопатогенного
действия — гепатоциты. Репродукция вируса в их цитоплазме приводит
к гибели клеток. Цитопатический эффект усиливают иммунные механиз­
мы, в частности ЫК-клетки, активированные ИФ Н , синтез которого инду­
цирует вирус. Маркёры репликации вируса — АТ (1§М и 1§С) к Аг ВГА
и вирусная РНК. Лечение и профилактика. Средства специфической про­
тивовирусной химиотерапии отсутствуют, лечение симптоматическое.
Разработанный сывороточный
предупреждает развитие заболевания
в течение 3 мес и значительно смягчает течение заболевания. Его приме­
няют для пассивной иммунизации людей, направляющихся в эндемичные
районы. Для активной иммунопрофилактики ВГА используются убитые
и рекомбинантные вакцины . Общие профилактические мероприятия
направлены на улучшение санитарной обстановки, включают соблюдение
карантинных мероприятий, улучшение условий водоснабжения и повы­
ш ение гигиенической культуры населения. • Г. В — инфекционное забо­
левание с кровеконтактным механизмом передачи, характеризующееся
поражением печени с развитием симптомокомплекса острого и хрониче­
ского гепатита. Вирус гепатита В (ВГВ) входит в состав рода Онкокерайпау&ш
семейства Нерайпаутйае. Вирионы ВГВ сферической формы 42 нм в диа­
метре имеют суперкапсид. Геном образован неполной (одна нить короче)
двухнитевой кольцевой молекулой ДНК. С короткой «плюс»-нитью Д Н К
связан праймерный белок. В состав сердцевины также входит Д Н К зависимая ДНК-полимераза. Для эффективной репликации необходим
синтез ДНК-полимеразы, так как вирусная Д Н К образуется на матрице
РНК; в динамике процесса вирусная Д Н К интегрирует в Д Н К клетки.
В крови больных г. В циркулируют частицы трех морфологических типов.
Лишь 7% частиц представлены комплексными двухслойными сферически­
ми образованиями с полной структурой — частицами Дейна, проявля­
ющими выраженную инфекционность. Их оболочку на 70% поверхности
образуют белки. Эпидемиология. Резервуар возбудителя — инфицирован­
ный человек. Механизм передачи инфекции — кровеконтактный. Основные
пути передачи ВГВ — инъекционный, гемотрансфузионный и половой.
Также показана возможность вертикальной передачи ВГВ от матери
к плоду. 7-10% инфицированных становятся хроническими носителями;
их число достигает 300 млн. Ежегодно заболевает не менее 50 млн человек.
В РФ отмечают 10—15% рост заболеваемости гепатитом. Основные группы
риска — медицинские работники; лица, получающие гемотрансфузии
или препараты крови; наркоманы, вводящие наркотики внутривенно;
больные гемофилией; лица, находящиеся на гемодиализе; дети матерей —
носительниц НВ5А§; половые партнеры носителей вируса. Антигенная
структура. Основные Аг частиц Дейна — поверхностный Н В5А§ и сердце­
винный НВСА§. АТ против НВ5А§ и НВсА§ появляются в течение заболе­
вания. Наличие АТ против НВ5А§ прямо связано с невосприимчивостью
к инфекции (постинфекционны й или поствакцинальный иммунитет).
• НВ5А§. Первый идентифицированный Аг ВГВ; впервые выделен из крови
австралийского аборигена, поэтому этот Аг также называют австралий­
ским. НВ5А§ появляется в крови через 1,5 мес после инфицирования;
постоянно циркулирует в сыворотке инфицированных лиц, а его очищ ен­
ные агрегаты входят в состав вакцины против ВГВ. • НВСА§. Сердцевинный
Н В ^ ё представлен единственным антигенным типом; его обнаруживают
только в сердцевине частиц Дейна. Аг маркирует репликацию вируса
в гепатоцитах. • НВеА§. Не входит в состав частиц Дейна, но связан с ними,
так как появляется в сыворотке в инкубационном периоде, сразу после
появления НВ5А§. НВеА§ можно расценивать как наиболее чувствительный
диагностический показатель активной инфекции. Обнаружение НВеА§ у
пациентов с хроническим гепатитом указывает на активацию процесса,
что представляет высокую эпидемическую опасность. • Н ВхАг — наименее
изученный Аг. Предположительно опосредует злокачественную трансфор­
мацию клеток печени. Патогенез поражений. ВГВ гематогенно заносится
в печень и размножается в гепатоцитах. Во второй половине инкубацион­
ного периода ( 4 0 - 180-е сутки) вирус выделяют из крови, спермы, мочи,
фекалий и секрета носоглотки. В патогенезе поражений важную роль игра­
ют аутоиммунные гуморальные и клеточные реакции, что подтверждает
связь между началом клинических проявлений и появлением специфиче­
ских АТ. Патологический процесс начинается после распознавания вирусиндуцированных Аг на мембранах гепатоцитов иммунокомпетентными
клетками. Осложнения хронической формы обусловлены хроническим
воспалением и некротическими процессами в паренхиме печени; основные
осложнения — цирроз и первичная карцинома печени. Цирроз обычно
отмечают у страдающих хроническим гепатитом; ежегодно регистрируют
более 10 ООО летальных исходов, обусловленных ВГВ. Карцинома печени.
Показана четкая связь между злокачественной трансформацией гепатоцитов и перенесенной ВГВ-инфекцией. В развитии опухолевого процесса
принимают участие кофакторы, многие из которых остаются неизвестны­
ми. Маркёры репликации ВГВ — НВеА§, АТ (1§М) к НВСА§, Д Н К вируса
и вирусная ДНК-полимераза. Лечение. Средства специфической терапии
отсутствуют, лечение в основном симптоматическое. Определенные пер­
спективы имеет применение ингибиторов ДНК-полимеразы (например,
ламивудина), а-И Ф Н и его индукторов. Несмотря на то что на терапию
И Ф Н реагируют менее 50% пациентов, показано достоверное исчезнове­
ние всех маркёров инфекции (Д Н К ВГВ, НВ5А§ и НВеА§) и увеличение
титров АТ к НВ5А§. Иммунопрофилактика. Пассивная иммунизация спе­
цифическим 1§ (НВ1§) показана людям, контактировавшим с инфициро­
ванным материалом и носителями НВ5А§ (включая половых партнеров
и детей, родившихся от НВ5А§-ноложительных матерей). Для активной
иммунизации разработаны два типа вакцин. Первые готовят из плазмы
пациентов, содержащей Аг ВГВ в количествах, достаточных для создания
вакцинны х препаратов. Главное условие — полная инактивация ВГВ.
Вторую группу составляю т реком бинантны е вакцины (например,
КесотЫуах В, Е щ епх В), полученные методом генной инженерии. Массовая
иммунизация — важнейший компонент борьбы с инфекцией. Взрослые
получают две дозы в течение месяца и бустерную иммунизацию через
6 мес. Дети получают первую дозу сразу после рождения, следующие —
через 1—2 мес и к концу первого года жизни. Если мать НВ.А§-положительна,
то ребенку вводят специфический 1§ одновременно с первой вакцинацией.
• Г. С характеризуется преимущественным развитием хронических форм г.
с исходом в цирроз и первичную карциному печени. Вирус гепатита С
(ВГС) входит в состав рода семейства П аут пйае. Вирионы сферической
формы, средний диаметр 35—50 нм. Геном образуется однонитевой + РНК.
Резервуар возбудителя — инфицированный человек. Основной путь пере­
дачи вируса — парентеральный. Основное отличие от эпидемиологии ВГВ —
более н изкая способность ВГС к передаче от беременной к плоду
и при половых контактах. Больной выделяет вирус за несколько недель
до появления клинических признаков и в течение 10 нед после начала
проявлений. Заболевание чаще регистрируют в СШ А (до 90% всех трансфузионных гепатитов) и в Африке (до 25%). Для клинической симптома­
тики ВГС характерно изменение консистенции и размеров печени.
При активном процессе печень обычно увеличена и болезненна при паль­
пации, ее консистенция умеренно плотная. Другие проявления включают
спленомегалию, диспепсический и астенический синдромы, желтуху,
артралгии и миалгии, кардиты, васкулиты, легочные поражения, анемии
и др. Осложнения хронического процесса — цирроз и первичная карцино­
ма печени. Маркёры репликации вируса — АТ (1§М) к Аг ВГС и вирусная
РНК. Лечение и профилактика. Средства этиотропной терапии отсутствуют;
при хронических инфекциях можно использовать а-И Ф Н . На фоне тера­
пии И Ф Н у 40—70% больных отмечают стихание воспалительного процес­
са (на что указывает снижение содержания концентрации аминотрансфераз в сыворотке), однако по окончании курса у 40—50% пациентов
наблюдают рецидив воспаления. Средства специфической иммунопрофи­
лактики не разработаны. Г. Б . Возбудитель дельта-г. — дефектный РН К содержащий вирус рода ОеНатгиз семейства То%атпйае. Его выделяют
только от пациентов, инфицированных вирусом г. В. Дефектность возбу­
дителя проявляется в полной зависимости от его передачи, репродукции
и наличия вируса г. В. Соответственно, моноинфекция вирусом г. Б (ВГО)
абсолютно невозможна. Вирионы ВГО имеют сферическую форму, диа­
метром 35—37 нм. Геном вируса образуется однонитевой кольцевой моле­
кулой РНК. Ее последовательности не имеют гомологии с Д Н К возбуди­
теля гепатита В, но суперкапсид ВГО включает значительное количество
НВ_А§ ВГВ. Резервуар возбудителя — инфицированный человек; вирус
передается парентеральным путем. Возможна вертикальная передача ВГБ
от матери к плоду. Патогенез. И нфицирование НВ5А§-положительных лиц
сопровождается активным размножением В ГБ в печени и развитием хро­
нического г. — прогрессирующего или фульминантного. Клинически про­
является только у лиц, инфицированных вирусом г. В. Может протекать
в двух вариантах: коинфекция — одновременное заражение вирусами гепа­
титов В и Б; суперинфекция — заражение вирусом гепатита Б человека,
инфицированного вирусом гепатита В. Маркёры репликации вируса — АТ
(1§М) к Аг ВГО и вирусная РН К . Лечение и профилактика. Средства спец­
ифической химиотерапии и иммунопрофилактики отсутствуют. Поскольку
репродукция вируса гепатита Б невозможна в отсутствие возбудителя гепа­
тита В, основные профилактические мероприятия должны быть направле­
ны на предупреждение развития гепатита В. • Г. Е — острое инфекционное
поражение печени, проявляющееся симптомами интоксикации и реже
желтухой. ВГЕ включен в род СаИстгш семейства СаИстгШае. Вирионы
сферической формы 27—38 нм в диаметре. Геном образован несегментированной молекулой + РНК. Резервуар возбудителя — человек. Эпидемиологизаболевания во многом напоминает гепатит А; возбудитель вызывает энде­
мичные вспыш ки. И нкубационны й период не превыш ает 2—6 нел
Заболевание проявляется общим недомоганием; желтуху наблюдают срав­
нительно редко. Инфицирование беременных, особенно в III триместре,
может закончиться фатально (смертность может достигать 20 %). Хронизаш
процесса не наблюдают. Выздоровление сопровождается формирование
стойкой невосприимчивости к повторным заражениям. Маркёры реплика­
ции вируса — АТ (1§М) к Аг ВГЕ и вирусная РНК. Лечение. Средств:
этиотропной терапии и специфической профилактики отсутствуют; про­
водят симптоматическое лечение. • Г. Г признают не все исследовател
• Г. 6 — острый или хронический г. (возбудитель условно относят к семей­
ству Р1ачтпйае\ геном вируса г. О (ВГО) образован несегментированнг
молекулой + РНК; предположительно, вирус гепатита С является дефект­
ным вирусом, и для его репродукции необходимо присутствие вирусгепатита С) в большинстве случаев протекает как микст-инфекция с вир,сом гепатита С. Резервуар возбудителя — больные острым или хрониче-
ским гепатитом О и носители ВГС. Регистрируют заболевание сравнитель­
но редко. В РФ частота выявления Р Н К ВГС колеблется от 2% в Москве
до 8% в Якутии. В то же время в сыворотке крови доноров частота выя