Министерство охраны здоровья Украины
Крымский государственный медицинский университет
им. С.И. Георгиевского
ОСНОВЫ ПАТОФИЗИОЛОГИИ
Ч.1 ОБЩАЯ ПАТОЛОГИЯ. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ
ПРОЦЕССЫ.
Учебное пособие для самостоятельной
внеаудиторной работы студентов
Специальности: 7.110101 «Лечебное дело»
7.110104 «Педиатрия»
7.110106 «Стоматология»
7.110201 «Фармация»
г.СИМФЕРОПОЛЬ – 2010 г.
2
АВТОРСКИЙ КОЛЛЕКТИВ:
Д.м.н., профессор А.В. Кубышкин, д.м.н., профессор В.З. Харченко, к.м.н.,
доценты П.Ф. Семенец, В.Ф. Веселов, Ю.И. Шрамко, к.м.н., ассистенты
И.И. Фомочкина, Л.Л. Алиев, Л.В. Анисимова, ассистенты В.В. Щербак,
С.В. Литвинова
Рекомендовано к изданию Координационным учебно-методическим Советом Крымского государственного медицинского университета
им. С.И. Георгиевского.
Рецензенты: доктор медицинских наук, профессор Шаповалова Е.Ю.
доктор биологических наук, профессор Евстафьева Е.В.
ОСНОВЫ
ПАТОФИЗИОЛОГИИ.
Ч1.Общая
патология.Типовые патологические процессы.
Учебное пособие для самостоятельной внеаудиторной работы
студентов.\ Под редакцией доцента П.Ф. Семенца.-Симферополь,
2010г.- 287с.
ОСНОВИ ПАТОФІЗІОЛОГІЇ. Ч.1Загальна патологія.Типові
патологічні прцеси
Навчальний посібник для самостійної позааудиторної роботи
студентів.\ Під редакцією доцента П.Ф. Семенця.-Симферополь,
2010 р.- 287с.
3
Предисловие.
Предлагаемое учебное пособие написано с целью помочь студентам в самостоятельной подготовке к занятиям и лицензионному экзамену "Крок-1" по
патофизиологии, которая является основой клинического мышления.
В сборнике освещены вопросы по основным разделам общего курса патофизиологии в соответствии с учебной программой для студентов медицинских ВУЗов, утвержденной Министерством охраны здоровья Украины (Киев,
2002,2003,2006).
Полученные знания должны способствовать успешному изучению клинических дисциплин.
АВТОРЫ
4
Пояснительная записка
Патологическая физиология – это наука о наиболее общих закономерностях возникновения, развития и исхода болезней и патологических процессов.
Среди основных задач патологической физиологии главное место занимает выяснение причин и условий возникновения болезней (этиология), определение ведущих механизмов их развития и отдельных проявлений на разных
уровнях организации живых объектов от молекулярного уровня до организма в
целом (патогенез).
Патологическая физиология как учебная дисциплина предусматривает
формирование теоретических основ для глубокого понимания этиологии, патогенеза, клинических проявлений, принципов предотвращения и лечения болезней. Эта цель означает особое место патофизиологии в системе подготовки
врача любой специальности и требует всестороннего интегрирования с другими
учебными дисциплинами. Такое интегрирование должно выполняться, минимум, в четырех направлениях.
Первое из них охватывает связи с дисциплинами, которые изучают свойства факторов окружающей среды, способных вызывать возникновение болезней. Речь идет о таких общетеоретических предметах, как физика, неорганическая и органическая химия, биология, микробиология, социология. Эти науки
дают студентам знания, необходимые для изучения этиологии.
Другую группу учебных дисциплин составляют предметы, которые дают
студенту понимание свойств организма и законов его деятельности в условиях
нормы (гистология, нормальная физиология, биохимия, иммунология, генетика). Эти науки являются основой для изучения патогенеза.
Третью группу предметов (патологическая анатомия, фармакология) студенты изучают одновременно с патологической физиологией. Связи с этими
дисциплинами способствуют формированию целостного представления о сущности болезней и возможных влияниях на них.
И, наконец, главное место в системе высшего медицинского образования
занимают клинические дисциплины, относительно которых патологическая физиология выступает в роли теоретической основы, поскольку она является
краеугольным камнем научного медицинского мировоззрения врача.
Кроме того, патологическая физиология определяет этиологические и патогенетические принципы профилактики, диагностики и лечения болезней, чем
объясняется ее важное прикладное значение для клиники.
По структуре патологическая физиология состоит из двух частей: общей
патофизиологии и патофизиологии органов и систем (частная патофизиология).
Общая патология, в свою очередь, включает общую нозологию – учение о болезни, этиологию – учение о внутренних и внешних причинах и условиях возникновения болезней, патогенез – учение о механизмах развития болезней и учение о типовых патологических процессах ( аллергия, типовые
нарушения местного кровообращения, воспаление, опухолевый рост, типовые
нарушения обмена веществ, отек, лихорадка, гипоксия и др.).
5
Патологическая физиология органов и систем предусматривает изучение общих закономерностей развития патологических процессов и патологических состояний в отдельных функциональных системах, а также этиологии и
патогенеза самых распространенных нозологических форм.
•
•
•
•
•
•
•
•
После окончания курса обучения студент ДОЛЖЕН ЗНАТЬ:
Суть основных понятий общей нозологии: болезнь, патологический процесс,
типовой патологический процесс, патологическая реакция, патологическое
состояние, этиология, патогенез;
Причины, механизм развития и проявления типовых патологических процессов;
Причины и механизм развития типовых нарушений обмена веществ;
Общие закономерности развития патологических процессов в отдельных органах и системах, понятие об их функциональной недостаточности;
Современные представления об этиологии и патогенезе самых распространенных болезней человека;
Этиологические и патогенетические принципы классификации патологических процессов и болезней;
Экспериментальные подходы к решению проблем этиологии и патогенеза;
Механизмы развития основных клинических проявлений патологических
процессов и самых распространенных болезней.
УМЕТЬ:
Анализировать роль факторов окружающей среды и внутренних факторов в
возникновении болезней.
• Отличать разрушительные явления и защитные (приспособительные и компенсаторные) реакции в развитии патологических процессов и болезней.
• Определять ведущее звено патогенеза болезней и причинно-следственные
связи между отдельными патогенетическими механизмами;
• На основании данных функциональных и биохимических исследований делать выводы о характере патологических изменений в организме и возможных механизмах их развития;
• Пользоваться приобретенными ранее знаниями о свойствах факторов окружающей среды и закономерностях жизнедеятельности здорового организма
для освещения проблем этиологии и патогенеза;
• Самостоятельно производить самые простые экспериментальные исследования на животных;
• Выполнять поиск необходимой научной информации в литературных источниках;
6
БЫТЬ ПРОИНФОРМИРОВАННЫМ О:
• Основных правилах планирования, проведения экспериментальных исследований и методах анализа полученных результатов;
• Современных методиках моделирования патологических процессов и болезней;
• Основных патогенетических способах предотвращения и лечения патологических процессов и самых распространенных болезней человека;
• Современных направлениях и тенденциях развития патофизиологических
исследований.
(Из программы по патофизиологии,
утвержденной МОЗ Украины.
Киев – 2002,2003,2006г.)
7
ПРЕДМЕТ, ЗАДАЧИ И МЕТОДЫ ПАТОФИЗИОЛОГИИ
Термин «патофизиология» происходит от двух слов: «патология» и
«физиология». « Патология» в переводе с греческого означает учение о болезни ( pathos-болезнь, страдание; logos – учение ) , «физиология» -учение о
природе организма ( physis – природа ; logos – учение ). Таким образом, «патофизиология» - это наука о жизнедеятельности больного организма.
Патофизиология – это наука, которая изучает общие закономерности
возникновения, развития и исхода болезней и патологических процессов.
Ряд важных понятий патофизиологии, такие как стресс, предболезнь,реактивность, резистентность, конституция, диатез - относятся к здоровым индивидам.
В уставе Всемирной Организации Здравоохранения (ВОЗ) патофизиологию определили как "основу медицинского профессионального интеллекта".
Патофизиология как фундаментальная наука и учебная дисциплина изучает общие закономерности, определяющие возникновение, течение и исход
болезни, раскрывает научные основы этиологии, патогенеза и саногенеза. Разнообразие возникающих нарушений жизнедеятельности клеток, органов и систем зависит от силы и качества чрезвычайного раздражителя, условий и реактивности организма. Основоположник отечественной патофизиологии как самостоятельной дисциплины В.В. Пашутин считал, что патофизиология есть
философия медицины. Она играет ведущую роль в борьбе с ненаучными концепциями в общей патологии.
Методологической основой предмета является метод диалектического
материализма. С диалектико-материалистических позиций патофизиология
раскрывает научные основы происхождения заболевания, диагноза и выздоровления (построение теории медицины), разрабатывает модели патологических явлений, принципы (симптоматический, этиологический, патогенетический, саногенетический) экспериментальной терапии.
Методологическими принципами патофизиологии являются принцип
биосоциального детерминизма, принципы единства анализа и синтеза, организма и внешней среды, структуры , функции и метаболизма, теории и практики, принцип сравнительной патологии.
Предметом изучения патофизиологии является жизнедеятельность больного организма на молекулярном, клеточном, органном, системном уровнях.
Предмет патофизиологии составляет совокупность знаний, накопленных при
изучении :
-сущности болезней;
-причин и условий возникновения болезней;
-механизмов развития болезней ;
-типических патологических процессов;
-закономерностей повреждения отдельных органов и систем.
8
Патофизиология – интегративная наука. Занимая особое место в системе
высшего медицинского образования, она обладает широкими теоретическими и
методологическими связями с другими дисциплинами.
Прежде всего, патофизиология опирается на науки, изучающие свойства
факторов окружающей среды, которые способны вызывать возникновение болезней, а также влиять на течение и исход. К ним относятся физика, химия,
биология, микробиология, социология. Эти дисциплины формируют теоретическую базу, необходимую для изучения этиологии.
Кроме того, для изучения патофизиологии необходимо обладать достаточно широким спектром знаний о структуре, функции и обмене веществ организма в условиях нормы. Этой цели служат такие дисциплины, как анатомия,
гистология, нормальная физиология, биохимия, генетика.
И, наконец, главное место в системе высшего медицинского образования
занимают клинические дисциплины, относительно которых патологическая
физиология выступает в роли теоретической основы, поскольку она является
краеугольным камнем научного медицинского мировоззрения врача.
Цель патофизиологии как учебной дисциплины заключается в формировании теоретических основ понимания этиологии, патогенеза, клинических
проявлений, принципов предотвращения и лечения болезней; получении фундаментальных сведений о сущности болезни и законах её развития; формировании и развитии, в конечном счете, клинического мышления.
Патофизиологический подход к решению задач клинической медицины
всё более активно внедряется в самые различные области практической медицины. Во всех направлениях современной медицины используются новейшие
методы лабораторной и инструментальной диагностики, анализ которых даёт
представление о патогенезе нарушений функций клеток, органов, систем больных. В процессе лечения применяют специальные лекарственные средства,
производят лечебные манипуляции. На базе патофизиологической теории формируются принципы и методы этиологической и патогенетической профилактики. Патофизиологические критерии лежат в основе определений и классификаций болезней. Ничто из перечисленного не может осуществляться без знания
патофизиологии.
Патофизиология состоит из 2 разделов:
1.Общая патология.
Она включает:
а/ общую нозологию – учение о сущности болезни.
Общая нозология
-формирует основные понятия и категории патологии ;
-разрабатывает принципы классификации и номенклатуры болезней;
-изучает социальные аспекты патологии.
б/ общую этиологию – учение о причинах и условиях возникновения
болезней.
Общая этиология изучает:
-общие свойства болезнетворных факторов;
-основные категории болезнетворных факторов;
9
-значение условий в возникновении болезней;
-принципы этиотропной профилактики и терапии.
в/ общий патогенез – учение о механизмах развития болезни. Учение о
патогенезе включает :
-механизмы устойчивости организма к действию патогенных факторов;
-общие механизмы развития болезней;
-механизмы выздоровления;
-механизмы умирания;
-принципы патогенетической профилактики и патогенетической терапии.
г/учение о наследственности, конституции и реактивности;
д/учение о типовых патологических процессах.
2. Частная патофизиология.
Рассматривает нарушения в отдельных органах и системах, а также этиологию и патогенез наиболее распространённых нозологических форм.
Задачи патофизиологии
Задачи медицинской науки состоят в обеспечении опережающего развития фундаментальных исследований и повышении эффективности прикладных
работ. В этой связи патофизиология призвана решить следующие задачи :
1. Установление сущности болезни.
2. Изучение причин и условий возникновения болезней.
3. Раскрытие механизмов развития болезни и отдельных ее проявлений, механизмов выздоровления и умирания.
4. Изучение основных закономерностей функционирования организма в условиях патологии.
5. Создание экспериментальных моделей патологических процессов.
6. Систематизация и аналитико-синтетическая обработка клинического экспериментального материала.
7. Разработка и внедрение эффективных методов ранней диагностики, мер
профилактики, принципов этиопатогенетической терапии.
8. Дальнейшее развитие клинической патофизиологии.
Патофизиология как учебная дисциплина формирует теоретические основы для глубокого понимания этиологии, патогенеза, клинических проявлений,
принципов лечения и профилактики болезней. Эта цель определяет особое место патофизиологии в системе подготовки врача любой специализации и требует всеобъемлющей интеграции с биологическими науками ( биология, биохимия, анатомия, гистология, физиология и др.), которые формируют фундамент патофизиологии, и клиническими дисциплинами ( терапия, хирургия, педиатрия и др.), относительно которых патофизиология выступает в роли теоретической основы.
В последние годы быстро развивается клиническая патофизиология ,
основанная на широком использовании современных высокоинформативных
биофизических, биохимических, электрофизиологических и других методах
исследования непосредственно в клинике. Это позволяет проводить безвред-
10
ные целенаправленные исследования на больных и использовать полученные
знания о патологических процессах, происходящих в организме, о характере
компенсаторно-приспособительных механизмов, о методах лабораторной и
функциональной диагностики для выбора рационального дифференцированного метода лечения.
В настоящее время представляется возможным выделить следующие задачи клинической патофизиологии:
1.
Исследование общих и частных закономерностей развития болезней у
больного человека. Установление их связи с реактивностью организма, а также
влиянием на их реализацию личности врача и содержания терапии.
2.
Изучение патогенеза конкретной болезни у конкретного больного с целью повышения эффективности терапии.
3.
Формирование патогенетических принципов терапии, адекватных современному уровню представлений о патогенезе.
Методы патофизиологии
В патологической физиологии используются два основных метода:
Метод клинического наблюдения. Он заключается непосредственно в
наблюдении течения болезни человека с помощью практически безвредных целенаправленных функционально-диагностических методик (биохимических,
иммунологических, электрофизиологических и т. д.). Преимущество этого метода заключается в отсутствии необходимости создания экспериментальной
модели. Исследователь наблюдает конкретную болезнь или патологический
процесс у постели больного. Однако, данный метод не даёт возможности изучить процессы, происходящие в организме человека в латентную и продромальную стадии. Кроме того, исследователь ограничен в оценке причины и
условий развития заболевания. Несмотря на это клиническая, патофизиология
является в определённом аспекте вершиной сравнительной патологии, ибо это
патология человека. Основной задачей клинической патофизиологии является
изучение наиболее общих вопросов этиологии и патогенеза болезней человека.
Эксперимент - основной метод патофизиологии. Эксперимент - это
моделирование болезней и патологических процессов на живых объектах. Такими объектами могут служить животные различных видов, отдельные органы,
культуры тканей, отдельные клетки и даже субклеточные структуры. .
Эксперимент включает моделирование патологических явлений на различных уровнях организации организма (молекулярном, клеточном, тканевом,
органном, системном, организменном), расширяет возможности познания сущности патологического процесса, способствует формированию у врача клинического мышления, умению использовать клинические знания в решении вопросов диагноза, лечения и профилактики различных заболеваний.
Для изучения нарушенных функций используются различные методы исследования:
-экспериментально-патофизиологические (электрофизиологические, биофизические, патохимические, иммунологические),
-клинико-патофизиологические (лабораторные, метод функциональных проб),
11
-статистические и др.
Эксперименты делятся на острые и хронические. Выбор осуществляется в
зависимости от цели исследования. Например, для изучения механизмов возникновения шоковых состояний применяют острый эксперимент, а эксперимент с привитием животному опухоли должен носить хронический характер.
Кроме того, все эксперименты в патофизиологии можно условно разделить на
синтетические и аналитические. При моделировании болезни или синдрома исследователь решает синтетическую задачу. Синтетические эксперименты проводятся, как правило, in vivo. При аналитическом эксперименте из болезни, как
целостного явления, вычленяется какой-то компонент, механизм, который чаще всего воспроизводится in vitro.
Патофизиологический эксперимент состоит из следующих этапов:
1.
Формирование рабочей гипотезы - определяет цель и задачи исследования.
2.
Выбор модели и метода, соответствующих поставленным задачам, обеспечение соответствующего контроля.
3.
Планирование эксперимента должно обеспечить корректное проведение
опыта, исключение посторонних и дополнительных воздействий на модель.
4.
Анализ полученных результатов и формулирование выводов. После
окончания эксперимента весь цифровой материал подвергается статистической
обработке для определения средних величин, их отклонений, достоверности.
Формулируются выводы.
Для изучения патологических процессов применяют следующие основные экспериментальные методики.
Метод выключения заключается в удалении какого-либо органа хирургическим путём или выключении его функции различными воздействиями (высокими или низкими температурами, ионизирующим излучением, фармакологическими препаратами, специфическими антителами). Эта методика применяется для моделирования заболеваний эндокринной и нервной систем, изучения
компенсаторных и пластических возможностей парных органов.
Метод включения может заключаться в введении в организм каких-либо
веществ (гормонов, антигенов, антител, ферментов) для моделирования эндокринологических, иммунологических заболеваний. Кроме того, к этой группе
относятся эксперименты с пересадкой органов.
Метод раздражения используется для изменения функциональной активности того или иного органа.
Метод изолированных органов даёт информацию о сущности патологических процессов в каждом конкретном органе или ткани.
Метод парабиоза оздание у двух животных единой системы кровообращения для исследования гуморальных механизмов различных воздействий.
Метод тканевых культур используется для получения информации о
сущности патофизиологических процессов, как в целом организме, так и в изолированном органе.
12
Сравнительно-эволюционный метод изучает, как правило, типические
патологические процессы в эволюционном аспекте. Патологический процесс,
как мы его наблюдаем у человека и животных, является результатом формирования соответствующих реакций в эволюции животного мира. Такие патологические процессы, как воспаление, лихорадка, возникали и усложнялись в эволюции в связи с усложнением и совершенствованием защитных и приспособительных реакций организмов на меняющиеся условия жизни. Вот почему правильный научный анализ реакций человека на патогенное воздействие требует
более полного знания путей и форм их становления. Это возможно только при
использовании исторического метода сравнительной патологии
Осуществление эксперимента сопряжено со многими трудностями, которые обусловлены сложностью патологических процессов, невоспроизводимостью ряда патологических процессов, наблюдаемых у человека и др.
В опытах на животных невозможно создать полную модель болезни человека. Организм человека много сложнее самых высокоорганизованных животных и подвергается воздействию социальных факторов. В этой связи возникает необходимость клинической патофизиологии, которая призвана существенно дополнить экспериментальные исследования клиническими, проведенными непосредственно на больных.
И тем не менее, благодаря патофизиологическому эксперименту накоплен
материал, составляющий содержание патологической физиологии. Болезни человека слишком сложны, и их механизмы спрятаны слишком глубоко. Раскрыть их трудно и даже невозможно, несмотря на применение самых современных технических средств. Простота опыта на животных является положительным моментом на определённом этапе научного анализа. Не в эксперименте на человеке, кролике, даже не на лягушке, а на прозрачной личинке морской
звезды И.И. Мечников наблюдал процесс фагоцитоза.
История развития патофизиологии.
Термин «патологическая физиология» впервые применил Й. Варандес в
XVII веке. В 1818г. Л. Гальо издаёт первое руководство по патофизиологии.
Большую роль в создании физиологического направления в изучении болезни
сыграл выдающийся французский учёный Клод Бернар, который заложил основы современной экспериментальной патологии. С его именем связано открытие гликогенобразовательной функции печени, липазы в секрете поджелудочной железы.
Известный немецкий патолог Вирхов является основателем клеточной
патологии, согласно которой всякий патологический процесс представляет собой сумму клеточных изменений. Он изучил и систематизировал ценный материал относительно клеточных изменений, лежащих в основе атрофии, гипертрофии, воспаления, опухолей. Его ученик Ю. Конгейм известен своими выдающимися экспериментальными исследованиями, посвященными эмболии, инфаркту, воспалению.
В России основоположником патологической физиологии как самостоятельной науки и предмета преподавания был В.В. Пашутин, возглавлявший с
13
1878 г. Санкт-Петербургскую Медико-хирургическую академию. С целью разработки проблем патологии В.В. Пашутин создал первый калориметр, предложил оригинальные методы исследования газообмена и теплообмена у человека
и животных. Используя эти методы, В.В. Пашутин и его ученики выполнили
капитальные исследования в области изучения различных видов голодания,
обмена веществ и теплообмена при патологических состояниях.
Одним из представителей отечественного общебиологического направления в патофизиологии и исследователей целлюлярной патологии является выдающийся биолог И.И. Мечников. Его исследования показали, как с повышением организации животных форм в процессе эволюции фагоцитознопищеварительная функция фагоцитоза переходит к приспособлению защитного
характера. С1898 по 1908гг. в лаборатории И.И. Мечникова были проведены
исследования по получению токсических сывороток и антител к разнообразным чужеродным клеткам и тканевым экстрактам.
В 1900г. В.К. Линдерман открыл нефролитическую сыворотку, способную вызывать у экспериментальных животных геморрагический нефрит. Так
была одна из первых экспериментальных моделей аутоиммунного заболевания.
А.В. Репрев известен как один из родоначальников отечественной эндокринологии. Его «Основы общей и экспериментальной патологии» способствовали развитию экспериментально-физиологического и физико-химического
направления в патологии. Работы Репрева по изучению патологии эндокринной
системы получили дальнейшее развитие в исследованиях представителей
Харьковской школы патофизиологов: Д.Е. Альперна, М.М. Павлова, Д.П. Гринева и др.
Начало формированию гистофизиологического направления в патологии
положил Н.А. Хржонщевский, возглавивший кафедру общей патологии в Киевском университете – автор физиологической инъекции красок для окрашивания органов и клеток. Его ученик В.В. Подвысоцкий, которого по праву считают основателем украинской школы патофизиологов, внёс значительный вклад
в изучение процессов регенерации эпителиальных тканей. Ярким представителем киевской школы являлся А.А. Богомолец. Его труды посвящены различным проблемам патофизиологии: роли реактивности в патологии, связи реактивности с эндокринной регуляцией и реакцией соединительной ткани, проблемам переливания крови, опухолевого роста. А.А. Богомолец выдвинул концепцию старения организма, связав этот процесс с изменениями физикохимических свойств тканей и межклеточного вещества, ослаблением трофической функции соединительной ткани. Он создал школу патофизиологов, из которой вышли Н.Н. Сиротинин, Н.А. Федоров, Н.Н. Зайко.
Основные труды Н.Н. Сиротинина посвящены вопросам сравнительной
патологии реактивности и резистентности организма. С помощью методов
сравнительной патологии Н.Н. Сиротинин показал, что филогенетически
наиболее древняя форма инфекции – простое размножение микроорганизмов в
тканях, а наиболее новой является выработка антител и спецификация фагоцитарного процесса. Кроме того, Н.Н. Сиротинину принадлежит ряд работ, по-
14
священных изучению патогенеза горной болезни и эволюции механизмов адаптации к гипоксии.
ОБЩАЯ НОЗОЛОГИЯ, ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Общая нозология
Нозология (греч. Nosos – болезнь) – учение о болезни. Патология – это
тоже учение о болезни, однако этот термин является более широким понятием,
чем «нозология», так как болезнь рассматривается во всех аспектах : морфологическом, функциональном, биохимическом и т.д.
Общая нозология- это часть патологии, которая рассматривает следующие
вопросы :
1. Номенклатуру и классификацию болезней.
2. Основные понятия и категории патологии.
3. Сущность болезни на разных этапах развития медицины.
4. Социальные аспекты патологии.
Номенклатура – это обширный перечень или каталог наименований известных болезней ( нозологических форм ). Классификация болезней – определенная система распределения болезней и патологических процессов на классы, группы , подгруппы и т.д. в соответствии с установленными критериями.
Наиболее принята в настоящее время Международная классификация и номенклатура болезней, предложенная ВОЗ.
ЗДОРОВЬЕ И БОЛЕЗНЬ
Ключевыми, наиболее общими категориями патологии являются понятия
здоровья и болезни. Обе эти категории являются особыми формами жизни.
Коренной особенностью существования .живых существ является приспособление (адаптация) организма к постоянно изменяющимся условиям
внешней среды. При этом реакции организма направлены на сохранение в нем
постоянства внутренней среды , активное равновесие внутренней среды, характеризующее ее стабильность, или гомеостаз. Включение адаптационных (приспособительных) механизмов, обеспечивающих гомеостаз, обусловливается
действием разнообразных факторов внешней среды. По силе различают воздействия трех видов:
1. Постоянные обычные воздействия на организм человека, в подавляющем большинстве являющиеся оптимальными и вызывающие многочисленные
отклонения в организме, которые вполне корригируются соответствующими
процессами ауторегуляции.
2. Сверхоптимальные воздействия, которые довольно часто встречаются в
жизни современного человека. Эти воздействия немедленно включают более
высокие уровни адаптивной регуляции.
3. Чрезвычайные (или экстремальные) воздействия, которые встречаются
все чаще. Для них характерны мобилизация нейроэндокринных механизмов и
развитие стресс-реакции.
15
Следует подчеркнуть, что названные реакции на воздействие -это реакции
здорового организма. Элементы, из которых состоит организм – это программные системы, они обладают значительной автономией , руководствуются в своем поведении эндогенными программами, которые могут отвечать на системные управляющие сигналы.Именно такое объединение элементов организма
делает состояние здоровья устойчивым.
В практической деятельности врача часто встречаются такие понятия, как
нормальная температура тела, нормальное давление крови и т.п.. При этом имеют в
виду средний результат измерений тех или других показателей в определенной
популяции.
Показатели, которые встречаются наиболее часто, принимаются за нормальные, а
человек, которых их имеет, считается здоровым. Всемирная организация
здравоохранения еще в 1946 году приняла такое определение здоровья:
“Здоровье- это состояние полного физического, психического и социального
благополучия, а не только отсутствие болезней или физических дефектов". Это
довольно обобщенное определение характеризует то, к чему следует стремиться,
видеть желательное соответствующей действительности.
Здоровье- это, прежде всего,состояние организма, в котором объединяются
соответствие структуры и функции, а также свойство регулирующих систем
поддерживать постоянность внутренней среды (гомеостаз). Здоровье – это
относительно совершенная и устойчивая форма жизнедеятельности,обеспечивающая
экономичные оптимальне механизмы приспособления к окружающей бреде и
позволяющая иметь функциональный резерв, используемый для ее изменения.
Здоровье состоит в том, что в ответ на действие ежедневных раздражителей
возникают адекватные реакции, которые по характеру, силе, времени и
продолжительности действия присущи большинству людей данной популяции. Вывод о
здоровье основывается на антропометрических, морфологических, физиологических
и биохимических исследованиях, а также учитываются и социальные критерии,
прежде всего степень участия человека в трудовой и общественной деятельности.
Физиологической мерой здоровья есть норма.
Таким образом, основными критериями здоровья следует считать :
- уравновешенность организма с внешней средой;
- соответствие структуры, функции и метаболизма;
- способность организма поддерживать гомеостаз;
- полноценное участие в трудовой деятельности.
Болезнь – особая форма жизнедеятельности организма или его
взаимоотношений с средой. Способность болеть – такое же неотъемлемое свойство
живого, как обмен веществ, размножение , смерть.Неживые объекты не болеют,
болезнь – свойство высокоорганизованных программных систем. Болезнь –
вынужденная неустойчивая форма жизнедеятельности организма, характеризующаяся
опасным приспособлением к условиям существования, при котором имеет место
рассогласование между реализуемой адаптивной программой и конкретной ситуацией.
Болезнь снижает эффективность функционирования системы и ее резервы.
Болезнь может быть следствием информационных нарушений ( патология
сигнализации и рецепции ), технических и технологических нарушений в программном
16
аппарате ( генетический материал ), повреждения исполнительного аппарата ( клетки,
органы, системы органов ).
Анализ классических и современных определений категории болезни позволяет
выделить основные содержание и признаки болезни :
1. Болезнь всегда имеет определенную первоначальную причину. Причиной болезни
может быть взаимодействие с фактором внешней среды ( экзогенный этиологический фактор). Причину заболевания может содержать и сам организм в виде своего
взаимодействия с внутренним этиологическим фактором.
2. Болезнь всегда системная, то есть реакция всего организма на действие этиологического фактора. Эту реакцию определяют : а) специфика, сила и длительность
действия этиологического фактора; б) реактивность организма. Реактивность организма – это генетически детерминированная и находящаяся под влиянием факторов
внешней среды совокупность свойств и качеств организма, определяющая характер
системной реакции на действие этиологического фактора.
3. На каждом этапе своего развития болезнь есть результат взаимодействия : а)
вызванных этиологическим фактором патологических реакций и патологических
процессов, которые могут привести к нарушениям гомеостаза или вызывают их;
б) защитных реакций в ответ на повреждение и нарушение гомеостаза; в) реакций
приспособления к повреждению, нарушению гомеостаза и расстройствам функциональных систем, связанных с болезнью; г) компенсации расстройств функциональных систем, связанных с заболеванием. Защитные, приспособительные и компенсаторные реакции составляют совокупность реакций саногенеза, направлены на
выздоровление и реабилитацию больных и используют для реализации резервы
самого организма, а также восполняются и повышаются оптимальной терапией.
Реакции саногенеза при нормальной реактивности, как правило, несколько избыточны относительно факторов, их вызвавших.Это обуславливает возможность их
превращения в звенья патогенеза болезней и патологических процессов. Естественный отбор дал организмам стереотипные приспособления. Их целесообразность
наивысшая в тех стандартных ситуациях , на которые они рассчитаны, а реальные
ситуации всегда в той или иной степени отличаются от стандартных.
При быстром изменении условий жизни, как это происходит с современным человечеством, рассогласование стереотипной программы и реальной ситуации сказывается на эффективности и цене адаптации. Например, активация симпатоадреналовой и ренин-ангиотензин-альдостероновой системы – стереотипный запрограммированный защитный механизм, направленный на адаптацию к острой кровои плазмопотере ( волюмосберегающая реакция). Она включается стереотипно при
снижении сердечного выброса через раздражение рецепторов крупных сосудов.
При острой кровопотере эта реакция приводит к уменьшению мочеотделения, задержке натрия и воды в организме, что позволяет организму поддерживать объем
циркулирующей крови на должном уровне.
При сердечной недостаточности в организме человека нет недостатка воды и солей. Уменьшение сердечного выброса связано слабостью сократительной функции
миокарда. Но в ответ на снижение сердечного выброса срабатывает стереотипная
программа задержки натрия и воды в организме. В результате формируются системные отеки.
17
4. При болезни меняются пределы функционирования нормальных механизмов
регуляции и действуют программы адаптации, которые не работали в
здоровом состоянии. Такие программы называются аварийными. Отличиями аварийных программ от физиологических являются их меньшая экономичность, малая эффективность, филогенетическая древность происхождения. Примером такой аварийной программы является дыхание Куссмауля,
наблюдаемое при глубоком ацидозе, в состоянии комы.
Аварийная регуляция заложена в программном наборе реактивности организма и архивируется при нормальной жизнедеятельности. Онкогены здоровых дифференцированных клеток кодируют архивированную программу
размножения и эмбрионального метаболизма. Под влиянием канцерогенных
факторов происходит ее несвоевременное или избыточное разархивирование, что приводит к малигнизации и развитию опухолевого процесса. Переход клетки на новые адаптивные механизмы оказывается вредоносным по
отношению к другим клеткам.
Таким образом « Болезнь человека… противоречивый процесс развития
повреждения и компенсации ( защиты ), не адекватный условиям среды, снижающий трудоспособность и способный прекратить существование организма
как целого « ( А. Д. Адо, 1985 ).
Болезнь – это динамическая интеграция патологических процессов, характеризующаяся диалектическим единством повреждения и защиты, ограничением защитно-приспособительных возможностей и понижением трудоспособности человека.Это определение следует из положения, что нормальная
жизнедеятельность есть результат постоянного приспособления организма к
беспрерывно меняющимся условиям окружающей среды.
Начало нарушения оптимального взаимодействия организма и внешней
среды получило название предболезни.
Патологическая реакция - кратковременная необычная реакция организма на какое-либо воздействие. Патологическая реакция по своим параметрам
выходит за границы нормы реагирования, свойственной данному организму.
Патологическая реакция- элемент патологического процесса, проявляющий одновременно и относительную адекватность и потенциальную патогенность. Таковыми являются фагоцитоз при воспалении, централизация кровообращения
при шоке, патологические рефлексы при патологических процессах с участием
ЦНС.
Патологический процесс – сложные мозаичные элементы болезней,
состоящие из неразрывно связанных общими механизмами патологических и
защитно-приспособительных реакций в поврежденных тканях, органах или организме, проявляющихся в виде морфологических, метаболических и функциональных нарушений.Примерами могут служить тромбоз, алкалоз, гипоксия,
воспаление, кома.
Часто различные патологические процессы и отдельные патологические
реакции клеток, тканей у человека и животных встречаются в виде постоянных
сочетаний или комбинаций, сформировавшихся и закрепленных в процессе
эволюции. Это типовые патологические процессы- процессы, которые раз-
18
виваются по одним и тем же законам, независимо от причины и локализации. К
ним относятся воспаление, отек, опухоль, гипоксия, лихорадка и др. Типовые
патологические процессы у человека и высших животных имеют много общего.
Воспаление, опухоли, отек, дистрофии встречаются как у позвоночных, так и у
беспозвоночных животных. Однако у последних они существенно отличаются
от таковых у человека и высших позвоночных.
Патологический процесс лежит в основе болезни, но не является ею. Отличия патологического процесса от болезни заключаются в следующем:
1. Болезнь всегда имеет одну главную этиологическую причину (специфический, производящий фактор), патологический процесс полиэтиологичен
2. Патологический процесс может быть локальным, затрагивать весь организм, болезнь – всегда системна..
3. Один и тот же патологический процесс может обусловливать различные картины болезней в зависимости от локализации.
4. Болезнь — часто комбинация нескольких патологических процессов.
5. Патологический процесс может не сопровождаться снижением приспособляемости организма и ограничением трудоспособности.
Патологическое состояние — это медленно (вяло) текущий патологический процесс. Может возникнуть в результате ранее перенесенного заболевания
(например, рубцовое сужение пищевода после ожоговой травмы; состояние после резекции почки, ампутации конечности и т. п.) или в результате нарушения
внутриутробного развития (плоскостопие, косолапость и пр.). Это как бы итог
закончившегося процесса, в результате которого стойко изменилась структура
органа, возникли атипические замещения в определенной ткани или части организма. В ряде случаев патологическое состояние может снова перейти в болезнь.
Разнообразные повреждения и приспособительные реакции во время болезни проявляются разными отклонениями жизнедеятельности от нормы. Эти
проявления болезни получили название симптомов, а совокупность симптомов,
которые характеризуют болезнь — синдромов.
В течении болезни выделяют 4 периода: 1) латентный (скрытый, инкубационный); 2) продромальный; 3) период разгара; 4) завершающий период - исход.
Латентный период (относительно инфекционных болезней - инкубационный
период) длится от момента влияния причины к появлению первых клинических
признаков заболевания. Этот период может быть короткой, как при действии боевых
ядовитых веществ, и довольно продолжительным, как при проказе (несколько лет).
Продромальный период - отрезок времени от первых признаков болезни
до полного ее проявления. Иногда этот период имеет яркие клинические проявления (крупозная пневмония, дизентерия), в других случаях есть нечеткие симптомы, например, беспричинная веселость (эйфория) при горной болезни.
Период выраженных проявлений, или разгара болезни характеризуется полным развитием клинической картины, появлением специфических симптомов и
синдромов заболевания: судороги при недостаточности паращитовидных желез,
19
лейкопения при лучевой болезни, типичная триада (гипергликемия, глюкозурия,
полиурия) у больных сахарным диабетом.
Исход болезни может быть следующий: выздоровление (полное и неполное), рецидив, переход в хроническую форму, смерть.
Выздоровление - процесс восстановления затронутой жизнедеятельности и
формирование нормальных взаимоотношений организма с окружающей средой,
для человека - прежде всего восстановления его трудоспособности. При полном выздоровлении следов болезни не остается, а при неполном - тот или иной дефект,
например, порок сердца, сохраняется, но он может быть на протяжении продолжительного времени компенсированным.
Рецидив (возвращение болезни) - это новое проявление болезни после временного исчезновения, ослабления или приостановки болезненного процесса (ремиссии). Рецидивирующее течение характерно для некоторых инфекционных заболеваний ( малярия, бруцеллез, брюшной и возвратный тиф ) и соматических заболеваний ( подагра, ревматизм, язвенная болезнь желудка).
При ослаблении защитных сил организма болезнь может затягиваться, острые ее
проявления исчезают, но выздоровление не наступает, болезнь приобретает хроническое течение. Для хронических заболеваний характерные периоды обострения и
ремисси.
Ремиссия - временное улучшение состояния больного, которое проявляется
частичным или полным исчезновением клинических проявлений заболевания. Тем не
менее после улучшения состояния закономерно наступает обострение болезни
(рецидив). Ремиссия может продолжаться от нескольких дней до нескольких лет.
Болезнь может закончиться инвалидностью или смертью больного.
Смерть – самый неблагоприятный исход заболевания. Она может быть естественной ( физиологическая от дряхлости, старения ) и преждевременной . Преждевременная смерть может быть насильственной ( убийство ) и от заболевания .Кроме того,
выделяют смерть мозговую ( внезапная гибель головного мозга на фоне всех здоровых
органов, поддерживаемых искусственной вентиляцией легких ) и соматическую,
наступающую вследствие необратимого, несовместимого с жизнью поражения органа,
органов или систем.
Прекращение жизненных функций происходит постепенно и динамично. В процессе умирания выделяют несколько фаз: преагония, агония, клиническая смерть,
биологическая смерть. Преагонию, агонию, клиническую смерть называют терминальными состояниями.
Преагония – характеризуется сумеречным помрачением сознания, которое, как
правило, сохранено, но может бать затемнено, спутано. АД- снижено, дыхание – резко
учащается. Затем тахикардия и тахипноэ сменяются брадикардией и брадипноэ.
Преагония заканчивается терминальной паузой – прекращение дыхания (апноэ) и резкое замедление сердечной деятельности вплоть до временной асистолии. Апноэ может
продолжаться от нескольких секунд до 3-4 минут. После терминальной паузы наступает агония.
Агония (agon; греч. – борьба) – терминальное состояние , характеризующееся глубоким нарушением функции высших отделов мозга, особенно коры больших полушарий, с одновременным возбуждения продолговатого мозга. Главным признаком агонии
20
служит появление после терминальной паузы первого самостоятельного вдоха. В дыхании участвует вспомогательные мышцы – мышцы шеи и лица, т.е. появляется
«гаспинг» - дыхание. Сознание отсутствует, иногда кратковременно проясняется, исчезают глазные рефлексы и реакция на внешние раздражители. Происходит расслабление
сфинктеров, непроизвольное выделение кала и мочи. Продолжительность агонии 2-4
минуты.
Клиническая смерть ( mors clinicalis ) – терминальное состояние, наступающее
после прекращения сердечной деятельности и дыхания и продолжающееся до наступления необратимых изменений в высших отделах ЦНС. В обычных условиях продолжительность клинической смерти у человека максимум 5-6 минут. Во время клинической смерти внешние признаки жизни (сознание, рефлексы, дыхание, сердечная деятельность) отсутствуют, но организм как целое еще не умер, в его тканях сохраняются
энергетические субстраты и продолжаются метаболические процессы, поэтому при
определенных воздействиях можно восстановить исходный уровень и направленность
метаболических процессов, а значит восстановить все функции.
Комплекс мероприятий по оживления организма, восстановлению и нормализации
нарушенных метаболических процессов и функций называется реанимацией. Восстановление жизнедеятельности организма возможно при использовании комплексного метода оживления и метода искусственного кровообращения. Восстановления структурно-функциональных отделов головного мозга при оживлении организма происходит в зависимости от устойчивости нейронов к действию гипоксического фактора (продолговатый мозг, варолиев мост, промежуточный мозг, кора). Функции коры больших полушарий головного мозга являются менее резистентными к гипоксии и, время сохранения жизнеспособности
корковых нейронов в период клинической смерти ограничивается 3-5 минутами. Главная задача реанимационных мероприятий - восстановление интеллекта (полноценная социальная реабилитация) больного, перенесшего терминальное состояние.
Биологическая смерть необратимое прекращение жизнедеятельности организма.
К абсолютным признакам биологической смерти относятся:
1. Трупное охлаждение – процесс понижения температуры трупа до уровня температуры окружающей среды.
2. Появление на коже трупных пятен.
3. Трупное окоченение – процесс посмертного уплотнения скелетных мышц и
гладкой мускулатуры внутренних органов.
4. Трупное разложение – процесс разрушения органов и тканей трупа под действием собственных протеолитических ферментов и ферментов, выделяемых микроорганизмами.
Общая этиология
Слово “этиология” означает учение о причине (от греч. aitia — причина,
logos - разум, учение). В древности это слово означало также учение о болезнях
вообще (Гален). В современном понимании этиология — учение о причинах
и условиях возникновения и развития болезней.
21
Причины болезней
Причиной болезни называют материальный фактор внешней
или
внутренней среды ( этиологический, производящий, специфический), который взаимодействует с организмом, вызывает заболевание и сообщает ему специфические черты. Например, причиной лучевой болезни является ионизирующая радиация, причиной инфекционной болезни — патогенные микробы.
Нередко, однако, возникновение болезни связано с воздействием не одного, а нескольких факторов. Например, крупозное воспаление легких возникает
не только под влиянием заражения человека пневмококком. Заболеванию способствуют также простуда,утомление, отрицательные эмоции, недостаточное
питание и другие предрасполагающие условия. Тем не менее без заражения
пневмококком все указанные факторы не смогут вызвать крупозное воспаление
легких. Поэтому причиной этого заболевания следует считать пневмококк. На
основании изложенного под причиной болезни нужно понимать такое воздействие, без которого развитие данного заболевания невозможно.
Однако иногда установить причину болезни трудно (некоторые опухоли,
психические болезни). Доказано, например, что язва желудка развивается как
от грубой пищи, так и от состояния невроза, нарушений функций вегетативной
нервной системы, эндокринных расстройств. Эти и многие другие наблюдения
послужили поводом для представлений о полиэтиологичности болезни. Положение это неверно. Оно возникло в результате недостаточности наших знаний о причинах некоторых болезней и их вариантов.
Как указывалось, каждая болезнь имеет свою, только ей свойственную причину. По мере накопления знаний о причинах всех видов и подвидов болезней
будут улучшаться их предупреждение и лечение. Многие болезни, когда информация об их подлинных причинах становится значительной, распадаются
на новые подвиды, каждый из которых имеет свою отдельную причину.
Например, раньше существовала болезнь “кровоточивость” (геморрагический
диатез). При изучении причин, вызывающих отдельные проявления этого заболевания, выявились новые, совершенно самостоятельные формы болезни, характеризующиеся кровоточивостью (цинга, гемофилия, геморрагическая пурпура и др.). Подобным образом распался на самостоятельные заболевания со
своими причинами нервно-артрический диатез (подагра, ревматизм, неинфекционный полиартрит и др.).
Различают причины болезней внешние ( экзогенный этиологический
фактор ) и внутренние( эндогенный этиологический фактор ). К внешним
причинам относят механические, физические, химические, биологические и социальные факторы, К внутренним - наследственность, конституцию, возраст, пол, реактивность. Следует указать, что формирование внутренних причин в процессе эволюции складывается также в тесном взаимодействии с
внешней средой. Поэтому название “внутренние причины” болезней в некоторой степени условно. Оно означает, что у данного человека болезнь развилась
без видимых влияний внешней среды.
22
Условия возникновения и развития болезней
Факторы, которые воздействуя на организм, сами по себе вызвать заболевание не могут, но влияют на возникновение и развитие болезней, называются
условиями (conditio – лат. условие ) возникновения болезни. В отличие от причинного фактора условия не являются обязательными для развития заболевания. При наличии причинного фактора болезнь может развиться и без участия
некоторых условий ее возникновения. Например, крупозная пневмония, вызываемая пневмококком сильной вирулентности, может развиться и без простуды,
без ослабления питания и других условий.
Различают условия, предрасполагающие к болезни или способствующие
ее развитию и препятствующие возникновению болезни и ее развитию. Как
способствующие, так и препятствующие развитию заболеваний условия могут
быть внутренними и внешними.
К внутренним условиям, способствующим развитию болезни, относят
наследственное предрасположение к заболеванию, патологическую конституцию (диатез), ранний детский или старческий возраст.
К внешним условиям, способствующим развитию болезней, относят
нарушения питания, переутомление, невротические состояния, ранее перенесенные болезни, плохой уход за больным.
К внутренним условиям, препятствующим развитию болезней, относят
наследственные, расовые и конституциональные факторы. К ним относится,
например, видовой иммунитет человека к некоторым инфекционным заболеваниям животных. Человек не болеет чумой собак и кошек, пневмонией рогатого
скота и многими другими инфекционными болезнями животных. Люди, страдающие серповидно-клеточной анемией, не болеют малярией.
К внешним условиям, препятствующим развитию болезней, относят
хорошее и рациональное питание, правильную организацию режима рабочего
дня, физкультуру, а в случае заболевания — хороший уход за больным.
Установление главного этиологического (производящего, специфического)
фактора, выделение условий, предрасполагающих к болезни или способствующих ее развитию, и условий, препятствующих возникновению болезни и ее
развитию, абсолютно необходимо для разработки эффективных мер профилактики заболеваний, снижения заболеваемости и оздоровления населения.
Факторы, не являющиеся непосредственной причиной болезни, но действие которых в популяции людей статистически достоверно увеличивает вероятность возникновения определенной болезни, называют факторами риска.
Так, например, факторами риска атеросклероза являются дислипопротеинемии,
артериальная гипертензия, курение, сахарный диабет, принадлежность к мужскому полу.
Общий патогенез
Термин "патогенез" происходит от двух слов: греч. pathos - страдание и
genesis -происхождение, развитие. Патогенез - это учение о механизмах развития, течения и исхода болезней, патологических процессов и патологических
состояний. Изучая патогенез, мы отвечаем на мопрос: как, каким образом воз-
23
никло заболевание, т.е. выясняем механизмы развития болезни и имеем дело
преимущественно с внутренними факторами.
Патогенез - это совокупность механизмов, включающихся в организме
при действии на него вредоносных (патогенных) факторов и проявляющихся в
динамическом стереотипном развертывании функциональных, биохимических
и морфологических реакций организма, обусловливающих возникновение, развитие и исход заболевания.
По широте охвата изучаемых вопросов различают:
а) частный патогенез, который изучает механизмы отдельных патологических реакций, процессов, состояний и заболеваний (нозологических единиц).
Частный патогенез изучают клиницисты, раскрывая механизм конкретных заболеваний у конкретных больных (например, патогенез сахарного диабета,
пневмонии, язвенной болезни желудка и т.д.). Частный патогенез относится к
конкретным нозологическим формам.
б) общий патогенез предполагает изучение механизмов, наиболее общих закономерностей, лежащих в основе типовых патологических процессов или отдельных категорий болезней (наследственных, онкологических, инфекционных, эндокринных и т.д.). Общий патогез занимается изучением механизмов,
приводящих к функциональной недостаточности какого-либо органа или системы. Например, общий патогенез изучает механизмы развития сердечной недостаточности у больных с патологией сердечно-сосудистой системы при пороках сердца, инфаркте миокарда, ишемической болезни сердца, заболеваниях
легких с легочной гипертензией.
Изучение патогенеза сводится к изучению так называемых патогенетических факторов, т.е. тех изменений в организме, которые возникают в ответ на
воздействие этиологического фактора и в дальнейшем играют роль причины и
развитии болезни. Патогенетический фактор вызывает появление новых расстройств жизнедеятельности в развитии патологического процесса, болезни.
Пусковым механизмом (звеном) любого патологического процесса, заболевания является повреждение, возникающее под влиянием вредоносного ( патогенного ) фактора.
Повреждения могут быть:
- первичными; они обусловлены непосредственным действием патогенного
фактора на организм;
- вторичными; они являются следствием влияния первичных повреждений на
ткани и органы, сопропождаются выделением биологически активных веществ
(БАВ), протеолизом, ацидозом, гипоксией, нарушением микроциркуляции,
микротромбозом и т.д.
Характер повреждения зависит от природы раздражителя (патогенного
фактора), видовых и индивидуальных свойств живого организма. Уровни повреждения могут быть различными: на молекулярном, клеточном, тканевом,
органном и организменном. Один и тот же раздражитель может вызвать повреждения на самых различных уровнях.
24
Одновременно с повреждением включаются защитно-приспособительные и компенсаторные процессы на тех же самых уровнях - молекулярном,
клеточном, тканевом, органном и организменном.
Повреждения на молекулярном уровне носят локальный характер и проявляются разрывом молекул, внутримолекулярными перестройками, что приводит к появлению отдельных ионов, радикалов, образованию новых молекул и
новых веществ, оказывающих патогенное действие на организм. Межмолекулярные перестройки способствуют появлению веществ с новыми антигенными
свойствами.
Но одновременно с повреждением включаются и защитно-приспособительные и компенсаторные процессы на молекулярном уровне. Например, при
наследственных заболеваниях первичное повреждение локализуется в генетическом аппарате на молекулярном уровне. Эта генная мутация вызывает нарушение синтеза белков, ферментов, что влияет на обменные процессы в организме, обусловливает нарушение структуры и функции органов и систем. При
таких повреждениях включаются и защитно-приспособительные и компенсаторные процессы, которые приводят к репарации генетического аппарата. При
соматических мутациях, например, в процессе онкогенеза, большую роль играет клеточное звено иммунитета, обеспечивающее лизис мутантных клеток.
Повреждения на клеточном уровне характеризуются структурными и метаболическими нарушениями, сопровождаются синтезом и секрецией биологически активных веществ: гистамина, серотонина, гепарина, брадикинина и др.
Многие из них оказывают патогенное действие, повышая проницаемость сосудов микроциркуляторного русла, усиливая экстравазацию и как следствие сгущение крови, нарастание ее вязкости, наклонность к сладжированию и микротромбозу, т.е. нарушению микроциркуляции. Повреждения на клеточном
уровне сопровождаются нарушением ферментативной активности: отмечается
ингибирование ферментов цикла Кребса и активация гликолитических и лизосомальных ферментов, что вызывает нарушение обменных процессов в клетке.
При повреждении клетки, особенно в условиях гипоксии, образуется большое
количество недоокисленных продуктов обмена, обусловливающих внутриклеточный ацидоз и нарушающих гомеостаз в целом. Структурные изменения
клетки характеризуются нарушением внутриклеточных органелл. Следствием
структурно-метаболических изменений может наступить перерождение клетки
вплоть до ее гибели.
Однако, образующиеся при повреждении или гибели клетки биологически активные вещества стимулируют процессы репаративной регенерации, что
обеспечивает нейтрализацию действия этиологического фактора, а функции
поврежденных и погибших клеток компенсируется за счет регенерации или гипертрофии оставшихся клеток. В других случаях дефект, вызванный повреждением клеток, замещается соединительной тканью.
Повреждения на тканевом уровне характеризуются нарушением основных функциональных свойств, развитием патологического парабиоза, перерождением тканей. Нарушение основных функциональных свойств сопровождается
25
снижением функциональной подвижности, уменьшением функциональной лабильности ткани.
Патологический парабиоз в отличие от физиологического не приводит к
восстановлению исходного состояния ткани. Он протекает по тем же стадиям,
что и физиологический, но при нем резко снижен уровень функциональной подвижности, отмечается ограничение функций, перерождение тканей (например,
жировая дистрофия сердечной мышцы, печени, коллагенозы и др.).
Защитно-приспособительные и компенсаторные процессы на тканевом
уровне проявляются включением ранее не функционировавших капилляров,
образованием новых микрососудов, что улучшает трофику поврежденных тканей.
Повреждения на органном уровне характеризуются снижением, извращением или потерей специфических функций органа, уменьшением доли участия поврежденного органа в общих реакциях организма. Например, при инфаркте миокарда, клапанных пороках сердца нарушается функция сердца и доля его участия в адекватном гемодинамическом обеспечении функционирующих органов и систем. Компенсаторные реакции и процессы при этом формируются на уровне органа, системы и даже организма в целом, что приводит,
например, к гипертрофии соответствующего отдела сердца, изменению его регуляции, что сказывается на гемодинамике - в итоге возникает компенсация
нарушенных функций.
При первичном повреждении на системном или организменном уровне
возникает генерализованное выпадение или ограничение той или иной функции, что особенно отчетливо наблюдается при заболеваниях ЦНС, эндокринных поражениях. При этом происходит сложная перестройка регуляторных
процессов, обмена веществ, что в ряде случаев позволяет организму сохранить
жизнь. К числу общих компенсаторных реакций, процессов при повреждении
на системном или организменном уровне относятся лихорадка,общий адаптационный синдром и т.д. Компенсаторно-приспособительные реакции направлены на защиту и восстановление нарушенных функций организма как целого.
Патогенное действие повреждающих факторов реализуется на уровне
функционального элемента органа. Функциональный элемент по А.М.Чернух это микросистема, представляющая собой упорядоченный структурно-функциональный комплекс, составляющий интегральное целое, состоящий из клеточных и волокнистых образований органа, включающий все его ткани, на основе
которого осуществляются обменные тканевые процессы. Более кратко это понятие звучит так: функциональный элемент - это совокупность паренхимы
клетки, микроциркуляторной единицы, нервных волокон и соединительной
ткани. Каждый функциональный элемент ткани состоит из:
- паренхимы клеток;
- артериол, прекапилляров, капилляров, посткапилляров, венул, лимфатических
капилляров, артериоло-венулярных анастомозов;
- нервных волокон с рецепторами;
- соединительной ткани;
- тканевых базофилов.
26
Функциональный элемент осуществляет:
а) транскапиллярный обмен питательных веществ ( нутриентов ), кислорода,
углекислоты и продуктов метаболизма тканей;
б) регуляцию системной и регионарной гемодинамики благодаря наличию в
нем резистивных и емкостных сосудов, артериоло-венулярных шунтов и резерных (не функционирующих в определенный момент) капилляров.
Функциональные элементы участвуют в местных и общих реакциях повреждения , защитно-приспособительных и компенсаторных процессах за счет
включения в работу резервных функциональных элементов ткани.
Причинно-следственные отношения патогенезе. Ведущее( главное ) звено
патогенеза. Порочный круг. Местные и общие, специфические и неспецифические реакции в патогенезе.
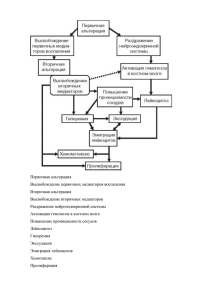
Каждый патологический процесс, заболевание рассматривается как длинная цепь причинно-следственных отношений, которая распространяется по типу цепной реакции. Первичным звеном в этой длинной цепи является повреждение, возникающее под влиянием патогенного фактора, и которое становится
причиной вторичного повреждения, вызывающего третичное и т.д. (Воздействие механического фактора травма-кровопотеря - централизация кровообращения -гипоксия - ацидоз - токсемия, септицемия - и т.д.).
В этой сложной цепи причинно-следственных отношений всегда выделяют основное (синонимы: главное, ведущее) звено. Под основным (главным)
звеном патогенеза понимают такое явление, которое определяет развитие процесса с характерными для него специфическими особенностями. Например, в
основе артериальной гиперемии лежит расширение артериол (это главное звено), что обусловливает ускорение кровотока, покраснение, повышение температуры гиперемированного участка, увеличение его в объеме и активацию обменных процессов. Главным звеном патогенеза острой кровопотери является
дефицит обьема циркулирующей крови (ОЦК), который обусловливает снижение артериального давления, централизацию кровообращения, шунтирование
кровотока, ацидоз, гипоксию и т.д. При устранении главного звена наступает
выздоровление.
Несвоевременное устранение главного звена приводит к нарушению гомеостаза и формированию порочных кругов ( circulus vitiosus ) патогенеза.
Они возникают тогда, когда появившееся отклонение уровня функционирования органа или системы начинает поддерживать и усиливать себя в результате
образования положительной обратной связи. Например, кровопотеря сопровождается патологическим депонированием крови - выходом ее жидкой части
из сосудистого русла - дальнейшим нарастанием дефицита ОЦК - углублением
артериальной гипотензии, что через барорецепторы активирует симпатоадреналовую систему.Это усиливает сужение сосудов и централизацию кровообращения - в конечном итоге, нарастает патологическое депонирование крови и дальнейшее уменьшение ОЦК; в результате этого патологический процесс прогрессирует.
27
Другой пример. При гипертонической болезни ускоряется развитие атеросклероза, что приводит к нарушению функции барорецепторов и понижению
их чувствительности к изменениям АД и в результате этого АД стабильно держится на высоких цифрах. Сужение сосудов почек вызывает гипоксию и включение системы ренин-ангиотензин, что еще больше усиливает спазм сосудов ,
повышает АД и способствует усилению секреции альдостерона.. Гиперсекреция альдостерона вызывает задержку натрия и гипернатриемию .Раздражение
осморецепторов активирует секрецию АДГ( антидиуретический гормон, вазопрессин) , что приводит к усилению реабсорбции воды в почечных канальцах увеличивается ОЦК и происходит дальнейшее нарастание АД.
Образование порочных кругов утяжеляет течение заболевания. Своевременная диагностика начальных стадий образования порочных кругов, предупреждение их становления и устранение главного звена патогенеза - залог
успешного лечения больного.
В сложной цепи причинно-следственных отношений выделяют местные
и общие изменения. Вопрос о взаимоотношении местных и общих явлений в
патогенезе болезни, патологического процесса остается достаточно сложным. В
целостном организме абсолютно локальных процессов не бывает. В патологический процесс, болезнь вовлекается весь организм. Как известно, при любой
патологии: пульпит, стоматит, локальный ожог, фурункул, аденома гипофиза страдает весь организм. И тем не менее, значение локальных и общих явлений в
патогенезе весьма вариабельно. Можно выделить 4 варианта взаимосвязи
местных и общих процессов в патогенезе:
1. Процесс начинается с местного повреждения органа или ткани в результате
действия внешних или внутренних факторов, затем включаются адаптивные
реакции, направленные на ограничение ( локализацию ) очага повреждения
(например, воспаление –экссудация,грануляционный вал, пиогенная капсула,
барьерная функция лимфоузлов).Участие общих реакций организма мобилизует локальные тканевые адаптивные механизмы, вследствие чего основные параметры гомеостаза (температура тела, количество лейкоцитов и лейкоцитарная
формула, СОЭ, обмен веществ) существенно не меняются.
2. Местный процесс через рецепторы и поступление в кровь и лимфу БАВ вызывает развитие генерализованной реакции организма и определенные сдвиги
параметров гомеостаза. Включаются приспособительные реакции, направленные на предупреждение развития общих патологических изменений в организме.
3. Генерализация местного процесса при его тяжелом течении отличается
максимальной напряженностью адаптивных и защитных реакций и процессов, а
также выраженностью патологических явлений на уровне организма. Возникает
общая интоксикация организма, сепсис. Параметры гомеостаза могут выйти за
рамки совместимых с жизнью изменений.
4. Локальные патологические изменения органов и систем развиваются вторично на основе первично генерализованного ( системного ) процесса
(шок – шоковые почки, шоковое сердце, шоковые легкие и т.д. ).
28
В любой болезни всегда можно различить специфические проявления,
характерные только для данного заболевания (генерализованная лимфоаденопатия при ВИЧ-инфекции), а также неспецифические, свойственные многим
болезням или даже всем. Эти признаки обусловлены стереотипными защитными реакциями, сформировавшимися в ходе эволюции. Эти реакции включаются
всякий раз, когда возникает патологическая ситуация, и связаны с изменениями
функций нервной и эндокринной систем. Таких неспецифических реакций по
крайней мере пять:
1. Парабиоз – застойное возбуждение, которое не распространяется и возникает в случае повреждения возбудимой ткани. Имеет значение в патогенезе некоторых блокад сердца.
2. Доминанта – наличие в центральной нервной системе очага возбуждения,
который подчиняет себе все другие центры. Может обусловливать развитие
психических заболеваний.
3. Нарушение кортико-висцеральной динамики обусловлено наличием тесной
связи между корой головного мозга и внутренними органами. Имеет значение в
развитии тиреотоксикоза.
4. Нарушении трофической функции нервной системы
5. Стресс. Разнообразные патогенные воздействия (высокие и низкие температуры, боль, интоксикация) вызывают стандартную неспецифическую реакцию, которая выражается в выбросе кортикотропина – гормона передней доли
гипофиза. На это надпочечники отвечают интенсификацией выработки гормонов, поддерживающих адаптативные реакции.
Патогенное действие этиологических факторов реализуется благодаря
трем механизмам патогенеза: прямого, гуморального и нейрогенного (нервнорефлеторного). Прямое повреждающее действие оказывают механические факторы, обладающие большим запасом кинетической энергии, физические ( тепловой -ожоги), химические (ожоги).
Гуморальные механизмы патогенеза опосредуются жидкими средами организма: кровью, лимфой, межклеточной жидкостью. Особая роль этим механизмам принадлежит в генерализации патологии (метастазирование, сепсис и
т.д.).
Нейрогенный механизм патогенеза опосредуется через нервную систему
вследствие нарушения регуляторных процессов.
Защитно-приспособительные, компенсаторные и восстановительные
процессы. Механизмы выздоровления.Патогенетические принципы терапии.
Важным выражением каждой болезни являются реактивные изменения со
стороны клеток, органов и систем, которые возникают всегда вторично, в ответ
на повреждение, вызванное болезнетворными причинами. К ним относятся такие процессы, как воспаление, лихорадка, отек и др.
Эти реактивные изменения в организме обозначаются как защитнокомпенсаторные процессы, или “физиологическая мера” защиты (И. П. Павлов), как “патологическая (или аварийная) регуляция функции”(В. В. Подвы-
29
соцкий, Н. Н. Аничков), как “целительные силы организма” (И. И. Мечников).
Н.Н. Зайко различает адаптацию, как проявление работы физиологических механизмов регуляции, и компенсацию, как результат работы аварийных механизмов.
В ходе развития болезни процессы повреждения и восстановления находятся
в тесном взаимодействии и, как указывал И. П. Павлов, часто трудно бывает
отделить один от другого.Эти процессы развиваются и протекают на различных
уровнях, начиная с молекулярного и заканчивая целым организмом больного
человека.
При изучении патогенеза заболевания особое внимание уделяется оценке
защитных механизмов, выработавшихся в процессе эволюции: барьерным образованиям, приспособительным и компенсаторным реакциям.
Барьеры - это морфологические и морфо-функциональные образования,
предохраняющие организм от патогенных факторов. К ним относятся кожа,
слизистые оболочки, костный покров черепа, передняя брюшная стенка, кишечник, ретикуло-эндотелиальная система - это все морфологические образования. К морфо-функциональным барьерам относятся гистогематический и гематоэнцефалический барьеры. Они представляют собой совокупность соединительнотканных элементов и капилляров, находящихся между кровью и тканями, а также между кровью, спинномозговой жидкостью и мозгом.
Гистогематический барьер обеспечивает постоянство состава и физикохимических свойств тканевой жидкости, а также задерживает переход в нее из
крови чужеродных веществ.
Гематоэнцефалический барьер защищает центральную нервную систему
от проникновения в ликвор чужеродных веществ, введенных в кровь, или продуктов нарушенного обмена веществ.
Барьерную функцию выполняют печень и буферные системы крови и
тканевой жидкости. Барьеры предупреждают возникновение и развитие болезни, а при их возникновении ограничивают распространение патогенного фактора, локализуют очаг повреждения.
Защитно-компенсаторные процессы включаются при действии патогенных факторов и представлены безусловными и условными рефлекторными реакциями. Безусловные реакции направлены на освобождение органа или ткани
от контакта с патогенным фактором (кашель, чихание, моргание, спазм сосудов
и др.).
При повторном контакте с патогенным фактором развивается условнорефлекторная реакция (учащение сердечных сокращений, подъем АД, слюнотечение, слезотечение) Действие условно-рефлекторных реакций более продолжительное.
Компенсаторные процессы направлены на компенсацию нарушений и
расстройств функциональных систем, вызванных патогенными факторами, и
характеризуются развитием гипертрофии органа, включением резервных клеток, репаративной регенерацией (ускоренное образование эритроцитов, лейкоцитов; новообразование миофибрилл и миюхондрий в кардиомиоцитах), усиле-
30
нием функции парного органа (почки, легкого) или органа и системы, смежных
по функции (печени и мочевыделительной системы).
Механизмы выздоровления ( саногенеза )
Выздоровление - это процесс восстановления нормальной жизнедеятельности организма после болезни, восстановление нарушенных функций больного организма и приспособление его к окружающей среде. Выделяют:
а) срочные неустойчивые (аварийные) механизмы. Они представлены защитными рефлексами - например, выделение катехоламинов при стрессе;
б) относительно устойчивые механизмы, действующие в течение всего периода заболевания: увеличение резервных клеток - лейкоцитоз, эритроцитоз; включение регуляторных систем -устанавливается пониженная теплопродукция при
повышении температуры окружающей среды;
в) продолжительно устойчивые механизмы - компенсаторная гипертрофия, репаративная регенерация, выработка антител, изменение пластических свойств
ЦНС, охранительное торможение, выработка условных рефлексов и усиление
безусловных рефлексов.
Знание механизмов патогенеза обеспечивает надежное лечение больного
и профилактику заболевания. Патогенетические терапия — комплекс мер,
направленных на прерывание цепи причинно-следственных отношений между
различными структурными, метаболическими и функциональными нарушениями, возникающими в организме вследствие воздействия главного этиологического фактора, путем устранения основного звена патогенеза. Например, стеноз
левого атриовентрикулярного отверстия служит основным звеном в цепи многих последующих нарушений: расширения левого предсердия, застоя крови в
малом круге, нарушения функции правого желудочка, а затем застоя в большом
круге кровообращения, кислородного голодания циркуляторного типа, одышки
и др. Устранение этого звена путем митральной комиссуротомии ликвидирует
все указанные нарушения.
Патогенетические терапия включают симптоматическую терапию, дезинтоксикационную и иммунодепрессивную терапию; лечение, направленное на
повышение резистентности организма; десенсибилизацию и терапию типовых
патологических процессов.
Основные направления учения о болезни
Современные представления в патологии сложились в результате исторического развития медицины.
В соответствии со взглядами некоторых древних философов на природу,
которую они считали состоящей из основных элементов – воды, огня, воздуха и
земли, основоположник медицины Гиппократ полагал, что организм человека
построен из жидкостей четырех родов: крови, слизи, желтой и черной желчи.
Он считал, что правильное смешение этих жидкостей обеспечивает здоровье.
Болезни же возникают в результате внешних воздействий, вызывающих неправильное смешение или загрязнение этих жидкостей. Таким было гуморальное
направление в понимании сущности болезней.
31
На основе учений Демокрита возникло солидарное направление учения
о болезни. Согласно этой теории, сущность болезни заключается в изменении
плотности тела, расположения твердых составных частей тела вследствие хаотического движения атомов.
Указанные взгляды древних философов уже содержали элементы материалистического объяснения сущности болезни и пользовались признанием в течение многих столетий.
Немецкий ученый Вирхов первый обратил внимание на то, что при болезни изменяются не только органы в целом, но и клетки, из которых построены ткани этих органов. Всякую болезнь он стал объяснять исключительно изменениями в клетках. На этом основании он создал целлюлярную патологию.
Вирхов изучил и систематизировал ценный фактический материал относительноклеточных явлений, лежащих в основе атрофии, гипертрофии, воспаления,
опухолей и др. патологических процессов.
Основные направления развития учения об этиологии.
Представления об этиологии болезней также значительно эволюционировало.
Монокаузализм – механистическая концепция, признающая значимость
только причин в развитии болезни и отрицающая роль условий. Возникновение
этого учения обусловлено бурным развитием микробиологии и представлений
об инфекционном процессе. Согласно этой теории, для возникновения болезни
достаточно проникновения инфекционного агента в организм, а реактивность
организма и его восприимчивость не имеют никакого значения.
Конституционализм отрицает объективную причинность возникновения
болезней. Согласно этой концепции причиной болезни является сумма равноценных по значению условий. Это учение базируется на формальной генетике:
если есть дефект в генах, то болезнь обязательно проявится. Оно оправдывает
многие неэтические действия врачей (стерилизацию неполноценных людей).
Конституционалисты считали, что медицина сохраняет жизнь неполноценных
людей и увеличивает банк патологических генов.
Кондиционализм (лат. Conditio – условие) отрицает объективную причинность возникновения болезней и подменяет категорию причины суммой
равноценных по значению условий. Эти утверждения базировались на тех
наблюдениях, когда проникновение патогенных микробов в организм человека
не всегда сопровождалось развитием заболевания.
Психосоматическая концепция (греч. Psyche – душа, soma – тело) –
направление, согласно которому определяющее значение придается межличностным и внутриличностным конфликтам, формирующимся обычно в раннем
детском возрасте и впоследствии проявляющимся в виде различных соматических расстройств. Ряд сторонников этого направления пытается установить зависимость между характером соматического заболевания и специфическими
чертами личности больного, типом эмоционального конфликта. Эта концепция
строится в основном на произвольных, обычно, психоаналитических толкованиях.
32
ПАТОГЕННОЕ ДЕЙСТВИЕ ФАКТОРОВ ВНЕШНЕЙ СРЕДЫ
Самые разнообразные факторы внешней среды, с которыми человек постоянно сталкивается в процессе жизнедеятельности, могут стать для него болезнетворными. Это зависит от их интенсивности, длительности воздействия, а
также от адаптационных возможностей и реактивности организма. Патогенными могут стать механические (механическая травма), физические воздействие
термических факторов, ионизирующего и других видов излучений, высокого и
низкого атмосферного давления, электромагнитных волн), химические (кислоты, щелочи, лекарственные препараты, алкоголь, наркотические вещества, промышленные и сельскохозяйственные яды), биологические факторы (бактерии,
вирусы, грибы, разнообразные виды паразитов). Кроме того, учитывая двойственную биосоциальную сущность человека, патогенными могут стать психические и социальные факторы.
Механическая травма – это повреждающее воздействие на ткани твердыми телами или взрывной волной. Характер повреждения может быть различным и местно проявляется в виде переломов, разрывов, ушибов, раздавливания или сочетания указанных видов повреждений. Особую группу механических повреждений составляют огнестрельные ранения, характер которых
зависит от специфики ранящего снаряда, его скорости и массы. Местные последствия травм могут сопровождаться тяжелыми общими нарушениями, вызванными кровопотерей, повреждением нервных стволов. В тяжелых случаях
эти нарушения носят характер травматического шока (см. тему «Патофизиология экстремальных состояний. Шок»).
Патогенное действие термических факторов может быть общим и
местным. Общее действие высокой температуры может привести к развитию
гипертермии, а низкой – гипотермии. При местном воздействии высокой
температуры возникают ожоги. Однако неверно рассматривать ожоги как исключительно местный процесс. Нередко опасность общих нарушений превышает значение местных. Это имеет место при развитии ожоговой болезни.
Гипертермия развивается в результате задержки тепла в организме
из-за нарушения теплоотдачи. Развитию гипертермии способствуют высокая
температура и влажность атмосферного воздуха, когда теплоотдача затруднена и осуществляется только при напряжении механизмов физической терморегуляции. При повышении температуры атмосферного воздуха до 33º (температура кожи) теплоотдача путем проведения и излучения становится неэффективной, а при повышенной влажности затрудняется отдача тепла путем
испарения. В этих условиях нарушается равновесие между образованием тепла в организме и его отдачей во внешнюю среду, что приводит к задержке
тепла и перегреванию. Развитию гипертермии способствуют также непроницаемая для влаги одежда, излишне развитая подкожная клетчатка, физическая
работа.
Стадия компенсации характеризуется сохранением нормальной температуры тела, что достигается усиленной работой механизмов теплоотдачи.
33
В этот период наблюдаются расширение кожных сосудов, усиление потоотделения, учащение дыхания. Перенапряжение механизмов терморегуляции приводит к их истощению, а наблюдающееся вслед за этим повышение температуры тела свидетельствует о наступлении второго периода гипертермии –
стадии декомпенсации. Она характеризуется резким возбуждением центральной нервной системы, учащенным поверхностным дыханием, повышенной частотой сердечных сокращений (до 140 ударов в минуту). Усиленное потоотделение приводит к нарушениям водно-электролитного обмена, повышению вязкости крови, что повышает нагрузку на систему кровообращения.
Дальнейшее повышение температуры тела и перевозбуждение нервных центров могут закончиться их истощением. Наблюдаются нарушение сознания,
судороги, снижение рефлекторной деятельности. Смерть наступает в результате прекращения деятельности центров регуляции дыхания и кровообращения.
Тепловой удар возникает в результате острого перегревания. Он характеризуется резким расстройством функций центральной нервной системы –
беспокойством, ощущением сильного жара, рвотой, судорогами, потерей сознания (гипертермическая кома). Повышается частота сердечных сокращений,
возникает сильная одышка, понижается артериальное давление. Температура
тела может повышаться до 42 - 43º. Смерть может наступить через несколько
часов при явлениях коллапса, которые могут возникнуть и с самого начала.
После перенесенного теплового удара нарушения функций центральной нервной системы могут сохраняться длительное время.
Солнечный удар возникает под воздействием солнечных лучей на поверхность головы. При солнечном ударе наблюдаются явления сильного раздражения центральной нервной системы: общее возбуждение, иногда психические и нервные расстройства. В менее выраженных случаях отмечаются интенсивные головные боли, раздражительность. В оболочках и ткани головного
мозга развивается гиперемия, возможны кровоизлияния.
Ожог – результат местного воздействия высокой температуры. Ожоги
характеризуются развитием местных деструктивных и реактивных изменений.
Выделяют четыре степени тяжести ожогов:
I.
Характеризуется покраснением кожи (эритема), слабой воспалительной реакцией. Целостность кожных покровов сохранена.
II. Проявляется отслоением эпидермиса с образованием пузырей, развитием экссудативного воспаления.
III. Сопровождается некротическими изменениями кожных покровов,
образованием язв.
IV. Обугливание кожи и подлежащих тканей.
Ожоги значительной площади и степени тяжести приводят к выраженным общим нарушениям. Развивается ожоговая болезнь. В ее течении различают следующие стадии:
1. Ожоговый шок. Ведущую роль в его патогенезе играет интенсивная болевая импульсация, которая вызывает выраженное раздражение и последующее истощение центральной нервной системы. Это приводит к нару-
34
шению регуляции сосудистого тонуса, дыхания и сердечной деятельности.
Кроме того, ожоговый шок сопровождается выраженной интоксикацией денатурированными белками и токсическими продуктами его ферментативного
гидролиза. Значительную роль в патогенезе ожогового шока играет гиповолемия и повышение вязкости крови, связанные с потерей жидкости, обусловленной повышением проницаемости сосудов на месте поражения.
2. Ожоговая инфекция всегда сопровождает ожоговую болезнь.
Входными воротами для инфекции служит ожоговая поверхность. Кроме того, источником инфекции является содержимое кишечника. Это связано с
нарушением барьерной функции кишечной стенки при шоке. Инфекционные
осложнения усугубляют интоксикацию и тормозят процессы регенерации.
3. Ожоговое истощение. Прогрессируют кахексия, анемия, отеки,
дистрофические изменения во внутренних органах. С нарушением функции
иммунной системы связано развитие пневмоний, пиелонефритов.
4. Выздоровление характеризуется полным отторжением некротических тканей, развитием грануляций, рубцеванием, эпителизацией.
Гипотермия – нарушение теплового баланса, сопровождающееся понижением температуры тела. Оно может возникнуть в результате повышения
теплоотдачи при снижении температуры окружающей среды, снижения теплопродукции или сочетания этих факторов. Гипотермия может возникнуть
при длительном пребывании в среде, температура которой ниже температуры
тела всего на 15º, особенно при пребывании в воде. Переохлаждению способствует высокая влажность атмосферного воздуха, так как вода обладает большей теплопроводностью, а также высокая скорость движения воздуха.
Первый период гипотермии – стадия компенсации – характеризуется
активацией адаптационных механизмов терморегуляции. Температурные рецепторы кожи воспринимают холодовое раздражение и посылают импульс в
гипоталамус, где расположен центр терморегуляции, а также в высшие отделы
центральной нервной системы. Отсюда по двигательным нервам поступают
сигналы к скелетным мышцам, в которых развивается терморегуляторный тонус и дрожь. По симпатическим нервам возбуждение достигает мозгового вещества надпочечников, где усиливается секреция адреналина. Последний вызывает сужение периферических сосудов и снижение интенсивности потоотделения, что приводит к уменьшению теплоотдачи, а также стимулирует распад гликогена в печени и мышцах.
Важным фактором является включение в процессы терморегуляции гипофиза, а через его тропные гормоны – щитовидной железы и коры надпочечников. Глюкокортикоиды стимулируют глюконеогенез, мобилизируя тем самым пластический материал для покрытия повышающихся энергетических
потребностей. Тиреоидные гормоны повышают обмен веществ, разобщают
окисление и фосфорилирование, что приводит к повышению теплопродукции.
Этот механизм способствует экстренному согреванию, однако, связан со снижением синтеза макроэргов, необходимых для осуществления различных
35
функций. Следовательно, разобщение окислительного фосфорилирования не
способно обеспечить длительную адаптацию к условиям холода.
Длительная адаптация к низким температурам достигается путем повышения мощности митохондриальной системы, повышения активности ферментов цикла трикарбоновых кислот. Биогенез митохондрий стимулируется
воздействием тиреойдных гормонов и связан с активизацией генетического
аппарата клетки, увеличением синтеза нуклеиновых кислот и белка.
В условиях продолжительного и интенсивного воздействия низких температур наблюдается истощение механизмов терморегуляции. Температура
тела снижается, и наступает вторая стадия гипотермии – стадия декомпенсации. В этот период отмечается снижение скорости обменных процессов и потребления кислорода, угнетение жизненно важных функций. Нарушение дыхания и кровообращения приводит к развитию гипоксии, нарушению функции
центральной нервной системы вплоть до развития гипотермической комы (см.
Патофизиология экстремальных состояний). Угнетение функции центральной
нервной системы является, в какой-то степени, защитным механизмом, так
как снижается чувствительность нервных клеток к гипоксии и дальнейшему
падению температуры тела. Снижение обмена веществ уменьшает потребность тканей в кислороде.
Интересен тот факт, что в состоянии гипотермии организм становится
менее чувствительным к разнообразным патогенным воздействиям внешней
среды – голоду, гипоксии, инфекции, ионизирующему излучению. На этом
основано применение искусственной гипотермии (гибернации) с лечебной целью, например при операциях на сердце и крупных сосудах. В этих случаях
температура тела поддерживается на низком уровне с помощью охлаждения и
применения препаратов, тормозящих функцию терморегуляторных центров
(наркотические препараты, ганглиоблокаторы).
Действие ионизирующего излучения. К ионизирующим излучениям
относят лучи высокой энергии (рентгеновские и γ-излучение), также ά- и βчастицы (радионуклиды). Все виды ионизирующих излучений обладают способностью проникать в облучаемую среду и производить ионизацию. Облучение организма может быть внешним и внутренним (инкорпорированное облучение, вызванное поступлением радионуклидов во внутренние среды организма). Возможно комбинированное облучение.
Чувствительность различных тканей к воздействию ионизирующего
излучения неодинакова. Самой высокой радиочувствительностью обладают те
ткани, в которых процессы деления клеток протекают наиболее интенсивно.
Это, в первую очередь, тимус, половые железы, кроветворная и лимфойдная
ткань. Следующей в этом ряду стоит эпителиальная ткань и эндотелий сосудов. Хрящевая, костная, мышечная и нервная ткань являются относительно
радиорезистентными. Нервные клетки не способны к делению и гибнут только при действии на них больших доз радиации (интерфазная гибель).
Механизм прямого повреждающего воздействия ионизирующего излучения на организм заключается в ионизации, возбуждении, разрыве наименее прочных связей, прежде всего, высокомолекулярных соединений. Пер-
36
вичной мишенью могут стать белки, липиды, нуклеиновые кислоты, нуклеопротеидные комплексы, липопротеиды.
Из всех радиохимических реакций наибольшее значение имеет радиолиз воды, продуктами которого являются свободные радикалы (OHֹ, Hֹ). Последние способны вступать с возбужденными молекулами воды, кислородом
тканей и дополнительно образовывать перекись водорода (H2O2), радикал
гидропероксида (HO2ֹ), атомарный кислород (O). Продукты радиолиза воды
обладают высокой биохимической активностью и способны вызывать реакцию окисления по любым химическим связям. Следующие друг за другом реакции окисления нарастают, приобретая характер цепных разветвленных реакций. Доказательством большого значения продуктов радиолиза воды в патогенезе воздействия ионизирующего излучения является высокая радиоустойчивость порошкообразных ферментов по сравнению с их водными растворами.
Механизм опосредованного повреждающего воздействия ионизирующего излучения заключается в повреждении клеточных структур свободными
радикалами и перекисями. Они способны вызывать радиохимическое окисление пиримидиновых и пуриновых оснований, нарушая тем самым структуру
нуклеиновых кислот. Продукты радиолиза воды окисляют свободные жирные
кислоты и аминокислоты, в результате чего образуются, соответственно, липидные и хиноновые радиотоксины.
Радиотоксины способны угентать синтез нуклеиновых кислот, непосредственно повреждать структуру ДНК, изменять активность ферментов. Радиотоксины хиноидного ряда действуют подобно самой радиации на главные
мишени — ДНК ядер клеток (радиомиметический эффект). Липидные радиотоксины повреждают главным образом биологические мембраны, в том числе
мембраны митохондрий и лизосом. Это влечет за собой "энергетический кризис в клетке", высвобождение лизосомальных ферментов. Нарушается ферментативное окисление, появляются вторичные радиотоксины (особые белки,
пептиды, биогенные амины и др.), которые сами вызывают повреждение биологических структур и усиливают образование первичных радиотоксинов —
образуются порочные круги патогенеза лучевого поражения.
Воздействие ионизирующего излучения приводит к нарушению всех
процессов жизнедеятельности клетки. Могут наблюдаться любые виды поломок генетического аппарата (генные, хромосомные, геномные мутации). Митотическая активность клетки угнетается. Повреждаются все органеллы клетки. Ионизирующее излучение повреждает внутриклеточные мембраны – мембраны ядра, митохондрий, лизосом, эндоплазматического ретикулума. Из лизосом высвобождаются ферменты, повреждающие внутриклеточные структуры (нуклеиновые кислоты, цитоплазматические и ядерные белки). В митохондриях нарушается окислительное фосфорилирование, что приводит к энергодефициту.
К наиболее уязвимым системам для воздействия ионизирующего излучения относится система крови. После облучении отмечается уменьшение количества всех форменных элементов крови, а также их функциональная
37
неполноценность. В первые часы после облучения наблюдается лимфопения,
позднее – недостаток гранулоцитов и тромбоцитов, еще позже – эритроцитов.
Возможно опустошение костного мозга.
Снижается иммунная реактивность. Угнетается активность фагоцитоза
и антителообразование. Последнее во многом обусловлено подавлением синтетических процессов радиотоксинами. Часто развиваются тяжелые инфекционные осложнения (пневмония, некротическая ангина, пиелонефрит и т. д.).
Бурно развивается инфекция в кишечнике, что наряду с нарушением барьерной функцией кишечника способствует выраженной интоксикации и септическим состояниям.
Для лучевой болезни характерно развитие геморрагического синдрома.
Это связано со снижением уровня тромбоцитов в периферической крови,
нарушением их агрегационной способности из-за нарушения микроструктуры
мембран. Развитию геморрагического синдрома способствуют также нарушение синтеза факторов свертывания в печени и повышение активности противосвертывающей системы. В периферической крови повышается количество
гепарина, высвобождающегося при дегрануляции тканевых базофилов.
Кроме того, в патогенезе геморрагического синдрома важную роль играют патологические изменения сосудистой стенки. Слущивается эндотелий,
лизосомальными ферментами повреждаются соединительнотканные элементы. Под воздействием биологически активных веществ происходит паралитическое расширение сосудов, повышение их проницаемости. С выходом жидкой части крови за пределы сосудистого русла развивается истинный капиллярный стаз, который усугубляет дистрофические изменения в тканях.
Не смотря на относительную устойчивость нервной ткани к воздействию ионизирующего излучения, практически всегда наблюдаются признаки
нарушения функций центральной нервной системы. Это обусловлено воздействием на рецепторы продуктов радиолиза воды и распада тканей.
Импульсы поступают в нервные центры, нарушая их функциональную
активность. Под действием облучения в высоких дозах развивается интерфазная гибель нейронов.
Острая лучевая болезнь может развиваться в одной из четырех
форм, в зависимости от поглощенной дозы тотального облучения.
1. Костномозговая форма (0,8 – 10 Гр). Различают четыре клинических периода. Период первичных реакций представляет собой реакции нервных и гуморальных механизмов на облучение: возбуждение, головная боль,
лабильность артериального давления и пульса, нарушения функций внутренних органов. Отмечается кратковременный лейкоцитоз с лимфопенией. Период мнимого благополучия характеризуется сохранением лабильности пульса и
артериального давления, лейкопенией. Период развернутых клинических проявлений сопровождается панцитопенией, развитием инфекционных осложнений (некротическая ангина, пневмония), геморрагическим синдромом. Период
исходов. Выздоровление начинается с нормализации картины крови. Долгое
время сохраняются астения, неустойчивость гемопоэза, ослабление иммунитета.
38
2. Кишечная форма (10 – 20 Гр) характеризуется массовой интерфазной гибелью клеток кишечного эпителия, нарушением его барьерной и моторной функций. Наблюдаются рвота, боль по ходу кишечника, возможно
развитие паралитической кишечной непроходимости.
3. Токсемическая форма (20 – 80 Гр) сопровождается выраженной интоксикацией продуктами жизнедеятельности кишечных бактерий и биологически активными веществами.
4. Церебральная форма (более 80 Гр). Наблюдаются структурные изменения и гибель нейронов коры больших полушарий, грубые повреждения
эндотелия сосудов. Тяжелые необратимые нарушения в центральной нервной
системе приводят к развитию судорожно-паралитического синдрома. Смерть
наступает в ходе самого облучения или через несколько минут после него.
Хроническая лучевая болезнь является результатом повторных
облучений небольшими дозами. Различают три степени тяжести хронической
лучевой болезни.
1. Наблюдаются обратимые функциональные расстройства наиболее
чувствительных систем. В периферической крови обнаруживаются нестойкая
лейкопения, тромбоцитопения.
2. Развиваются выраженные изменения со стороны кроветворения и
нервной системы, геморрагический синдром, иммунодефицит. В анализе крови отмечаются стойкая лейкопения, лимфопения, тромбоцитопения.
3. Наблюдаются глубокие необратимые дистрофические изменения в
органах. Истощена функция эндокринных желез. В нервной системе выражены признаки органического поражения. Резко угнетено кроветворение, тонус
сосудов понижен, а проницаемость их стенки повышена.
В настоящее время выделяют две группы эффектов воздействия
ионизирующего излучения:
-пороговые или нестохастические эффекты – имеющие порог вредного
воздействия (острая и хроническая лучевая болезнь, лучевые ожоги);
-беспороговые или стохастические эффекты – не имеющие количественного порога (мутагенное, канцерогенное, эмбриотропное действие).
Одного кванта энергии достаточно для мутации, а последствия единственной
мутации могут оказаться для организма трагическими, особенно в тех случаях, когда имеет место нарушение функции репаразной системы или клеточного иммунного ответа.
Известно, что малые дозы излучения, не вызывающие в ранние сроки видимых функциональных и морфологических нарушений, могут вызывать патологические изменения в организме в отдаленные сроки, в частности, повышать
частоту новообразований.
На всех уровнях организации в ответ на воздействие ионизирующего излучения возникают компенсаторно-приспособительные реакции. Воздействию
свободных радикалов противостоят системы антиоксидантной защиты.
В клетке функционируют ферменты репарации поврежденной ДНК, ингибиторы и инактиваторы биологически активных веществ. Способность кле-
39
ток реперировать повреждения ДНК – один из основных факторов, определяющих устойчивость организма к радиации.
Коррекция лучевого поражения направлена на борьбу с интоксикацией,
инфекционными осложнениями, геморрагическим синдромом. Лечение предполагает терапевтическую коррекцию нарушений функций эндокринной и
нервной систем, желудочно-кишечного тракта. Особое значение имеет восстановление гемопоэза. Успешно применяются средства, перехватывающие
активные радикалы, антиоксиданты.
Действие инфракрасного излучения во многом связано с его термическим эффектом. При незначительной интенсивности инфракрасного излучения возникает гиперемия кожи. Интенсивное облучение инфракрасными
лучами может вызвать ожоги кожного покрова. Кроме того, воздействие инфракрсного излучения на поверхность головы может привести к нагреванию
мозговых оболочек и развитию теплового удара.
Действие ультрафиолетового излучения. Ультрафиолетовые лучи активируют обменные процессы в тканях, что способствует повышению количества образующихся продуктов метаболизма и биологически активных веществ, вызывающих расширение сосудов. Возникает эритема, которая может
сопровождаться болевыми ощущениями. Ультрафиолетовое излучение способно вызывать пролиферацию клеток эпидермиса. Острая передозировка
УФО сопровождается дерматитом, повышением температуры тела. Особенно
опасно развитие фотоофтальмии (поражение роговицы и сетчатки). Хроническая передозировка УФО вызывает общее снижение резистентности организма, обострение хронических заболеваний. Длительная инсоляция – фактор
риск развития рака кожи.
Действие радиоволн сверхвысоких частот. Источниками СВЧ– волн
являются радиолокаторы, микроволновые печи, мобильные телефоны. СВЧволны оказывают негативное действие на центральную нервную систему, вызывая развитие астено-вегетативного синдрома, оказывают повреждающее
воздействие на клетки с высокой митотической активностью. Наблюдаются
нарушения кроветворения, функций органов эндокринной системы.
Действие электрической энергии зависит от характера электрического
тока (постоянный или переменный), напряжения, частоты, направления и длительности воздействия. Механизм действия электрического тока возможен в
трех направлениях: электролиз, электротермическое и электромеханическое
воздействия.
Электролиз вызывает биохимические и коллойдные изменения в тканях. Проходя через биологические объекты, ток производит поляризацию
атомов и молекул, изменяет пространственную ориентировку заряженных частиц и усиливает их движение.
Электротермическое действие обусловлено переходом электрической
энергии в тепловую, вследствие чего возникают ожоги, а электромеханическое выражается в переходе электрической энергии в механическую.
Электромеханическое действие приводит к нарушению целостности
тканей вплоть до разрывов и даже переломов.
40
Переменный ток опаснее постоянного при относительно низком
напряжении и частоте, так как сопротивление тканей переменному току слабее, чем постоянному.
Большую роль играет направление тока. Если электрический ток проходит через голову, смерть может наступить в результате паралича дыхательного центра в продолговатом мозге. В случае прохождения тока через сердце
наступают тяжелые нарушения электрической активности миокарда, и развиваются фатальные нарушения сердечного ритма (фибрилляция желудочков,
асистолия). Нарушения функции сердца и асистолия могут возникнуть без
прохождения тока через сердечную мышцу. Такие явления могут быть результатом рефлекторного нарушения коронарного кровотока или повышения
тонуса блуждающего нерва.
Степень нарушений, вызванных электрическим током, зависит также от
продолжительности действия. Известно, что ток даже высокого напряжения
и большой силы не является смертельным, если он действует менее 0,1 секунды.
Действие высокого атмосферного давления человек испытывает при
погружении под воду во время проведения водолазных и кесонных работ. При
резком повышении атмосферного давления возможен разрыв легочных альвеол. Кроме того, в условиях гипербарии человек дышит воздухом или другой
газовой смесью под повышенным давлением, в результате чего в крови и тканях растворяется большее количество газов (сатурация). Наибольшее значение имеет азот. В условиях высокого атмосферного давления азот накапливается в тканях, богатых липидами. Поскольку липиды в большом количестве
содержатся в нервной ткани, на первый план выходят симптомы поражения
центральной нервной системы.
На начальных этапах развивается эйфория, ослабление способности
концентрировать внимание, позднее наблюдается депрессия, различные степени нарушения сознания. Для предупреждения этих состояний дыхательные
приборы заправляются кислородно-гелиевыми смесями, так как гелий менее
тропен к нервной ткани.
Токсичен в больших количествах для организма и кислород. Это связано с тем, что ткани в первую очередь утилизируют кислород, растворенный в
плазме крови, количество которого в условиях гипербарии возрастает. Затрудняется диссоциация оксигемоглобина. Количество восстановленного гемоглобина, необходимого для выведения углекислоты недостаточно. Развивается своеобразное удушье. Кроме того, гипероксия вызывает образование
свободных радикалов и перекисей, повреждающих липиды клеточных мембран, нуклеиновые кислоты, белки.
Во время возвращения в условия нормального атмосферного давления
(декомпрессия) наблюдается десатурация – выведение избыточного количества растворенных газов крови легкими. Декомпрессия должна проводиться
медленно, чтобы скорость выхода газов из растворенного состояния не превышала возможности легких выводить их. В противном случае развивается
декомпрессионная болезнь, вызванная множественной газовой эмболией.
41
При этом наблюдаются кожный зуд, боль в суставах, в тяжелых случаях нарушения зрения, потеря сознания, параличи.
Действие низкого атмосферного давления человек испытывает на
больших высотах. Патологические изменения, развивающиеся в таких условиях, обусловлены экзогенной гипоксией (снижение парциального давления
кислорода во вдыхаемом воздухе) и декомпрессией (непосредственное снижение атмосферного давления).
При снижении атмосферного давления газы, находящиеся во внутренних средах организма расширяются. В связи с этим при снижении атмосферного давления развиваются высотный метеоризм (расширение кишечных газов), болевые ощущения в лобных пазухах, носовые кровотечения. На высоте
19000м при разгерметизации кабины летательного аппарата практически
мгновенно наступает смерть. Это вызвано тем, что на этой высоте кровь закипает при температуре тела.Во время быстрого перепада атмосферного давления развивается синдром взрывной декомпрессии. При этом наблюдаются
баротравмы легких (разрыв альвеол и легочных сосудов, который приводит к
развитию газовой эмболии), сердца и крупных сосудов.
Действие на организм факторов космического полета. На старте и во
время приземления космонавт испытывает воздействие перегрузок, вибрации,
шума, высокой температуры. Во время орбитального полета на человека воздействуют невесомость и гипокинезия.
Перегрузка – сила, которая действует на организм во время движения с
ускорением. Основным механизмом перегоезки является смещение органов и
жидких сред организма в направлении, противоположном движению. Большое значение в патогенезе воздействия ускорения имеет нарушение внешнего
дыхания, легочного кровотока и газообмена. Не менее важным является раздражение интерорецепторов и интенсивная афферентная импульсация, вызванные смещением внутренних органов.
Действие невесомости во время длительного космического полета приводит к перестройке систем организмана новый уровень функционирования.
Значительные изменения претерпевает система кровообращения. В результате
выпадения гидростатического компанента артериального давления происходит перераспределение крови с увеличением кровенаполнения сосудов верхней половины тела. Раздражение волюморецепторов приводит к снижению
выделения вазопрессина и альдостерона и перестройке водно-электролитного
обмена.
Происходят значительные изменения в опорно-двигательном аппарате.
Из костной ткани выводятся кальций и фосфор, возникает остеопороз.
Наблюдается уменьшение массы скелетных мышц, снижается сила их сокращений, что является следствием гипокинезии и нарушения нервная нервной
трофики мышц. Последнее развивается в результате снижения интенсивности
афферентной импульсации.
42
Патогенное действие химических факторов. Химические вещества
могут оказывать различное действие, нередко являясь причиной отравлений.
Отравления могут быть вызваны веществами, поступающими в организм из
вне (экзогенные яды) или образующимися в самом организме (эндогенные
яды). Аутоинтоксикация (вызванная эндогенными ядами) возникает в результате нарушения функции выделительных органов, барьерной функции кишечника, врожденных ферментопатий (фенилкетонурия).
Отравляющее действие ядов проявляется нарушением разнообразных
функций. На этом основании различают химические вещества общеядовитого
действия (цианиды), гепатотоксические (толуилендиамин, флоридзин, четыреххлористый углерод), нейротоксические (стрихнин, мышьяк) и т. д.
Токсичность – способность химических соединений вызывать изменения процессов клеточного метаболизма, приводящие к нарушению функций и
гибели клетки.
Молекулы инородных соединений, проникшие в организм, подвергаются распределению. Они или равномерно разносятся по организму, или, в силу
своих химических свойств, связываются с определенными структурами, в результате чего эти соединения накапливаются в различных органах (например,
связывание ионов тяжелых металлов промежуточным веществом соединительной ткани). При равномерном распределении токсичное вещество разводится, и его концентрация во внутренней среде организма снижается. При неравномерном распределении этого не происходит. Например, радиоактивные
вещества аккумулируются в промежуточном веществе костей и в коллойде
щитовидной железы, откуда и оказывают вредное воздействие. При выведении чужеродных соединений почками там их концентрация может превышать
таковую в крови.
Следующим этапом является трансформация чужеродного соединения.
До недавнего времени существовало мнение, что она приводит к образованию
веществ менее активных, чем исходные. Сегодня известно, что может сложиться и противоположная ситуация.
Элиминация – совокупность процессов, с помощью которых чужеродные соединения выводятся из организма. Часто в целях элиминации трансформированная молекула подвергается последующим биосинтетическим изменениям (конъюгация).
Попадая в ткань, яд, кроме прямого действия на различные системы,
может вызывать раздражение рецепторов различных областей организма, особенно синокаротидной и аортальной зон.
Повторное введение химических веществ, нередко сопровождается привыканием к ним. Это объясняется постепенным снижением проницаемости
поверхности кожи и слизистых оболочек (мышьяк), ускорением разрушения
вещества (этанол), а также ускорением выведения (атропин) или снижением
чувствительности к ним.
Патофизиологические аспекты алкоголизма.
Эндогенный этанол относится к незаменимым метаболическим факторам.
В норме его концентрация в крови составляет 0,01-0,03% (0,01-0,03 г/л). В ор-
43
ганизме этанол подвергается быстрому и эффективному окислению, преимущественно в печени (до 80%), примерно 10% метаболизируется в других тканях и
столько же выделяется легкими и мочой. Основным ферментом, принимающим
участие в метаболизме этанола является алкогольдегидрогеназа. Наибольшее
количество фермента находится в печени, хотя он присутствует во всех тканях.
С помощью алкогольдегидрогеназы окисляется 75-90% этанола, поступающего
в организм. Окисление этанола осуществляется также микросомальной этанолокисляющей системой (МЭОС), которая локализуется на мембранах гладкого
цитоплазматического ретикулума и представляет собой фрагмент общей детоксицирующей системы микросомальной фракции печени. В здоровой печени эта
система окисляет около 10-20% этанола.
Несмотря на быструю утилизацию этанола в организме, у больных алкоголизмом обнаруживается обширный спектр нарушений обмена веществ. Это
объясняется тем, что алкоголь распадается до Н20 и С02 только при небольшом
его потреблении (примерно до 20 г/сут). при превышении этой дозы в организме накапливается избыток этанола и продуктов его распада (прежде всего ацетальдегида), вторые вызывают патологические эффекты.
В организме у этанола существует много точек приложения. Это связано с его физико-химическими особенностями. В связи с повышенным сродством этанола к липидам, при отравлении этиловым спиртом страдают прежде
всего органы, богатые ими. При введении этанола в кровоток, если его концентрацию в крови принять за единицу, то в печени она составит 1,5, а в головном
мозге — 1,75. Взаимодействие этанола с гидрофобными участками липопротеидов, составляющих структурную основу мембран клеток, лежит в основе повреждающего действия на них этилового спирта. В результате такого взаимодействия в последних происходят конформационные изменения, сопровождающиеся нарушением работы мембранных насосов и других мембранных ферментных систем.
Патогенетические механизмы наркомании и токсикоманий. В организме человека обнаружены эндогенные морфиноподобные вещества, выполняющие роль антистрессоров, анальгетиков и эйфоригенов. К ним относятся
мет- и лей-энкефалины и эндорфины. В настоящее время обнаружено еще несколько эндогенных морфиноподобных соединений,свойства которых изучаются. Производные опия и морфина способны стимулировать опиатные рецепторы гипоталамуса.
Наркотические и токсические вещества, взаимодействуя с липопротеидными компонентами клеточных мембран, вызывают их конформационные
изменения, что нарушает нормальное функционирование нейронов за счет
нарушения мембранных насосных систем. Воздействуя в дозах, не вызывающих тяжелых поражений нейронов, эти вещества нарушают прежде всего самые сложные нервные процессы, протекающие с взаимосвязанным участием
многих нервных клеток. К ним относятся многие процессы активного коркового торможения, процессы тонкой регуляции работы отдельных корковых зон и
подкорковых центров. При нарушениях такого рода возможно растормажива-
44
ния "центра удовольствия" (индукция эйфории), несбалансированность работы
чувствительных центров (индукция галлюцинаций).
В более высоких концентрациях наркотики и токсические вещества подавляют корковую активность в целом, что клинически проявляется состоянием
оглушенности. Наконец, еще более высокие концентрации этих соединений
угнетают работу подкорковых жизненно важных центров (дыхательного и сосудодвигательного),что и является нередко причиной смерти при передозировке наркотиков и токсических веществ. В своем развитии наркоманы проходят
через фазу привыкания к наркотику. Организм автоматически отвечает повышением толерантности к этому веществу (за счет активации метаболизирующих систем, снижения чувствительности рецепторов к этим веществам, синтеза
медиаторов, проявляющих конкурентный или функциональный антагонизм к
ним). В результате этого наркоманы и токсикоманы вынуждены увеличивать
дозы наркотических веществ.
Роль свободных радикалов в патологических процессах. Свободные
радикалы – это молекулярные частицы, имеющие непарный электрон на
внешней орбитали и поэтому обладающие высокой окислительной активностью. Свободные радикалы – физиологические метаболиты и образуются в
эндоплазматическом ретикулуме в ходе работы микросомальной окислительной системы цитохрома P 450, при функционировании митохондрий с участием убисемихинона, в лизосомах и пероксисомах под действием мембранных НАДФН-зависимых оксидаз. В нормальных условиях электроны от субстратов окисления передаются по дыхательной цепи на молекулярный кислород с образованием воды. Но при торможении дыхательной цепи в результате
интоксикации или повреждения митохондрий, электроны передаются по одному на молекулярный кислород от радикала коэнзима Q; при этом образуется супероксидный радикал (ֹОО ֿ◌). В норме супероксидный радикал под воздействием фермента супероксиддисмутазы превращается перекись водорода
(H2O2). Фагоциты используют перекись водорода, превращая ее с помощью
миелопероксидазы в гипохлорит (CLO ֿ◌) – соединение, разрушающее бактериальную стенку. Избыток перекиси водорода удаляется под действием глутатион-пероксидазы или каталазы.
В условиях патологии могут произойти нарушения либо системы защитных ферментов (в частности, снижение активности супероксиддисмутазы), либо ферментных систем, связывающих ионы железа в плазме крови (церулоплазмин, трансферрин). В этом случае супероксидные радикалы и перекись водорода вступают в альтернативные реакции, продуктами которых являются радикалы гидроксила (HOֹ), которые способны повреждать нуклеиновые кислоты и инициируют реакции перекисного окисления липидов.
Радикал гидроксила вступает во взаимодействие с полиненасыщенными
жирными кислотами клеточных мембран. При этом образуются липидные радикалы: HOֹ + LH = H2O + Lֹ
45
Липидный радикал (Lֹ) вступает в реакцию с молекулярном кислородом.
В результате образуется новый свободный радикал – радикал липоперекиси: Lֹ
+ LH = LOOֹ
Этот радикал атакует соседнюю молекулу липида с образованием гидроперекиси липида и нового липидного радикала:
LOOֹ + LH = LOOH + Lֹ Таким образом реакция приобретает цепной
характер.
В присутствии ионов двухвалентного железа происходит разветвление
цепей при их взаимодействии с гидроперекисями липидов:
Fe 2+ + LOOH = Fe 3+ + HO ֿ◌ + LOֹ Образующиеся радикалы LOֹ инициируют новые цепи окисления липидов.
Кроме непосредственного участия в процессе перекисного окисления
липидов свободные радикалы способны вызывать повреждение ДНК, остановку ее репликации, что приводит к тератогенному, канцерогенному, цитостатическому эффекту.
Патогенному действию свободных радикалов противостоят механизмы
антиоксидантной защиты. Основными разрушителями активных кислородных
радикалов служат ферменты каталаза, глутатионпероксидаза, супероксиддисмутаза. Каталаза и глутатионпероксидаза восстанавливают АКР (перекись водорода), провоцирующий цепной свободно-радикальный процесс, до неактивного состояния. Супероксиддисмутаза восстанавливает супероксидный анион
до менее активной перекиси водорода, разрушаемой каталазой. Основными
антиоксидантными субстратами клеток являются тиоловые соединения (глутатион, цистеин). Другая группа веществ, используемая клетками для защиты
от свободных радикалов – это витамины. Для восстановления и реактивации
глутатиона необходима аскорбиновая кислота. Сильнейшим антиоксидантом
является токоферол, который служит для инактивации липоперекисей. Витамин Е способен улавливать свободный электрон и не участвует в дальнейшей
цепи окислительных реакций. Липидными антиоксидантами считаются женские половые стеройды.
Патогенное действие биологических факторов. К основным патогенным биологическим факторам относят инфекционных возбудителей и паразитов.
Инфекционный процесс – типический патологический процесс, характеризующийся комплексом взаимосвязанных функциональных, морфологических, иммунобиологических, биохимических изменений, возникающих под
действием микроорганизмов.
К возбудителям инфекционных болезней относят простейшие, грибы,
бактерии, вирусы и прионы. Каждый из перечисленных возбудителей обусловливает специфические черты инфекционного процесса. В значительной
мере они определяются природой микроорганизма. Важным отличительным
свойством микроорганизмов-паразитов является патогенность.
Патогенность – видовой признак микроорганизма, обусловливающий
способность вызывать определенную инфекционную болезнь. Патогенность
обеспечивает проникновение микроорганизма в макроорганизм (инфектив-
46
ность), размножение в нем, развитие болезни с патогенезом, характерным для
данного заболевания. Мерой патогенности является фенотипическое свойство
– вирулентность.
Вирулентность – свойство, характеризующее степень болезнетворности данного микроорганизма.
Значительную роль в развитии инфекционного процесса играет снижение резистентности макроорганизма, которое может быть вызвано разнообразными факторами (врожденные дефекты иммунного ответа, голодание, перерохлаждение, интоксикации, лечение иммунодепрессантами и т. д.).
Кроме того, необходимым условием развития инфекционного процесса
является наличие входных ворот инфекции – места проникновения микробов
в макроорганизм. Такими воротами могут быть:
• Кожные покровы (возбудители сыпного тифа, кожного лейшманиоза);
• Слизистые оболочки дыхательных путей (грипп, корь, скарлатина);
• Слизистая оболочка мочеполовых органов (возбудители гонореи, трихомониаза);
• Стенки кровеносных и/или лимфатических сосудов (при укусах членистоногих и животных, инъекциях);
• Раневые поверхности .
Известны следующие пути распространения бактерий:
• По межклеточному пространству;
• По лимфатическим капиллярам;
• По кровеносным сосудам;
• По жидкостям серозных полостей и спинномозгового канала.
Большинство возбудителей имеет тропность к определенным тканям
организма. Это определяется наличием молекул адгезии у микроорганизмов и
специфических рецепторов у клеток макроорганизма.
Стадийность течения инфекционных болезней является одной из патогномоничных их особенностей. При развитии инфекционной болезни выделяют несколько периодов:
1. Инкубационный период – интервал времени от инфицирования до
первых клинических признаков болезни. В этот период происходит размножение и избирательное накопление микроорганизмов в
определенных органах и тканях, а также мобилизация защитных
механизмов организма.
2. Продромальный период – этап инфекционного процесса от появления первых клинических проявлений до полного развития
симптомов. В этот период происходит снижение эффективности
защитных реакций организма, нарастание степени патогенности
возбудителя (размножение, выделение экзо- и эндотоксинов).
3. Период основных проявлений характеризуется развитием типичных для данной болезни признаков, которые зависят от специфических патогенных свойств возбудителя и характера ответных ре-
47
акций организма, фрмирующихся на фоне недостаточности его
адаптативных механизмов.
4. Период завершения, который имеет несколько вариантов: выздоровление, гибель организма, развитие осложнений, бактерионосительство.
Основными общими звеньями инфекционного процесса являются лихорадка, воспаления, гипоксия, нарушения обмена веществ, а также расстройства функций органов и систем. Степень их выраженности определяет проявления инфекционного процесса (см. соотв. темы).
Одним из прогностически неблагоприятных осложнений инфекционного процесса является сепсис. Сепсис – синдром, обусловленный постоянным
или периодическим поступлением микроорганизмов из очага гнойного воспаления в кровь. Характерно образование метастатических очагов гнойного воспаления в различных органах и тканях. В патогенезе преобладают тяжелые
полиорганные нарушения, тогда как местные воспалительные изменения выражены слабо. Происходит глубокое нарушение обмена веществ вследствие
выраженной интоксикации, преобладание процессов катаболизма. Тяжелые
дистрофические изменения дополнительно ухудшают функции органов, что
даже при отсутствии в них гнойных метастазов приводит к полиорганной недостаточности, характерной для поздних необратимых стадий сепсиса.
РОЛЬ НАСЛЕДСТВЕННОСТИ, КОНСТИТУЦИИ, ВОЗРАСТА
И ПОЛА В ПАТОЛОГИИ.
ИНФОРМАЦИОННЫЙ МАТЕРИАЛ
Наследственные болезни - это болезни, этиологическим фактором которых
являются мутации (генные, хромосомные,геномные ).
Врожденные болезни проявляются при рождении ребенка. Они могут быть
обусловлены как наследственными, так и экзогенными факторами (например,
действие экзогенных факторов на организм эмбриона, плода).
Фенокопии - ненаследственное изменение каких-либо признаков организма
под влиянием окружающей среды, копирующее фенотипическое проявление
мутаций, отсутствующих в генотипе данной особи.
Мутагены – вещества любой природы, обуславливающие развитие мутаций.
Мутагены делят на:
1.Экзогенные:
- химические (пестициды, уретан, формальдегид, бензол, соединения ртути,
кофеин, цитостатики, некоторые пищевые добавки и др.);
- физические (ионизирующая радиация ультрафиолетовые лучи).
- биологические - вирусы.
2. Эндогенные:
- перекись водорода;
- липидные перекиси;
48
- свободные кислородные радикалы.
Мутация - это внезапно возникающее стойкое скачкообразное изменение
генетической информации, не связанное с рекомбинацией генетического материала.
В зависимости от этого, в каких клетках возникают мутации в половых
или соматических мутации делятся на:
- половые
- соматические.
В зависимости от уровня, на котором произошла мутация, они делятся на:
- геномные
- хромосомные
- генные.
Генная мутация – связана с изменением структуры гена или нескольких
генов, она возникает в результате изменения последовательности нуклеотидов в
молекуле ДНК.
Хромосомные мутации — изменение структуры хромосом.
Геномные мутации — изменения числа хромосом в геноме, не сопровождаемые изменением их структуры.
Моногенными наследственными болезнями называют те болезни, которые возникают в результате формирования одного патологического гена,
например - фенилкетонурия, гемофилия, недостаточность глюкозо-6фосфатдегидрогеназы.
Виды генных мутаций.
По характеру изменений в составе гена выделяют следующие виды генных мутаций:
Трансзиционная мутация – это изменение нуклеотидной последовательности
ДНК, состоящее из замены одного пурина и другой или одного пиримидина на
другой.
Трансверзионная мутация – мутация, заключающаяся в замене одного пурина
на пиримидин и наоборот.
Инсерционная мутация – мутация, возникшая в результате вставки фрагментов
ДНК от одного нуклеотида до целого гена.
Делеция – мутация, возникшая в результате утраты сегмента ДНК от одного
нуклеотида до гена.
Проявления генных мутаций.
В результате генных мутаций происходит изменение либо структуры
белка, либо его количества. Регуляция синтеза белка происходит на нескольких
уровнях (претранскрипционный, транскрипционный, трансляционный) и на
всех этих этапах, осуществляемых соответствующими ферментами могут возникать наследственные аномалии. Генные мутации могут привести к отсутствию какого-либо фермента (альбинизм, алкаптонурия, фенилкетонурия),
транспортного (цистинурия, семейный гипофосфатемический рахит) или рецепторного (семейная гипохолестеринемия, тестикулярная феминизация) белка.
Мутации, которые вызывают наследственные болезни, могут затрагивать лю-
49
бые белки: структурные, транспортные, ферменты. Каждый ген может мутироваться и обусловливать другое строение белка, а каждое звено в цепи биохимических реакций осуществляется каким-либо ферментом, следовательно, контролируется определенным геном в соответствии с правилом «один ген – один
фермент». Таким образом, каждый ген контролирует биосинтез, специфичность
и функцию только одного определенного фермента, а развитие наследственных
признаков происходит по следующей схеме: ген – фермент - метаболиты –
клетки – ткани – органы – организм. Исходя из теории регуляции биосинтеза
белка, в клетке имеется несколько видов генов:
1. Структурные гены (и них иРНК считывает информацию), которые определяют последовательность аминокислот в полипептидной цепи.
2. Контролирующие гены:
а) ген-регулятор, отвечающий за синтез белка-репрессора, который контролирует активность оперона.
б) ген-оператор, который в зависимости от ситуации «разрешает» иРНК или
«не разрешает» считывать информацию со структурного гена.
Белок-репрессор может связываться с определенным участком ДНК и тем
самым препятствовать связыванию РНК-синтезирующих ферментов. Репрессор
выключает определенный ген или группу смежных генов, поэтому транскрипция закодированной в них информации становится невозможной, а клетка при
этом не может синтезировать и соответствующие белки. В результате мутация
структурного гена приводит к формированию качественно нового белка, а мутация контролирующего гена – к количественным изменениям. Например, качественные изменения гена – серповидно-клеточная анемия, количественные –
гемофилия, агаммаглобулинемия.
Типы наследования:
• Аутосомно-доминантный — фенотипически патологическое состояние
обнаруживается у гетерозиготном состоянии. В их основе лежит нарушение синтеза структурных белков или белков, выполняющих специфические функции. Действие мутантного гена проявляется почти в 100%.
Вероятность развития болезни в потомстве 50%. Один из родителей
больного ребенка обязательно болен.
• Аутосомно-рецессивный — фенотипически патологическое состояние
обнаруживается в гомозиготном состоянии. Болезнь проявляется тогда,
когда дети получают патологический ген от обоих родителей, т.е. в гомозиготном состоянии. Сами же родители, являясь гетерозиготными носителями признака, остаются фенотипически здоровыми. Вероятность
рождения больного ребенка составляет 25%. Большое значение для проявления этих болезней у потомства имеет кровное родство родителей, у
которых есть большая вероятность обладания одинаковым рецессивным
патологическим геном.
• Х-сцепленное наследование — патогенный ген локализован в половой Ххромосоме.
По Аутосомно-доминантному типу наследуются:
50
скелетные аномалии (короткопалость, многопалость, сросшиеся и искривленные пальцы)
• близорукость, дальнозоркость, астигматизм.
• врожденная катаракта,
• отосклероз,
• некоторые формы мышечной атрофии (хорея Гетингтона, ахондроплазия).
• множественный полипоз толстой кишки,
• нейрофиброматоз (болезнь Реклингаузена).
• Синдром Марфана
• Наследственный сфероцитоз
По Аутосомно-рецессивному типу наследуются:
• дефекты аминокислотного обмена (фенилкетонурия, альбинизм, алкаптонурия),
• врожденная глухонемота,
• микроцефалия,
• пигментопатии.
• Серповидно-клеточная анемия
Х-сцепленное наследование по данному типу наследуются:
• гемофилия
• мышечная дистрофия Дюшена.
•
Полигенные болезни – это болезни, признаки которой определяются не
одним, а несколькими генами. Они имеют сложный характер наследования и
зависят от влияния окружающей среды. К полигенным относятся болезни с
наследственным предрасположением: гипертоническая болезнь, атеросклероз, сахарный диабет и др.
Наследственная склонность к болезням.
Болезни с наследственным предрасположением это те болезни, для которых наследственность является этиологическим фактором, но для пенетрантности мутированных генов необходимо соответствующее состояние
организма, обусловленное вредным влиянием среды (подагра, сахарный
диабет, атеросклероз – их проявление зависит от питания). Такие заболевания обычно проявляются с возрастом под действием внешних факторов.
Антигенасоциированые болезни.
Это болезни предрасположенность, к которым определяется определенными антигенами, кодируемыми генами из системы главного комплекса
лейкоцитарных антигенов человека (HLA) и системы главного комплекса
антигенов тканевой гистосовместимости. К ним относятся: аутоиммунный
тиреоидит, ревматоидный артрит, болезнь Аддисона, рассеянный склероз,
болезнь Бехтерева, псориаз, миастения.
51
Хромосомные болезни.
Все хромосомные болезни делят на две группы:
1. Вызванные геномными мутациями, т.е. изменением числа хромосом (полиплоидии, анеуплоидии) при сохранении нормальной структуры хромосом
2. Обусловленные хромосомными мутациями, т.е. изменением структуры
хромосомы (транслокации, инверсии, делеции, дупликации).
Полиплоидия – кратное увеличение полного набора хромосом (диплоидии,
триплоидии и т.д.) которые приводят к нежизнеспособности организма, мертворождениям и спонтанным абортам.
Анеуплоидия – изменение числа хромосом в одной или нескольких парах.
Транслокация – обмен сегментами между хромосомами.
Инверсия - поворот участка хромосомы на 180о.
Делеция - выпадение участка хромосомы.
Дупликация – удвоение участка хромосомы.
Синдром Патау. Трисомия по 13 паре хромосом. У больного наблюдается умственная отсталость, микроцефалия, микроофтальмия, срединная расщелина верхней губы («заячья губа») и неба («волчья пасть»), аномалии сердца,
аномальная стопа, полидактилия
Синдром Эдвардса. Трисомия по 18 паре хромосом. У больного наблюдается умственная отсталость, повышенная подвижность пальцев, выступающий затылок, низко-посаженные уши, короткая шея, аномалии внутренних органов, аномальная стопа.
Синдром Дауна. Трисомия по 21-й хромосоме. Общее число хромосом
47. Но может быть и 46, что означает, что лишняя 21-я хромосома слилась в одной из крупных, например 15-й. У больного наблюдается умственная отсталость, характерная внешность — низкий рост, короткопалые руки и ноги, короткая средняя фаланга и маленький мизинец, монголоидный разрез глаз, открытый рот, большой торчащий язык. Задержка физического развития, аномалии внутренних органов, особенно сердца. Синдром Дауна встречается относительно часто — один случай на 500-600 родов.
Синдром Клаинфельтера. Частота данного заболевания его составляет
1:1000. Общее количество хромосом 47 (кариотип XXУ, но встречаются 48,
XXXУ, 49, ХХХХУ). Наружные половые органы сформированы по мужскому
типу. Характерны высокий рост, астеническое телосложение, длинные ноги,
снижение сперматогенеза, признаки феминизации (геникомастия, высокий голос, отсутствие характерного оволосенеения на теле и лице). Как и для других
хромосомных болезней свойственна умственна отсталость. В соматических
клетках обнаруживается половой хроматин (тельца Барра).
Синдром Шерешевского-Тернрнера Общее количество хромосом 45
(кариотип 45, ХО). Наружные, половые органы сформированы по женскому типу. Характерны низкий рост широкая щитоподобная грудная клетка недостаточное физическое и половое развитие. Внутренние половые органы недоразвиты, яичники представлены фиброзными тяжами, первичная аменорея. В
клетках слизистой оболочки рта отсутствует половой хроматин (тельца Барра).
52
Фенотипические проявления хромосомных аберраций, т.е. клиническая картина синдрома, зависит от многих факторов:
- генотипа организма
- индивидуального вовлечения в аберрацию хромосомы или ее участка (набора
генов)
- типа аберрации
- размера недостающего (при делеции) или избыточного (при частичной трисомии) материала
- степени мозаичности организма по аберрантным клеткам
- зависимости от условий среды
- зависимости от стадии онтогенеза и возраста больных.
Для изучения наследования как нормальных, так и патологических признаков человека, медицинская генетика использует такие методы:
Генеалогический метод. Метод родословных, т.е. прослеживание болезни (или признака) в семье или роду с указанием родственных связей между
членами родословной. Этот метод относится к наиболее универсальным методам генетики человека. Суть этого метода сводится к выяснению родственных
связей и к прослеживанию признака или болезни среди родственников. Данный
метод состоит из двух этапов: составления родословных и генеалогического
анализа. Составление родословных начинается от пробанда (лицо, которое первым попало в поле зрения исследователя). Обычно родословную составляют по
одному или нескольким признакам. Родословная может быть полной и ограниченной. Генеалогический метод позволяет установить такие генетические закономерности как, наследственный характер признака и тип наследования.
Объективное установление типа наследования того или иного заболевания
возможно лишь при массовых обследованиях семей.
Близнецовый метод - исследование генетических закономерностей на
близнецах. Производися сопоставление монозиготных близнецов с дизиготными, партнеров монозиготных пар между собой, результатов анализа близнецовой выборки и общей популяции.
Монозиготными (однояйцовыми, идентичными) близнецами называются индивиды, выросшие из одной зиготы, разделившиеся на ранних стадиях дробления на две части и обладающие, поэтому одинаковыми генотипами. Дизиготные (двуяйцовые, неидентичные) близнецы возникают за счет оплодотворения
двух одновременно развившихся яйцеклеток. Какой-либо качественный признак может встречаться либо у обоих близнецов данной пары, либо у одного из
них. Данный метод применяется для:
- оценки соотносительной роли наследственности и среды в развитии признака;
- установления наследственного характера признака и определения пенетрантности гена;
- оценки действия некоторых внешних факторов: лекарственных препаратов,
методов воспитания, обучения.
Метод состоит из следующих этапов:
53
1. Сопоставление близнецовой выборки.
2. Установление зиготности
3. Сопоставление пар и групп близнецов по рассматриваемым признакам.
Диагностика основывается на анализе наиболее изученных моногенных полиморфных признаков.
Популяционно-статистический метод - заключается в исследовании
наследственных признаков в больших группах населения из одной или нескольких популяций, в одном или нескольких поколениях. Этот метод используется для изучения:
- частоты генов в популяции, включая частоту болезней,
- мутационного процесса,
- роли наследственности и среды в возникновении различных болезней, особенно с наследственным предрасположением,
- роли наследственности и среды в формировании фенотипического полиморфизма по нормальным признакам,
- значения генетических факторов в антропогенезе.
Ццтогенетический метод основан на микроскопическом изучении
хромосом.
Этот метод используется для:
- диагностики хромосомных болезней,
- составления карт хромосом,
- изучения мутационного процесса,
- изучения нормального хромосомного полиморфизма в человеческой популяции.
С этим методом связано открытие всех форм хромосомных болезней. С его
помощью изучаются частота хромосомных и геномных мутаций и частота хромосомных аберраций, механизмы мутагенеза.
Биохимические методы используют в случае, подозрения на наследственные болезни обмена веществ и на те формы наследственных болезней, при которых установлены дефекты первичного генного продукта или патогенетическое звено развития заболевания.
Иммунологические методы используются для диагностики такой группы
наследственных болезней как гликогенозы, гемоглобинопатии, ихтиоз и ряд
других.
Метод изучения рельефных узлов на коже, образуемых папиллярными
линиями и гребешками, или дерматоглифика (дерма — кожа, глифе - гравировать) базируется на индивидуальном характере папиллярного рисунка, который
находится под генетическим контролем.
Экспериментальное моделирование наследственных болезней основано
на искусственном размножении мутантных линий животных, имеющих те или
иные наследственные дефекты, аналогичные таковым у человека (ахондроплазия у кроликов, гидроцефалия и дефекты губы у мышей, гемофилия у собак).
54
Принципы лечения и профилактики наследственных заболеваний.
Принципы профилактики наследственных заболеваний
Профилактика должна быть направлена на:
а) вновь возникающие наследственные болезни, как результат спонтанной
мутации в зародышевых клетках родителей;
б) болезни, унаследованные от предыдущих поколений;
в) заболевания, развивающиеся в результате наследственного предрасположения и действия повреждающих факторов внешней среды.
В настоящее время разработана система генетических и гигиенических
мероприятий по исключению мутагенных факторов.
1. Массовое «просеивание» новорожденных на наследственные дефекты
обмена выществ. ( выявление фенилкетонурии, гипотиреоза, галактоземии, муковисцедоза и т.д.)
2. Перинатальная диагностика – комплексное исследование, основанное на
использовании лабораторных и инструментальных методов.
3. Медико-генетическое консультирование.
4. Контроль за мутагенной опасностью окружающей среды.
Принципы лечения наследственных заболеваний:
1. Симптоматическое лечение — хирургическое коррекция при расщелине верхней губы и твердого неба, врожденных пороках сердца, поли- и
синдактилии, корригирующие линзы и др..
2. Патогенетическое лечение — воздействие и коррекция тех механизмов, которые формируют наследственное заболевание:
- заместительная терапия – введение инсулина при сахарном диабете, факторов свертывания крови при гемофилии и т.д.
- диетотерапия - назначение диеты исключающую фенилаланин при фенилкетонурии,
- медикаментозное лечение, направленное на удаление продуктов, избыточно накапливающихся в организме
3. Этиологическое лечение. Исходя из успехов молекулярной биологии и
генной инженерии о ней можно говорить как о перспективе. Например,
включение искусственно полученных генов в геном человека, использование
направленного химического мутагенеза, позволяющего индуцировать специфические мутации в строго определенном локусе (получение обратных
мутаций — от патологического аллеля к нормальному) и т.д.
Конституция – совокупность физиологических, морфологических, биохимических, психических особенностей организма, сформировавшаяся на
наследственной основе под действием факторов внешней среды.Особенности конституции оказывают большое влияние на реактивность,
а, следовательно, на предрасположенность к тем или иным заболеваниям,
приспособительные возможности организма, течение физиологических и патологических процессов.
55
С древних времен и до наших дней было предложено множество названий и различных классификаций конституциональных типов. Одна из первых
классификаций была предложена Гиппократом. В основу своей классификации он положил особенности темперамента.
Холерик – вспыльчив, порывист, раздражителен, иногда необуздан. Работоспособность высокая, непостоянная. Склонен к заболеваниям печени, пищеварительной и выделительной систем.
Флегматик – медлительный, спокойный, устойчивый. Склонен к заболеваниям мочеполовой системы
Сангвиник – общительный, подвижный, живой, эмоциональный. Склонен
к заболеваниям сердечнососудистой системы и крови.
Меланхолик – замкнутый, подавленный, нерешительный. Склонен к заболеваниям нервной системы, легких.
К.Сиго была предложена классификация в основу, которой положены
морфологические особенности по преимуществу развития той или иной физиологической системы. В формировании конституционального типа основным он
считал раннее хроническое влияние условий окружающей среды.
Дыхательный тип
Пищеварительный тип
Мышечный тип
Мозговой тип
Э.Кречмер связал в своей классификации физиологические, морфологические, психические особенности с частотой возникновения психических заболеваний и выделил следующие конституциональные типы:
Астенический тип характеризуется долихоморфизмом, с длинной плоской грудной клеткой, острым эпигастральным углом, длинной шеей тонкими и
длинными конечностями, узкими плечами. Для этого типа характерна большая
длительность и стабильность внутренних переживаний при небогатом внешнем
выражении, замкнутость, аффективность, аутический стиль поведения. Астеники чаще болеют шизофренией.
Атлетический тип характеризуется выраженным развитием мышечной
системы. Для них характерна стойкость и малоподвижность психических процессов, высокая стрессоустойчивость, упорядоченность, мелочность, педантизм. Атлетики склонны к развитию эпилепсии.
Пикнический тип – характеризуется брахифорностью, широкой коренастой фигурой, круглой головой, короткой шеей, широкой грудной клеткой с тупым эпигастральным углом, выступающим животом и выраженной подкожножировой клетчаткой. Для них характерна эктравертированность, цикличная подвижнойсть эмоциональных состояний. Пикники чаще склонны к маниакальнодепрессивным психозам.
А.А. Богомолец в основу своей классификации положил особенности активной мезенхимы и выделил следующие конституциональные типы:
Астенический тип характеризуется преобладанием тонкой нежной соединительной ткани
56
Фиброзный тип характеризуется соединительной тканью более плотной
и волокнистой
Липоматозный тип характеризуется развитием жировой ткани, способностью элементов мезенхимы к жировой инфильтрации.
Пастозный тип характеризуется преобладанием отечной, рыхлой соединительной ткани.
И.П.Павлов по уровню силы, подвижности и уравновешенности возбуждения и торможения выделял следующие конституциональные типы:
Слабый тормозной тип
Сильный уравновешенный спокойный
Сильный неуравновешенный возбудимый
Сильный уравновешенный живой
Диатезы – своеобразная аномалия конституциональных признаков, которая характеризуется патологической реакцией организма на физиологические и
патологические раздражители.Состояние диатеза это не заболевание, предрасположение к нему.
Классификация диатезов
Лимфатико-гипопластический диатез характеризуется гиперплазией
тимико-лимфатического аппарата и гипоплазиеий надпочечников, хромаффинной ткани, щитовидной железы, половых органов, сердца, аорты, гладкомышечных органов. Для данного диатеза характерны пониженные приспособительные возможности организма, слабая устойчивость к стрессам. Клинически
у лиц с данным диатезом отмечается тимомегалия, увеличение миндалин, фолликулов языка, селезенки, лимфоцитоз и нейтропения.
Нервно-артритический диатез – состояние, которое характеризуется
повышенной возбудимостью и высокой лабильностью нейровегетативной регуляции, сильным неуравновешенным возбудимым типом высшей нервной деятельности, высокой интенсивностью пуринового обмена и гиперурикемией.
Данный диатез предрасполагает в развитию дискенезии желудочно-кишечного
тракта. Для лиц с данным диатезом характерны периодические ацетонемические состояния, а также предрасположенность к развитию сахарного диабета,
мигрени, ХПН, невралгии, атеросклероза, подагры.
Экссудативно-катаральный диатез. Для него характерны следующие
признаки: географический язык, увеличение периферических лимфоузлов,
склонность к диспепсии, пастозная, рыхлая и бледная кожа, лабильность водного обмена, тенденция к гипергликемии, эозинофилии. В основе развития данного вида диатеза лежат атопические особенности иммунологической реактивности организма. У лиц с этим диатезои отмечается избыток Т-хелперов, повышена продукция ИЛ-5 и ИЛ-10, низкая выработка гама-интерферона и ИЛ-2 и ИЛ4, значительная активности кининовой системы и преобладание на поверхности клеток Н1-гистаминовых рецепторов. В совокупности эти факторы определяют у таких лиц повышенный риск анафилактических реакций и гиперергическое течение воспаления.
57
Астенический диатез – характеризуется общей адинамией, лабильностью сосудистых реакций.
Значение возрастных факторов в патологии.
Патология внутриутробного развития – патологические состояния, которые сформировались во внутриутробном периоде, т.е. периоде закладки генетической программы развития под действием патогенных факторов.
Нарушения внутриутробного развития в зависимости от времени воздействия патогенного фактора делятся на:
Гаметопати – формируются в результате неполноценности половых клеток. В результате образуется зигота, которая погибает или дает аномальный
плод.
Бластопатии – формируются в первые две недели развития зародыша
(бластоцисты – зародыша первых 15 суток после оплодотворения) когда происходит дробление зародыша.
Эмбриопатии – формируются с 16 суток до 8-9 недели беременности
внутриутробного развития, т.е. в период с начала дифференцировки эмбриобласта до конца закладки органов.
Фетопатии – нарушения развития плода. Выделяют ранние фетопатии,
которые характеризуют образование тонких структур до достижения плодом
жизнеспособности (с 12 недели до 7 месяцев) и поздние фетопатии когда происходит становление функций плода (с 7 месяцев до родов).
Причины нарушений внутриутробного развития (тератогенные факторы):
Химические – никотин; алкоголь; пестициды, лекарственные препататы.
Физические – ионизирующая радиация.
Биологические – вирусы (краснуха, корь, грипп), простейшие (токсоплазмоз).
Влияние любых патогенных факторов в определенной стадии развития
приводят аномалии развития тех структур, которые имеют наибольшую чувствительность к повреждению в данный момент, т.е. проходят критический период развития. Знание этих критических периодов позволяет дифференцировать врожденные и наследственные пороки развития.
Развитие патологии внутриутробного развития напрямую зависит от болезней и вредных привычек матери. Курение вызывает спазм сосудов матки и
плода, что приводит к гипоксии и гипотрофии плода, при употреблении алкоголя развивается алкогольный синдром плода (задержка роста, развития, энцефалопатия). При повреждении эндометрия происходит разрыв рефлекторный
связей между маткой и яичниками, что приводит к снижению влияния эстрогенов на матку в результате нарушается, процессы имплантации и плацентации,
последняя приводит к формированию плацентарной недостаточности. Нарушение плацентарного кровообращения ведет к нарушению всех ее функций – дыхательной, трофической, защитной, выделительной, гормонообразующей. В
результате происходит нарушение трофики плода и формирование его физиологической незрелости у пода развивается гипотрофия, а затем гипоксия.
58
Механизмы развития внутриутробных пороков развития (ВПР)
Механизмы развития ВПР заключаются в искажениях межмолекулярных и
межклеточных взаимодействий, а также в нарушениях морфогенетических
процессов.
Расстройства межмолекулярных и межклеточных взаимодействий приводят к нарушениям синтеза биологически активных веществ (гормонов, цитокинов и др.), структуры белков (например, ферментов или компонентов мембран,
энергетического обеспечения реакций метаболизма и жизненно важных процессов, искажающих дифференцировку и функции клеток, тканей и органов.
Нарушения морфогенетических процессов (пролиферация, миграция,
дифференцировка и гибель клеток) приводят к аплазии (отсутствие органа при
наличии его сосудистой ножки) или гипоплазии (недоразвитию) органа или его
части, задержке слияния эмбриональных структур (например, расщелины нёба,
губы; спинномозговые и черепно-мозговые грыжи), персистированию эмбриональных структур, к атрезии и гетеротопии (наличие клеток и/или тканей в другом органе или в тех зонах органа, где их в норме не должно и т.д. и т.п.
Виды ВПР
Агенезия — полное отсутствие органа (например, тимуса, почки, глаз).
Аплазия и гипоплазия — отсутствие или значительное уменьшение органа
при наличии его сосудистой ножки и нервов (например, одной почки, селезёнки, лёгкого, конечности, кишечника).
Атрезия — полное отсутствие канала или естественного отверстия (например,
атрезия наружного слухового прохода, пищевода, ануса).
Гетеротопия — перемещение клеток, тканей или части органа в другую ткань
или орган (например, клеток поджелудочной железы в дивертикул Меккеля,
хромаффинных клеток в ткань лёгких).
Персистирование — сохранение эмбриональных структур, исчезающих в норме к определённому этапу развития (например, открытый артериальный проток
у годовалого ребёнка, крипторхизм)
Стеноз — сужение просвета отверстия или канала (например, клапанного отверстия сердца, привратника желудка, фрагмента кишечника).
Удвоение (утроение) — увеличение числа органов или его части (например,
удвоение матки, мочеточников).
Эктопия — необычное расположение органа (например, почки в малом тазу,
сердца — вне грудной клетки).
РОЛЬ РЕАКТИВНОСТИ В ПАТОЛОГИИ. ПАТОЛОГИЯ
ИММУНОЛОГИЧЕСКОЙ РЕАКТИВНОСТИ
Понятие о реактивности - одно из ключевых в патологической физиологии и клинической медицине. Этот подход детально разрабатывался такими известными патологами, как И.И. Мечников, А.А. Богомолец, А.Д.Сперанский, Г.
Селье и многими другими.
59
Под реактивностью понимают способность живого организма определенным образом изменять свои функциональные параметры и структурные характеристики в ответ на изменение воздействующих на организм факторов
внешней среды обитания.
Реактивность включает в себя весь набор доступных организмов адаптативных ответов:
1)наследственно определенные нормы реагирования.
2)ненаследуемые стереотипы программные ответы связанные с индивидуальным онтогенетическим опытом и сохраняемые иммунологической памятью и
нейропамятью.
Норма реакции – доступный индивиду диапазон ответа при разных условиях среды.
Реактивность – это атрибутивное свойство живых систем, присущая всем
уровням организации живого – от молекулярного до уровня организма.
В случаях, когда внешние воздействия могут существенно изменить
функциональные и морфологические характеристики организма, но его основные показатели останутся в пределах физиологической нормы, принято говорить об адаптации или приспособляемости.
Если организм не может приспособиться, то в нем развивается патологический процесс – такие взаимодействия называются патогенными.
Если среда постоянно агрессивна, чем создается непосредственная угроза
гибели организма, то факторы, действию которых подвергается организм (пострадавший), принято называть критическими или экстремальными.
Очень важно помнить, что для различных индивидов одни и те же воздействия могут оказаться или адаптивными или патогенными. Подобным же
образом может меняться реактивность одного и того же индивида в зависимости от особенностей его жизнедеятельности на каждом этапе его жизни.
Особенности реактивности могут зависеть от характеристики жизненных
процессов на разных уровнях организации и функционирования: молекулярном,
клеточном, органном, системном и организменном.
Молекулярный уровень.
Особые представления реактивности, присущие больным серповидноклеточной анемией, фенилкетонурией, галактоземией, гемофилией и прочие
выявляются на молекулярном уровне дефектами строения гемоглобина или
определенных ферментов.
Клеточный уровень.
Реактивность, ответственная за развитие онкологических заболеваний,
иммунодефицитов, аллергий, во многом формируется на клеточном уровне.
Органный уровень.
Сформированные пороки сердца, цирроз печени, фиброз легких или обструкция дыхательных путей и многие другие заболевания проявляются в основном на органном уровне.
Проявления синдромов, например, различные варианты шоков, должны трактоваться, как системные нарушения реактивности.
60
Нарушения на организменном уровне могут проявляться в форме неврозов, психических заболеваний, некоторых нарушений обмена веществ и т.д.
Конечно, подобные характеристики уровней реактивности весьма условны, всегда можно при достаточно детальном рассмотрении выявить молекулярноклеточные основы патологии. Например, бластоматозный рост по современным
данным является следствием активации онкогенов, иммунодефицит может зависеть от нарушений энергетики этих клеток; обструкция бронхов определяется на уровне регуляции Ig Е, острая сосудистая недостаточность - шок - следствием дефицита ингибиторов ферментов протеолиза в организме и т.п. Однако
подобный подход, связывающий, понятие реактивности с определенными
структурами и функциями может быть полезен для анализа конкретных клинических ситуаций.
Те или иные варианты реактивности могут быть присущи различным
группам индивидов или отдельным организмам.
Реактивность бывает:
а) видовой, групповой и индивидуальной.
Видовая (биологическая) реактивность присуща всем особям данного вида,
групповая - определенной группе особей,
индивидуальная - конкретному индивидууму;
б) неспецифической и специфической
Неспецифическая (первичная, простая) реактивность проявляется при действии разнообразных факторов на организм. В её основе - генетически запрограммированные стандартные ответы на действие высокой и низкой температур, кислородное голодание и т.д.
Специфической называется иммунологическая реактивность - способность
организма отвечать на действие генетически чужеродных агентов
(антигенов) реакциями, направленными на узнавание антигена, связывание,
элиминацию или изоляцию антигена, формирование иммунологической памяти.
в) физиологической и патологической.
Физиологическая реактивность - реакция здорового организма. Патологической называют качественно измененную реактивность при действии патогенных факторов на организм.
Общепризнанным является представление о видовой или биологической
реактивности. Известно, что различные биологические виды по-разному воспринимают внешние воздействия, например, температуру внешней среды, различные варианты питания, количество доступной для потребления воды, контакт с теми или иными инфекционными возбудителями. Например, есть полярные животные и животные тропиков, обитатели пустынь и водоемов и т.д., для
которых перемена внешней среды не совместима с жизнью. Люди не болеют
чумой собак, инфекционной анемией рогатого скота, а большинство животных
- венерическими болезнями или лепрой. Животные не болеют психическими
болезнями, хотя срывы высшей нервной деятельности - неврозы у них могут
проявляться.
61
Групповая реактивность. Это реактивность, обусловленная местом проживания, климатическими условиями, характером питания, генетическими особенностями, например особенностями, маркируемыми группами крови или особенностями антигенов гистосовместимости. Хорошо известны тропические болезни, болезни холодного и влажного климата, болезни болотистых местностей.
Жители Заполярья, питающиеся жирным мясом морских животных, легко переносят нагрузки холестерина, патогенные для жителей средней полосы или
тропиков.
Люди, имеющие группу крови В (Ш) устойчивы против чумы, а у лиц,
гомозиготных по группе 0 (I), в несколько раз чаще, чем у остальных, встречается язва желудка. У людей - носителей антигенов гистосовместимости - HLAB8 и HLА-В15 чаще встречается юношеский диабет, а у носителей НLА-В27 ревматоидный артрит, у HLA-В13, HLA-В17 и HLA-BW8 - псориаз.
Хорошо известны особенности реактивности, формирующиеся в связи с
профессиональной принадлежностью и проявляющиеся устойчивостью или
склонностью к определенным заболеваниям: моряки или летчики устойчивы к
кинетозам. профессиональные таежные охотники не чувствительны к простудным болезням, врачи-рентгенологи склонны к онкологическим заболеваниям и
т.д.
Известны особенности реактивности, обусловленные половой принадлежностью. Женщины, поскольку для них характерен более низкий уровень основного обмена, лучше переносят голодание, кровопотери, гипоксию. Но у
мужчин, в 5 раз реже, чем у женщин встречается холелитиаз.
Очень существенны особенности реактивности, обусловленные возрастом. Например, у новорожденных недостаточно развиты нервная, эндокринная,
иммунная системы, несовершенны тканевые барьеры. Имеет место существенное недоразвитие рецепторного аппарата клеточных мембран, в результате чего
клетки не реагируют даже при наличии достаточных регуляторных стимулов.
Для старческого возраста характерно резкое изменение гормонального профиля, в частности дефицит половых гормонов и СТГ и прогрессирует иммунодефицит.
С учетом всего вышесказанного мы приходим к чрезвычайно важному
для клинициста понятию индивидуальной реактивности. Индивидуальная реактивность складывается на основе всего вышесказанного и, что чрезвычайно
существенно, последующих конституциональных характеристик. Правильно
поставить диагноз и выбрать терапевтическую тактику можно только при условии глубокого понимания индивидуальных особенностей реактивности каждого конкретного больного.
Одним из проявлений реактивности организма является его резистентностъ, то есть устойчивость к различного рода повреждающим воздействиям.
Резистентность -это устойчивость организма к действию патогенных
факторов.
Виды резистентности
Резистентность бывает пассивной и активной, неспецифической
62
и специфической. В основе специфической резистентности лежит иммунологическая реактивность
1.
2.
3.
4.
Пассивная и активная резистентность
Пассивная резистентность — это нечувствительность к действию патогенного фактора, невосприимчивость его. Она возникает в том случае, когда
невозможно или затруднено взаимодействие организма с патогенным фактором. Пассивная резистентность является энергонезависимой и может быть обусловлена следующими механизмами:
1) существованием преград для взаимодействия патогенного фактора
со структурами организма (биологические барьеры);
2) отсутствием или поломом структур организма, способных взаимодействовать с патогенным фактором, например, отсутствием рецепторов к патогенным вирусам;
3) уничтожением патогенного фактора механизмами, не связанными с реакцией организма на действие этого фактора (например, уничтожение холерного вибриона желудочным соком);
Активная резистентность (сопротивляемость) — это устойчивость, которая обеспечивается комплексом защитно-компенсаторных реакций, направленных на уничтожение патогенного фактора и последствий его действия. Активная резистентность является энергозависимой, ее основу составляют механизмы реактивности, например, фагоцитоз, синтез антител, реакции клеточного
иммунитета.
Взаимосвязь между реактивностью и резистентностью является сложной. Возможны разные ее варианты.
Увеличение реактивности вызывает повышение активной резистентности. Например, при повышении температуры тела увеличивается образование
антител, что повышает активную резистентность к инфекциям.
Увеличение реактивности вызывает уменьшение активной резистентности. Так, увеличение образования антител может быть «причиной аллергии,
при которой уменьшается устойчивость организма к действию веществ антигенной природы.
Уменьшение реактивности приводит к уменьшению активной резистентности. Например, уменьшение образования антител уменьшает активную резистентность к инфекциям.
Уменьшение реактивности сопровождается увеличением пассивной
резистентности. Так, при гипотермии увеличивается пассивная резистен-тность
к инфекциям, интоксикациям и действию других патогенных факторов
(например, у животных во время зимней спячки).
В качестве одного из важнейших аспектов реактивности необходимо рассматривать иммунную резистентность, то есть способность организма противостоять инфекционным и онкологическим заболеваниям.
В организме человека постоянно происходят мутации. Их суммарное количество в расчёте на один клеточный цикл составляет примерно 1х106. Часть
мутаций сопровождается синтезом новых белков, обладающих антигенными
63
свойствами. Происходящие в связи с этим структурные и функциональные изменения могут привести к существенные расстройствам жизнедеятельности
клеток, тканей, органов и организма в целом. Кроме того, организм постоянно
подвергается атаке вирусов, бактерий, риккетсий, грибов, паразитов, способных
вызвать различные болезни. В связи с этим в ходе эволюции сформировалась
высокоэффективная система клеточных и неклеточных факторов распознавания
собственных и чужих структур — система иммунобиологического надзора
(ИБН).
Биологическое значение системы иммунобиологического надзора ИБН
заключается в контроле (надзоре) за индивидуальным и однородным клеточно-молекулярным составом организма.
Обнаружение носителя чужеродной генетической или антигенной информации (молекулы, вирусы, клетки или их фрагменты) сопровождается его
инактивацией, деструкцией и, как правило, элиминацией. При этом клетки иммунной системы способны сохранять «память» о данном агенте.
Повторный контакт такого агента с клетками системы ИБН вызывает развитие эффективного ответа, который формируется при участии как специфических — иммунных механизмов защиты, так и неспецифических факторов резистентности организма
Неспецифическая защита организма(«врожденный, естественный, иммунитет, или неспецифическая резистентность».)
Неспецифический иммунитет -совокупность защитных факторов,
направленных на элиминацию широкого круга патогенов. Эти факторы являются универсальными, детерминируются многими генами, передаются по наследству, являются видовыми (человек не болеет чумой плотоядных).
Неспецифическую защиту организма осуществляют кожные и слизистые покровы, внутренние барьеры организма, лимфатические образования во всех тканях и
в виде самостоятельных органов — лимфатических узлов, фагоцитирующие клетки и естественные киллеры, а также гуморальные факторы — лизоцим, белки
острой фазы, комплемент, интерферон и другие цитокины. Важнейшим фактором неспецифической защиты является нормальная микрофлора кожи и слизистых . Часть
упомянутых факторов действует постоянно (лизоцим), другие — только после активации (комплемент), либо после стимуляции продуцирующих их клеток (интерферон).
Внешние баръеры.Кожа и слизистые оболочки
Для большинства микроорганизмов, в том числе патогенных, неповрежденная кожа и слизистые оболочки служат барьером, препятствующим их проникновению внутрь организма. Постоянное слущивание верхних слоев эпителия, секреты
сальных и потовых желез способствуют удалению микроорганизмов с поверхности кожи. Однако кожа представляет собой не только механический барьер, она
обладает также бактерицидными свойствами, связанными с действием молочной и
жирных кислот, различных ферментов, выделяемых потовыми и сальными железами. Поэтому микроорганизмы не входящие в число постоянных обитателей кожных покровов быстро исчезают с ее поверхности.
64
Еще более выраженными защитными функциями обладают конъюнктива
глаза, слизистые оболочки носоглотки, дыхательного, желудочно-кишечного и мочеполового трактов. Слезы, моча и секреты, выделяемые слизистыми, слюнными и
пищеварительными железами, не только смывают микроорганизмы с поверхности
слизистых оболочек, но и оказывают бактерицидное действие, обусловленное содержащимися в них ферментами, в частности лизоцимом.
Защитные функции кожи и слизистых не ограничиваются неспецифическими
механизмами. На поверхности слизистых, в секретах кожных, молочных и других
желез присутствуют секреторные иммуноглобулины, обладающие бактерицидными свойствами и активирующие местные фагоцитирующие клетки. Кожа и слизистые принимают активное участие в антиген-специфических реакциях приобретенного иммунитета. Их относят к самостоятельным компонентам иммунной системы.
Внутренние баръеры
К внутренним барьерам организма относится система лимфатических сосудов и лимфатических узлов. Микроорганизмы и другие чужеродные частицы, проникшие в ткани, фагоцитируются на месте или доставляются фагоцитами в лимфатические узлы или другие местные лимфатические образования, где формируется воспалительный процесс, направленный на уничтожение возбудителя. В тех
случаях, когда местная реакция оказывается недостаточной, процесс распространяется на следующие регионарные лимфоидные образования, служащие новым
барьером проникновения возбудителя во внутреннюю среду организма.
Существуют функциональные гисто-гематические барьеры, препятствующие проникновению возбудителей и чужеродных субстратов из крови в
головной мозг, репродуктивную систему, глаз.
Мембрана каждой клетки также служит барьером для проникновения в нее
посторонних частиц и молекул.
Клеточные факторы
1.Фагоцитирующие клетки
Защитная роль подвижных клеток крови и тканей была впервые обнаружена
И.И. Мечниковым в 1883 г. Он назвал эти клетки фагоцитами и сформулировал
основные положения фагоцитарной теории иммунитета.
Все фагоцитирующие клетки организма, по И.И. Мечникову, подразделяются
на макрофаги и микрофаги. К микрофагам относятся полиморфноядерные гранулоциты крови: нейтрофилы, эозинофилы и базофилы. Макрофаги различных тканей организма (соединительной ткани, печени, легких и др.) вместе с моноцитами
крови и их костномозговыми предшественниками (промоноциты и монобласты)
объединены в особую систему мононуклеарных фагоцитов (СМФ). СМФ филогенетически более древняя по сравнению с иммунной системой. Она формируется в
онтогенезе достаточно рано и имеет определенные возрастные особенности.
Микрофаги и макрофаги имеют общее миелоидное происхождение — от полипотентной стволовой клетки, которая является единым предшественником
грануло- и моноцитопоэза. В периферической крови содержится больше гранулоцитов (от 60 до 70% всех лейкоцитов крови), чем моноцитов (от 8 до 11%). Вместе
с тем длительность циркуляции моноцитов в крови значительно больше (полупе-
65
риод 22 ч), чем короткоживущих гранулоцитов (полупериод 6,5 ч). В отличие от
гранулоцитов крови, являющихся зрелыми клетками, моноциты, покидая кровяное
русло, в соответствующем микроокружении созревают в тканевые макрофаги.
Внесосудистый пул мононуклеарных фагоцитов в десятки раз превышает их число
в крови. Особенно богаты ими печень, селезенка, легкие.
Все фагоцитирующие клетки характеризуются общностью основных
функций, сходством структур и метаболических процессов. Наружная плазматическая мембрана всех фагоцитов является активно функционирующей структурой.
Она отличается выраженной складчатостью и несет множество специфических рецепторов и антигенных маркеров, которые постоянно обновляются .Фагоциты
снабжены высокоразвитым лизосомным аппаратом, в котором содержится богатый арсенал ферментов. Активное участие лизосом в функциях фагоцитов обеспечивается способностью их мембран к слиянию с мембранами фагосом или с
наружной мембраной. В последнем случае происходит дегрануляция клеток и сопутствующая секреция лизосомных ферментов во внеклеточное пространство.
Фагоцитам присущи три функции:
-защитная, связанная с очисткой организма от инфекционных агентов, продуктов распада тканей и т.д.;
- представляющая, заключающаяся в презентации лимфоцитам антигенных
эпитопов на мембране фагоцита;
-секреторная, связанная с секрецией лизосомных ферментов и других биологически активных веществ — цитокинов, играющих важную роль в иммуногенезе.
Различают следующие последовательно протекающие стадии фагоцитоза.
1. Хемотаксис (приближение ).
2. Адгезия (прикрепление,прилипание).
3. Эндоцитоз (погружение).
4. Переваривание.
1. Хемотаксис — целенаправленное передвижение фагоцитов в направлении
химического градиента хемоаттрактантов в окружающей среде. Способность к хемотаксису связана с наличием на мембране специфических рецепторов для хемоаттрактантов, в качестве которых могут выступать бактериальные компоненты, продукты деградации тканей организма, активированные фракции системы комплемента — С5а, СЗа, продукты лимфоцитов — лимфокины.
2. Адгезия (прикрепление) также опосредована соответствующими рецепторами, но может протекать в соответствии с законами неспецифического физикохимического взаимодействия. Адгезия непосредственно предшествует эндоцитозу
(захвату).
3.Эндоцитоз является основной физиологической функцией так называемых
профессиональных фагоцитов. Различают фагоцитоз — в отношении частиц с
диаметром не менее 0,1 мкм и пиноцитоз — в отношении более мелких частиц и
молекул. Фагоцитирующие клетки способны захватывать инертные частицы угля,
кармина, латекса обтеканием их псевдоподиями без участия специфических рецепторов.В то же время фагоцитоз многих бактерий, дрожжеподобных грибов рода
66
СапсИёа и других микроорганизмов опосредован специальными маннозофукозными рецепторами фагоцитов, распознающими углеводные компоненты поверхностных структур микроорганизмов. Наиболее эффективным является фагоцитоз, опосредованный рецепторами, для Fс-фрагмента иммуноглобулинов и для СЗфракции комплемента. Такой фагоцитоз называют иммунным, так как он протекает
при участии специфических антител и активированной системы комплемента, опсонизирующих микроорганизм. Это делает клетку высокочувствительной к захвату
фагоцитами и приводит к последующей внутриклеточной гибели и деградации. В
результате эндоцитоза образуется фагоцитарная вакуоль — фагосома.
4. Внутриклеточное переваривание начинается по мере поглощения бактерий
или других объектов. Оно происходит в фаго-лизосомах, образующихся за счет
слияния первичных лизосом с фагосомами. Захваченные фагоцитами микроорганизмы погибают в результате осуществления механизмов микробоцидности этих
клеток.
Различают кислородзависимые механизмы микробоцидности, связанные с
«окислительным взрывом», и кислороднезависимые механизмы, опосредованные катионными белками и ферментами (в том числе лизоцимом), попадающими
в фагосому в результате ее слияния с лизосомами.Так называемый окислительный взрыв проявляется усилением потребления кислорода и глюкозы с одновременным выбросом биологически активных нестабильных кислородных радикалов,
участвующих в микробоцидности фагоцитов.Внутриклеточная участь захваченных фагоцитами микроорганизмов может быть различной в зависимости от их вирулентности и способности к внутриклеточному паразитизму. Авирулентные и
низковирулентные бактерии погибают и перевариваются в фаголизосомах лизосомными гидролазами.
Незавершенный фагоцитоз. Многие вирулентные бактерии часто не погибают и могут длительно персистировать внутри фагоцитов.Факультативно и облигатно внутриклеточные паразиты после эндоцитоза сохраняют жизнеспособность и
размножаются внутри фагоцитов, вызывая их гибель и разрушение.
Выживание фагоцитированных микроорганизмов могут обеспечивать различные механизмы. Одни патогенные агенты способны препятствовать слиянию
лизосом с фагосомами (токсоплазмы, микобактерии туберкулеза). Другие обладают
устойчивостью к действию лизосомных ферментов (гонококки, стафилококки,
стрептококки группы А и др.). Третьи после эндоцитоза покидают фагосому, избегая действия микробоцидных факторов, и могут длительно персистировать в цитоплазме фагоцитов (риккетсии и др.). В этих случаях фагоцитоз остается незавершенным.
Презентативная, или представляющая, функция макрофагов состоит в
фиксации на наружной мембране антигенных эпитопов микроорганизмов и других
чужеродных агентов. В таком виде они бывают представлены макрофагами для их
специфического распознавания клетками иммунной системы — Т-лимфоцитами .
Секреторная функция заключается в секреции фазоцитами биологически
активных веществ — цитокинов. К ним относятся вещества, оказывающие регулирующее действие на пролиферацию, дифференциацию и функции фагоцитов,
лимфоцитов, фибробластов и других клеток. Особое место среди них занимает
67
интерлей-кин-1 (ИЛ-1), который секретируется макрофагами. Он активирует многие функции Т-лимфоцитов, в том числе продукцию интерлейкина-2 (ИЛ-2). ИЛ-1
и ИЛ-2 являются клеточными медиаторами, участвующими в регуляции иммуногенеза и разных форм иммунного ответа. Одновременно ИЛ-1 обладает свойствами эндогенного пирогена, поскольку он индуцирует лихорадку, действуя на
ядра переднего гипоталамуса.
Макрофаги продуцируют и секретируют такие важные регуляторные факторы,
как простагландины, лейкотриены, циклические нукле-отиды с широким спектром
биологической активности.
Наряду с этим фагоциты синтезируют и секретируют ряд продуктов с преимущественно эффекторной активностью: антибактериальной, антивирусной и цитотоксической. К ним относятся кислородные радикалы , компоненты комплемента,
лизоцим и другие лизосомные ферменты, интерферон. За счет этих факторов фагоциты могут убивать бактерии не только в фаголизосомах, но и вне клеток, в ближайшем микроокружении.
Рассмотренные функции фагоцитирующих клеток обеспечивают их активное
участие в поддержании гомеостаза организма, в процессах воспаления и регенерации, в неспецифической противоинфекционной защите, а также в иммуногенезе и
реакциях специфического клеточного иммунитета (ГЗТ). Раннее вовлечение фагоцитирующих клеток (сначала — гранулоцитов, затем — макрофагов) в ответную
реакцию на любую инфекцию или какое-либо повреждение объясняется тем, что
микроорганизмы, их компоненты, продукты некроза тканей, белки сыворотки крови, вещества, секретируемые другими клетками, являются хемоаттрактантами для
фагоцитов. В очаге воспаления происходит активация функций фагоцитов. Макрофаги приходят на смену микрофагам. В тех случаях, когда воспалительной реакции с участием фагоцитов оказывается недостаточно для очищения организма от
возбудителей, тогда секреторные продукты макрофагов обеспечивают вовлечение
лимфоцитов и индукцию специфического иммунного ответа.
2.Естественные клетки-киллеры (ЕК, natural killer-NK-клетки)
В организме человека и животных функционирует популяция лимфоцитоподобных клеток, обладающих естественной цитотоксичностью по отношению к
клеткам-«мишеням». Они получили название естественных киллеров (ЕК). ЕК являются клетками с эффекторной противоопухолевой, противовирусной и противопаразитарнои активностью. Они способны спонтанно, без предварительного контакта с антигеном убивать опухолевые клетки, а также клетки, зараженные некоторыми вирусами или паразитами. По-видимому, основной функцией ЕК является
противоопухолевый «надзор». Эта система неспецифической клеточной защиты,
вероятно, является филогенетически более древней по сравнению со специфическими Т-клеточными механизмами иммунитета.
Морфологически ЕК представляют собой большие гранулосодержащие лимфоциты. Характерные для них азурофильные цитоплазматические гранулы являются
аналогами лизосом фагоцитирующих клеток. Однако ЕК фагоцитарной функцией
не обладают. Неспецифический характер их цитотоксического действия отличает
эти клетки от антигенспецифических Т-киллеров и от К-клеток, опосредующих
68
антителозависимую цитотоксичность . Среди лейкоцитов крови человека ЕК составляют до15% (подробнее см.иммунокомпетентнык клетки).
Гуморальные факторы
1.Лизоцим
Лизоцим представляет собой термостабильный белок типа муколитического фермента. Он содержится в тканевых жидкостях животных и человека - в слезах,
слюне, перитонеальной жидкости, плазме и сыворотке крови, в лейкоцитах, материнском молоке и др.
Лизоцим продуцируется моноцитами крови и тканевыми макрофагами. Он
вызывает лизис многих сапрофитных бактерий, оказывая менее выраженное литическое действие на ряд патогенных микроорганизмов и не активен в отношении
вирусов.
Механизм бактериолитического действия лизоцима состоит в гидролизе связей в полисахаридных цепях пептидогликанового слоя клеточной стенки бактерии.
Это приводит к изменению ее проницаемости, сопровождающемуся диффузией
клеточного содержимого в окружающую среду, и гибели клеток.
Заживление ран в области слизистых оболочек, имеющих контакт с большим количеством различных микроорганизмов, в том числе и патогенных, в известной
степени объясняется наличием лизоцима.
2.Система комплемента
Системой комплемента называют многокомпонентную самособирающуюся
систему белков сыворотки крови, которая играет важную роль в поддержании гомеостаза. Она способна активироваться в процессе самосборки, т.е. последовательного присоединения к образующемуся комплексу отдельных белков, которые называются компонентами, или фракциями комплемента. Таких фракций известно
девять. Они продуцируются клетками печени, мононуклеарными фагоцитами и содержатся в сыворотке крови в неактивном состоянии.
Процесс активации комплемента может запускаться (инициироваться) двумя
разными путями, получившими названия классический и альтернативный .
При активации комплемента классическим путем инициирующим фактором является комплекс антиген-антитело (иммунный комплекс). Причем антитела только
двух классов IgG и IgМ в составе иммунных комплексов могут инициировать активацию комплемента.При присоединении С1 к комплексу антиген-антитело образуется фермент (С1-эстераза), под действием которого формируется энзиматически активный комплекс (С4b, С2а), называемый СЗ-конвертазой. Данный фермент расщепляет СЗ на СЗа и СЗb. При взаимодействии субфракции СЗb с С4 и С2 образуется пептидаза, действующая на С5. Если инициирующий иммунный комплекс
связан с клеточной мембраной, то самособирающийся комплекс С1, С4, С2, СЗ
обеспечивает фиксацию на ней активированной фракции С5, а затем С6 и С7. Последние три компонента совместно способствуют фиксации С8 и С9. При этом
два набора фракций комплемента — С5а, С6, С7, С8 и С9 — составляют мембраноатакующии комплекс, после присоединения которого к клеточной мембране
клетка лизируется из-за необратимых повреждений структуры ее мембраны
69
Таким образом, при активации комплемента классическим путем ключевыми
компонентами являются С1 и СЗ, продукт расщепления которого СЗb активирует
терминальные компоненты мембраноатакующего комплекса (С5-С9).
Особенность альтернативного пути активации комплемента ( ключевым компонентом является СЗ,) состоит в том, что инициация может происходить без участия комплекса антиген-антитело за счет полисахаридов и липополисахаридов бактериального происхождения — липополисахарида (ЛПС) клеточной стенки грамотрицательных бактерий, поверхностных структур вирусов, иммунных комплексов,
включающих IgА и IgЕ.
В альтернативном пути активации комплемента необходимо участие сывороточного белка, названного пропердином, который активен лишь в присутствии ионов
М§2+.
Как видно из описанных каскадов реакций, многие компоненты комплемента
при активации проявляют активность протеиназ или эстераз, работающих только
внутри системы. При этом в процессе активации комплемента появляются продукты протеолиза компонентов С4, С2, СЗ и С5. Одни из них (фрагменты С4b, С2b,
СЗb, С5b) участвуют непосредственно в самосборке и активации самой системы
комплемента. В отличие от них низкомолекулярные фрагменты СЗа и С5а,
названные анафилатоксинами, по совокупности биологических эффектов освобождение гистамина из тучных клеток, хемотаксис фагоцитов, нарушение
проницаемости сосудов, сокращение гладких мышц и др. -играют существенную
роль в патогенезе болезней иммунных комплексов и других заболеваний, при которых резко усиливаются связывание и активация комплемента в организме.
Фракции комплемента при их активации классическим или альтернативным путем выполняют ряд эффекторных функций:
- мембраноатакующий комплекс опосредует цитолитическое и цитотоксическое
действие специфических антител на клетки-«мишени»;
- анафилотоксины участвуют в иммунопатологических реакциях;
- компоненты комплемента изменяют физико-химические свойства иммунных
комплексов; уменьшают степень агрегации и эффективность их фагоцитоза ;
- фрагмент СЗb способствует связыванию и захвату иммунных комплексов фагоцитами, опсонизируя объекты фагоцитоза;
- фрагменты СЗb, С5а , обладающие свойствами хемоаттрактантов, участвуют в
развитии воспаления.
3. Белки острой фазы
В ходе развития защитных воспалительных реакций после инфицирования
или повреждения, а также при онкогенезе и беременности в организме начинается
усиленная продукция белков острой фазы. Так назвали большую группу белков,
обладающих антимикробным действием, способствующих фагоцитозу, активации
комплемента, формированию и ликвидации воспалительного очага. Белки острой
фазы продуцируются в печени при действии цитокинов, в основном ИЛ-1, ФНО-а
и ИЛ-6. Основную массу белков острой фазы составляют С-реактивный белок и
сывороточные амилоиды А и Р. Другие группы белков острой фазы составляют
факторы свертывания крови, металлосвязывающие белки, ингибиторы протеаз,
компоненты комплемента и некоторые другие. При воспалении содержание в кро-
70
ви большинства белков многократно возрастает, и определение С-реактивного
белка входит в число общепринятых методов диагностики воспалительных процессов.
С-реактивный белок получил название вследствие способности присоединять и преципитировать С-полисахарид. Далее было установлено, что Среактивный белок (СРБ) присоединяется к фосфатидилхолину - компоненту клеточной мембраны любых клеток. Он способен присоединяться к микроорганизмам, активированным лимфоцитам, поврежденным клеткам разных тканей, активируя при этом комплемент. Присоединяясь к нейтрофильным фагоцитам, СРБ
усиливает фагоцитоз и элиминацию объектов фагоцитоза. Вместе с этим СРБ подавляет продукцию супероксида и освобождение из гранул фагоцитов ферментов,
защищая тем самым ткани от повреждения.
Сывороточный амилоид Р близок по структуре к СРБ, обладает способностью
к активации комплемента.
Сывороточный амилоид А - липопротеин, обладающий способностью к хематтракции нейтрофилов, моноцитов и лимфоцитов. Повышенный уровень этого
белка в крови наблюдается при туберкулезе и ревматоидном артрите.
К факторам свертывания крови относятся фибриноген и фактор фон Виллебранда, способствующие образованию сгустков в сосудах зоны воспаления.
Другую группу белков острой фазы составляют белки, связывающие железо гаптоглобин, гемопексин, трансферрин - и тем самым препятствующие размножению микроорганизмов, нуждающихся в этом элементе.
Уровень ингибиторов протеаз в крови возрастает при воспалении в 2-3 раза.
Антитрипсин, антихимотрипсин и макроглобулин препятствуют разрушению
тканей протеазами нейтрофилов в очагах воспаления.
4.Цитокины
Медиаторы межклеточных взаимодействий, именуемые цитокинами,
определяют как реакции врожденного и приобретенного иммунитета, так и ряд
других жизненно необходимых функций организма, значение которых выходит
за рамки иммунологии.
Цитокинами называют гормоноподобные медиаторы, продуцируемые
разными клетками организма и способные повлиять на функции других или этих
же групп клеток. Цитокины - пептиды или гликопротеиды, действующие как
аутокринные, паракринные или межсистемные сигналы. Цитокины формируются
как активированными или поврежденными клетками, так и клетками без дополнительной стимуляции. Регуляторами продукции цитокинов могут быть другие
цитокины, гормоны, простагландины, антигены и многие другие агенты, воздействующие на клетку. Некоторые закономерности цитокиновой регуляции могут
быть сформулированы следующим образом:
• Каждая клетка продуцирует разные цитокины.
• Каждый цитокин может быть продуктом разных видов клеток.
• Один цитокин обладает разными эффектами действия.
• Цитокин может стимулировать или подавлять активность клетки-«мишени».
71
• Каждая клетка имеет рецепторы к разным цитокинам и, следовательно, может
подвергаться одновременному или разновременному воздействию нескольких
цитокинов.
• Взаимодействие нескольких цитокинов на клетку может быть синергичным
или антагонистичным.
• Рецепторы цитокинов могут отделяться от клетки и взаимодействовать с цитокинами вне клетки. В этих условиях свободные рецепторы связывают соответствующие цитокины, что препятствует их контакту с клеточными рецепторами.
• Цитокины, их рецепторы на клетках и во внеклеточных средах составляют
сложную функциональную сеть, результат действия которой зависит от взаимодействия этих факторов между собой и другими цитокинами.
• Цитокины действуют в низких концентрациях порядка 0,001 мкг/мл. Для воздействия на клетку достаточно, чтобы цитокин связался с 10% клеточных рецепторов к нему.
Цитокины составляют обширный класс медиаторов различного происхождения,
обладающих разными свойствами. Их классификация носит условный характер,
так как многие их них обладают одновременно несколькими свойствами и могут
быть отнесены к разным группам. Цитокины объединены в группы в зависимости
от их происхождения (лимфокины, монокины), от характера эффекта (провоспалительные, противовоспалительные). Цитокины, регулирующие взаимодействия
лейкоцитов между собой и другими клетками, называют интерлейкинами (ИЛ).
Большинство цитокинов именуется по действию, которое было впервые обнаружено.
Группа интерлейкинов включает 17 цитокинов, большинство из которых играет ключевую роль в развитии специфического иммунного ответа.
Продуцируемый макрофагами и моноцитами ИЛ-1 обуславливает пролиферацию
лимфоцитов при индукции иммунного ответа, а также активирует Т-лимфоциты,
увеличивает продукцию антител. ИЛ-1 действует на нейтрофилы, способствуя хемотаксису, активации метаболизма, выходу из клеток лизоцима и лактоферрина.
Этот цитокин - эндогенный пироген, вызывающий лихорадку за счет воздействия на
гипоталамический центр терморегуляции.
ИЛ-2 продуцируется Т-лимфоцитами (в основном Тх1), активированными антигеном, собственным ИЛ-2, другими интерлейкинами: ИЛ-1, ИЛ-6, интерфероном,
фактором некроза опухоли (ФНО). Без ИЛ-2 позитивный иммунный ответ на антиген не возникает, стимулированный антигеном лимфоцит гибнет, что может привести к развитию толерантности к данному антигену. Интерлейкины ИЛ-4 и ИЛ-10
подавляют продукцию ИЛ-2. Это способствует развитию эффекторов гиперчувствительности замедленного типа (ГЗТ), формированию киллеров из СD8+ лимфоцитов,
усилению действия ЕК. Все это стимулирует противоопухолевый иммунитет и позволяет рекомендовать рекомбинантный ИЛ-2 для лечения онкологических больных.
72
Факторы роста — большая группа гликопротеинов, контролирующих пролиферацию и созревание потомков стволовой кроветворной клетки. Они продуцируются разными видами клеток и действуют на разные этапы их развития.
Колониестимулирующие факторы (КСФ) получили свое название благодаря
тому, что было обнаружено их свойство способствовать дифференцировке введенных
мышам клеток костного мозга в зрелые гранулоциты и/или моноциты с образованием в селезенке животных колоний соответствующих клеток. Гранулоцитарный КСФ
обеспечивает дифференцировку предшественников гранулоцитов в зрелые нейтрофилы. Моноцитарный КСФ способствует созреванию моноцитов и макрофагов из клеток-предшественников, а гранулоцитарно-моноцитарный КСФ стимулирует формирование гранулоцитов и макрофагов из их общих предшественников.
Интерфероны (ИФ) были открыты как противовирусные агенты. Затем
были обнаружены их иммунорегулирующие свойства. Существует три разновидности ИФ
У здоровых людей ИФ в крови не обнаруживаются. Их уровень повышен при
красной волчанке, ревматоидном артрите, склеродермии. Наличие интерферона в
крови этих больных увеличивает резистентность к вирусным инфекциям и опухолям, но неблагоприятно сказывается на развитии аутоиммунных процессов, свойственных этим заболеваниям.
Препараты интерферонов используются для лечения лейкемий и некоторых других онкологических процессов. Для усиления противовирусной защиты используют
средства, повышающие продукцию собственного интерферона (интерфероногены).
В качестве индукторов эндогенного интерферона применяют противовирусные вакцины, препараты РНК и ДНК.
Цитотоксины. Такое название получили цитокины группы факторов некроза
опухолей (ФНО), который был впервые обнаружен как компонент сыворотки крови животных, стимулированных бактерийным токсином, вызывающий некротические процессы в опухолевой ткани. ФНО служит медиатором ответа организма на
микробную инвазию. Эндотоксины (липиполисахариды) микробов стимулируют
клетки-продуценты к образованию ФНО, который, в свою очередь, обеспечивает
хемотаксис фагоцитов в инфицированную ткань и усиливает фагоцитоз возбудителей. В настоящее время известно, что ФНО составляют по крайней мере две группы
(альфа и бета) медиаторов, продуцируемых активированными макрофагами, естественными киллерами, а также лимфоцитами, нейтрофилами и тучными клетками.
Завершая рассмотрение цитокинов и их эффектов, необходимо подчеркнуть, что
в механизмах иммунитета участвуют две группы противоположно действующих цитокинов. Одна группа — провоспалительные цитокины (ИЛ-1, ИЛ-6, ИЛ8 и другие лимфокины, ФНО-а, а также ИФ), стимулируя разные клетки и механизмы, усиливают врожденную неспецифическую защиту, воспаление, способствуют развитию специфических иммунных реакций. Вторая функциональная
группа — противовоспалительные цитокины (ИЛ-4, ИЛ-10, ИЛ-13, ТРФ) подавляет развитие как неспецифических, так и специфических иммунных реакций.
Адгезины. Среди факторов, определяющих прямые контакты клеток организма
между собой и с представителями микрофлоры, существенную роль играют моле-
73
кулы адгезии или адгезины. Предполагается, что в эволюции живого появление
молекул адгезии сделало возможным возникновение многоклеточных организмов.
Более 90% микробов, составляющих нормальную микрофлору человеческого организма, обитают в нем благодаря молекулам адгезии. Блокирование адгезии патогенных микроорганизмов к клеткам и тканям организма — один из основных путей
антимикробной защиты. Молекулы адгезии экспрессируются на мембранах клеток,
определяя их способность контактировать с другими клетками и неклеточными субстратами. Рецепторами молекул адгезии в организме могут быть другие молекулы
адгезии на поверхности клеток, углеводные компоненты мембран, иммуноглобулины. Количество молекул адгезии и рецепторов к ним увеличивается при антигенной
или любой другой активации клеток.
В ходе иммунного ответа молекулы адгезии определяют контакты антигенпредставляющих клеток с лимфоцитами и лимфоцитов между собой. Молекулы адгезии входят в состав рецепторов иммунокомпетентных клеток и определяют тропность клеток иммунной системы к определенным тканям или органам — хомингэффект (англ. Ноте — дом).
Молекулы адгезии условно разделяют на группы: селектины, интегрины, молекулы суперсемейства иммуноглобулинов.
Селектины — семейство поверхностных молекул адгезии, определяющие присоединение клеток к углеводным компонентам других структур.
Интегрины — большая группа молекул, определяющая взаимодействия белокбелок.Интегрины играют роль в межклеточных контактах при воспалении, реакциях
иммунитета, аутоиммунных повреждениях тканей, процессах репарации. Интегрины экспрессируются на клетках опухолей и играют роль в процессах метастазирования. Их определение используется для диагностики разных видов злокачественных опухолей.
К молекулам суперсемейства иммуноглобулинов относится более 15 вариантов
молекул, которые обозначаются заглавными латинскими буквами, соответствующими обозначению их функции: адгезии клетка-клетка или белок-белок.
К молекулам адгезии суперсемейства иммуноглобулинов относятся СD4+, СD8+
молекулы Т-лимфоцитов, определяющие их контакты со структурами МНС II или I
класса и дифференцировку этих двух классов Т-клеток между собой.
Адгезины формально не относятся к системе цитокинов, но обладают многими
сходными с ними функциями и участвуют в межклеточной кооперации.
5.Белки теплового шока
При воздействии на микробные и эукариотические клетки неблагоприятных стрессовых факторов - повышенной температуры, голодания, токсинов, тяжелых металлов, вирусов, в них формируются защитные белки. Они получили название белков теплового шока , так как они были впервые обнаружены при тепловом
воздействии на клетки.В результате повышаются термоустойчивость и резистентность клеток за счет защиты и коррекции поврежденных стрессом клеточных белков
Помимо поддержания резистентности клеток к шоковым воздействиям БТШ
принимают участие в эндоцитозе вирусных частиц, процессинге антигенов, входят
в состав некоторых рецепторных комплексов (стероидные рецепторы).
74
Нормальная микрофлора человека
Нормальная микрофлора человека играет важную роль в защите организма от
патогенных микроорганизмов. Представители нормальной микрофлоры участвуют в
неспецифической защите заселенных ими участков желудочно-кишечного, дыхательного, мочеполового трактов, кожных покровов.
Обитающие в определенных биотопах микроорганизмы препятствуют адгезии и колонизации поверхностей тела патогенными микроорганизмами. Защитное
действие нормальной микрофлоры может быть обусловлено конкуренцией за питательные вещества, изменением рН среды, продукцией колицинов и других активных
факторов, препятствующих внедрению и размножению патогенных микроорганизмов.
Нормальная микрофлора способствует созреванию иммунной системы и
поддержанию ее в состоянии высокой функциональной активности, так как компоненты микробной клетки неспецифически стимулируют клетки иммунной системы. Существенная защитная и иммуностимулирующая роль нормальной микрофлоры выявляется тогда, когда гнотобионты (животные, выращенные в стерильных условиях) погибают после инфицирования непатогенными микробами. Лечение антибиотиками, при котором меняется состав нормальной микрофлоры, а иногда происходит полное ее исчезновение, вызывает тяжелые дисбактериозы , существенно осложняющие заболевание. В случаях нарушения состава биотопов или
при существенном снижении естественной иммунной защиты организма заболевания могут вызвать и представители нормальной микрофлоры организма.
Специфическая резистентность ( адаптивный иммунитет )
Осуществляется специализированной системой - иммунной системой,
назначение которой состоит в защите организма от биологической агрессии внешней (инфекции) и внутренней (опухоли).
Иммунная защита от биологической агрессии достигается триадой реакций, включающей :
- распознание чужеродных и измененных собственных макромолекул (антигенов -АГ);
- удаление из организма антигенов и несущих их клеток - элиминация;
- запоминание контакта с конкретными антигенами –формирование иммунологической памяти.
Иммунная система — комплекс органов и тканей, содержащих иммунокомпетентные клетки и обеспечивающая антигенную индивидуальность и однородность организма путём обнаружения и, как правило, деструкции и элиминации из него чужеродного АГ. Иммунная система состоит из центральных и
периферических органов.
К центральным (первичным) органам относят костный мозг и вилочковую железу. В них происходит антигеннезависимое деление и созревание лимфоцитов, которые впоследствии мигрируют в периферические органы иммунной системы.
75
К периферическим (вторичным) органам относят селезёнку, лимфатические узлы, миндалины, лимфоидные элементы ряда слизистых оболочек. В этих
органах происходят как антигеннезависимая, так и антигензависимая пролиферация и дифференцировка лимфоцитов. Как правило, зрелые лимфоциты впервые контактируют с АГ именно в периферических лимфоидных органах.
Заселение периферических органов иммунной системы T- и Bлимфоцитами, поступающими из центральных органов иммунной системы,
происходит не хаотически. Каждая популяция лимфоцитов мигрирует из кровеносных сосудов в определённые лимфоидные органы и даже в различные их
регионы. Так, B-лимфоциты преобладают в селезёнке (в её красной пульпе, а
также по периферии белой) и пейеровой бляшке кишечника (в центрах фолликулов), а T-лимфоциты — в лимфатических узлах (в глубоких слоях их коркового вещества и в перифолликулярном пространстве).
В организме здорового человека в процессе лимфопоэза образуется более
9
10 разновидностей однородных клонов лимфоцитов. При этом каждый клон
экспрессирует только один вид специфического антигенсвязывающего рецептора. Большинство лимфоцитов периферических органов иммунной системы не
закрепляются в них навсегда. Они постоянно циркулируют с кровью и лимфой
как между различными лимфоидными органами, так и во всех других органах и
тканях организма. Такие лимфоциты получили название рециркулирующих.
Биологический смысл рециркуляции T- и B-лимфоцитов:
- осуществление постоянного надзора за антигенными структурами организма;
- реализация межклеточных взаимодействий (кооперация) лимфоцитов и мононуклеарных фагоцитов, что необходимо для развития и регуляции иммунных
реакций.
Иммунокомпетентные клетки
К иммунокомпетентным клеткам относятся T- и B-лимфоциты, NKклетки и антигенпредставляющие клетки.
Т-лимфоциты развиваются в тимусе из клеток-предшественниц. Влимфоциты дифференцируются в печени плода и костном мозге взрослого организма. NK-клетки образуются из предшественников лимфоидных клеток в
костном мозге. Лимфоциты, как и другие лейкоциты, на своей поверхности
экспрессируют большое количество различных молекул, по которым при помощи моноклональных АТ идентифицируют их принадлежность к конкретной
клеточной популяции. Чаще всего с этой целью выявляют дифференцировочные антигены (CD- claster differentiation), являющиеся специфичными клеточными маркёрами.
B-лимфоциты
Эта субсистема образована различными клонами B-лимфоцитов. Название субсистемы отражает то обстоятельство, что лимфоциты, представляющие
её, формируются у птиц в сумке (bursa) Фабрициуса (впервые B-лимфоциты
были выявлены в лимфоидных органах птиц). У человека подобной бурсы нет,
B-лимфоциты созревают в костном мозге, а также, возможно, в пейеровых
бляшках, миндалинах, определённых зонах селезёнки и лимфоузлов. B-
76
лимфоциты берут начало от стволовых кроветворных клеток костного мозга. Bлимфоциты обеспечивают реализацию эффекторного звена гуморального иммунного ответа.
В мембране B-лимфоцита присутствует рецептор АГ - мономер IgM. Из
красного костного мозга B-лимфоциты мигрируют в тимус-независимые зоны
лимфоидных органов. Продолжительность жизни большинства B-лимфоцитов
не превышает десяти дней, если они не активируются АГ. Зрелые В-лимфоциты
(плазматические клетки) вырабатывают антитела(АТ)-иммуноглобулины(Ig )
всех известных классов. CD19, CD20 и CD22 — основные маркёры, используемые для идентификации B-клеток.В процессе формирования B-клеток выделяют антигеннезависимую и антигензависимую стадии.
Антигеннезависимая стадия созревания B-лимфоцитов происходит под
контролем локальных клеточных и гуморальных сигналов от микроокружения
пре-B-лимфоцитов и не определяется контактом с АГ. На этой стадии происходит формирование отдельных пулов генов, кодирующих синтез Ig, а также экспрессия этих генов. Однако, на цитолемме пре-B-клеток ещё нет поверхностных рецепторов - Ig, компоненты последних находятся в цитоплазме.
Образование B-лимфоцитов из пре-B-лимфоцитов сопровождается появлением на их поверхности первичных Ig, способных взаимодействовать с АГ.
Только на этом этапе B-лимфоциты попадают в кровоток и заселяют периферические лимфоидные органы. Сформировавшиеся молодые B-клетки накапливаются в основном в селезёнке, а более зрелые — в лимфатических узлах.
Антигензависимая стадия развития B-лимфоцитов начинается с момента
контакта этих клеток с АГ (в том числе - аллергеном). В результате происходит
активация B-лимфоцитов, протекающая в два этапа: пролиферации и дифференцировки.
Пролиферация B-лимфоцитов обеспечивает два важных процесса:
1.Увеличение числа клеток, дифференцирующихся в плазматические клетки,
продуцирующие АТ (Ig);по мере созревания B-клеток и их превращения в
плазматические клетки происходит интенсивное развитие белоксинтезирующего аппарата, комплекса Гольджи и исчезновение поверхностных первичных Ig.
Вместо них продуцируются уже секретируемые (т.е. выделяемые в биологические жидкости - плазму крови, лимфу, СМЖ и др.) антигенспецифические
АТ.Каждая плазматическая клетка способна секретировать большое количество
Ig-несколько тысяч молекул в секунду. Процессы деления и специализации Bклетки осуществляются не только под влиянием АГ, но и при обязательном
участии T-лимфоцитов-хелперов, а также выделяемых ими и фагоцитами цитокинов-факторов роста и дифференцировки.
2.Образование В-лимфоцитов иммунологической памяти. Эти клоны Bклеток представляют собой долгоживущие рециркулирующие малые лимфоциты. Они не превращаются в плазматические клетки, но сохраняют иммунную
«память» об АГ. Клетки памяти активируются при повторной их стимуляции
тем же самым АГ. В этом случае B-лимфоциты памяти (при обязательном участии T-клеток-хелперов и ряда других факторов) обеспечивают быстрый синтез
77
большого количества специфических АТ, взаимодействующих с чужеродным
АГ, и развитие эффективного иммунного ответа или аллергической реакции.
T-лимфоциты
Субсистема T-лимфоцитов представлена различными клонами Tлимфоцитов. Их пролиферация и дифференцировка происходит под контролем
вилочковой железы. В связи с этим их обозначают как T-клетки,или тимус-зависимые лимфоциты. T-клетки, как и B-лимфоциты, развиваются из
стволовых кроветворных клеток костного мозга. Отсюда в виде клетокпредшественниц T-лимфоциты попадают с кровью в тимус, где происходит их
антигеннезависимое созревание, сопровождающееся экспрессией на цитолемме
специфических (у каждого лимфоцита своего) рецепторов.
Т-лимфоциты ответственны за реализацию клеточного звена иммунного
ответа, а также участвуют в регуляции гуморального иммунного ответа.
Т-клетки состоят из функциональных подтипов CD4+ и CD8+.
Т-хелперы (Th) — CD4+ Т-клетки. При активации синтезируют и секретируют цитокины (ИЛ2, ИЛ4, ИЛ5, ИЛ6, ɤ-ИФН). В ходе иммунного ответа взаимодействуют с молекулами MHC класса II.
Цитотоксические T-лимфоциты (Tk) — CD8+ Т-клетки, уничтожают инфицированные вирусом, опухолевые и чужеродные клетки при помощи цитолитического белка — перфорина. Взаимодействуют с молекулой MHC класса I
плазматической мембраны клетки–мишени.
T-супрессоры (Ts) — представители CD8+ Т-клеток — регулируют интенсивность иммунного ответа, подавляя активность Th клеток; предотвращают
развитие аутоагрессивных иммунных реакций; защищают организм от нежелательных последствий иммунной реакции, от чрезмерного воспаления и аутоагрессии.
NK-клетки
NK-клетки (МНС-нерестригированные киллеры, естественные киллеры)
составляют до 15% всех лимфоцитов крови. Они не имеют поверхностных детерминант, характерных для T- и B-лимфоцитов, не имеют рецептора Тлимфоцитов. В типичных NK-клетках экспрессируются дифференцировочные
АГ CD2, СD7, CD56 и CD16 (рецептор Fc-фрагмента IgG). В плазматической
мембране активированных NK-клеток появляется гликопротеин CD69. NKклетки распознают и уничтожают опухолевые и вирус-инфицированные клетки. Механизм распознавания неясен. Существует представление о наличии поверхностноклеточных молекул, защищающих клетки организма от цитотоксического действия NK-клеток. Примером служит продукт гена HLA-C. Распознавание рецептором NK-клетки этой молекулы тормозит цитотоксическую активность NK-клеток и таким образом защищает клетку, экспрессирующую
HLA-C. Модификация продукта гена HLA-C вирусами или связанными с опухолью молекулами приводит к уничтожению этой клетки NK-клеткой. NKклетки, располагая рецептором IgG (CD16), способны также взаимодействовать
с клетками, окружёнными молекулами IgG, и уничтожать их (феномен
АТ-зависимой цитотоксичности). Активированные NK-клетки выделяют
ɤ-ИФН, ИЛ1. При активации (например, под влиянием ИЛ2) NK-клетки приоб-
78
ретают способность к пролиферации. Функция NK-клеток нарушена при синдроме Шедьяка–Хигаси. Дефект NK-клеток — причина хронических инфекций.
В отличие от цитотоксических T-лимфоцитов, способность NK-клеток к
цитолизу не связана с необходимостью распознавания молекул MHC на поверхности мишени. NK-клетки уничтожают клетку-мишень не путём фагоцитоза, а (после установления с ней прямого контакта) при помощи перфорина.
NK-клетки, наряду с макрофагами, нейтрофилами и эозинофилами,
участвуют также и в АТ-зависимом клеточно-опосредованном цитолизе. Для
этого NK-клетки экспрессируют на своей поверхности рецептор Fc-фрагмента
IgG (CD16). Fc-фрагмент этих АТ взаимодействует с рецептором Fc-фрагмента,
встроенным в плазматическую мембрану NK-клетки.
Антигенпредставляющие клетки
Антигенпредставляющие клетки (АПК) присутствуют преимущественно
в коже, лимфатических узлах, селезёнке и тимусе.К ним относятся макрофаги,
дендритные клетки, фолликулярные отростчатые клетки лимфоузлов и селезёнки, клетки Лангерханса, М-клетки в лимфатических фолликулах пищеварительного тракта, эпителиальные клетки вилочковой железы.
Эти клетки захватывают, перерабатывают и представляют АГ (эпитоп) на
своей поверхности другим иммунокомпетентным клеткам, вырабатывают ИЛ1
и другие цитокины, секретируют простагландин E2 (PGE2), угнетающий иммунный ответ.
Дендритные клетки происходят из костного мозга и образуют популяцию долгоживущих клеток, которые запускают и модулируют иммунный ответ.
В костном мозге их предшественники образуют субпопуляцию CD34+-клеток,
которые способны дифференцироваться в клетки Лангерханса для эпителия и
дендритные клетки для внутренней среды. Незрелые и неделящиеся предшественники дендритных клеток заселяют многие ткани и органы. Дифференцировку дендритных клеток поддерживают колониестимулирующий фактор гранулоцитов и макрофагов и ИЛ3. Дендритные клетки имеют звёздчатую форму и
в состоянии покоя несут на поверхности относительно небольшое количество
молекул МНС. В отличие от клеток Лангерханса, интерстициальные дендритные клетки способны стимулировать синтез Ig В-лимфоцитами. Все дендритные клетки могут вначале поступать в тимус-зависимую зону периферических
лимфоидных органов, где созревают в так называемые интердигитирующие
клетки.
Молекулы иммунной системы
Антигены (Аг) - химические вещества, свободные либо встроенные в
мембрану клетки, способные индуцировать иммунный ответ (ИО).
Мембранные АГ делятся на дифференцировочные (CD-АГ), HLA ((human
leukocyte antigen), относятся к главному комплексу гистосовместимости (ГКГС
или МНС - main histocompartibility complex), их три класса), детерминантные.
Полноценный антиген состоит из двух частей:
- носитель (стабилизирующая часть) - 97 - 99% молекулы антигена; это, как
правило, макромолекулы, инертные корпускулярные частицы;
79
- детерминантная группа (эпитоп) - олигосахариды или олигопептиды, располагаются как правило на поверхности молекулы ; на одном носителе может быть
несколько эпитопов, в связи с этим вводят понятие эпитопная плотность;детерминантная группа определяет специфичность антигена.
Свойства антигенов: способны вызывать иммунный ответ; способны к
специфическому взаимодействию с различными молекулами и клетками (эритроцитами и т.д.). Если реализованы оба указанных свойства, то такой антиген
называют полноценным, если реализовано только второе свойство, то такой антиген называют неполноценным или гаптеном. Гаптен может быть фиксирован
на специальные носители - адьюванты. Механизм действия адьювантов:
- создают депо антигенов;
- укрупняют молекулу;
- активируют лимфоидную ткань.
Классификация антигенов:
по чужеродности : ксеноантигены (гетеро-) - не принадлежат особям данного
вида; аллоантигены (гомо-) - принадлежат особям данного вида; аутоантигены
-собственные антигены, например "забарьерные" клетки - сперматозоиды,
клетки мозга; собственные клетки с иммунной активностью;
по типу вызываемого иммунного ответа :
- иммуногены;
- аллергены;
- толерогены;
- трансплантационные антигены;
по связи с вилочковой железой ( тимусом) : - Т- зависимые; - Тнезависимые.
по локализации в микроорганизме :
- О - антигены - липополисахариды (ЛПС) клеточной стенки, термостабильные,
высокоактивные, многообразны у разных микроорганизмов и даже у одного и
того же;
- Н - антиген - жгутиковый белок, термолабильный, достаточно активный, также разнообразен;
- К - антигены - капсульные гликопротеиды, иммуногенность зависит от химической природы; фимбриальные антигены; протоплазматические антигены; экзоаллергены;
по специфичности для микроорганизма - носителя :
- видовые - у всех особей вида;
- типовые - вариантные, у варов;
- групповые - общие для микроорганизмов разных видов и родов;
- стадийные - появляются на определенных стадиях развития;
- штаммоспецифичные.
Антитела
Антитела (иммуноглобулины) - продукты гуморального имунного ответа,
это глобулины, специфически реагирующие с антигеном, вызвавшим их образование. Это сложные белковые образования, мономеры или полимеры.
80
Структура иммуноглобулинов. Молекула иммуноглобулина состоит из
4 гликозилированных полипептидных цепей — 2 легких и 2 тяжелых, соединенных дисульфидными мостиками в симметричную структуру . Существует
5 классов иммуноглобулинов — IgA, IgG, IgM, IgD и IgE. Они различаются по
типу тяжелых цепей . Легкие цепи могут быть лишь двух типов — каппа и
лямбда. Каждая молекула иммуноглобулина состоит из тяжелых цепей одного
типа, соединенных с легкими цепями также только одного типа. Иммуноглобулины одного класса могут содержать как каппа-, так и лямбда-цепи. Мономерные иммуноглобулины, например IgG, состоят из одной молекулы, полимерные — IgM и IgA — из нескольких. Так, IgM состоит из 10 мю-цепей и
10 каппа- или лямбда-цепей. Помимо легких и тяжелых цепей молекулы полимерных иммуноглобулинов включают J-цепь, а молекулы IgA — секреторный
компонент.
Разнообразие антител. Для распознавания всего многообразия антигенов
окружающей среды иммунная система должна вырабатывать не менее
108 антител разной специфичности. Специфичность антител, то есть способность распознавать какой-либо один антиген, определяется аминокислотной
последовательностью вариабельных областей легких и тяжелых цепей . Разнообразие антител обеспечивается уникальным строением участков ДНК, кодирующих вариабельные области. В зрелом B-лимфоците участок ДНК, кодирующий вариабельную область тяжелой цепи, состоит из трех генов, которые
обозначаются V, D и J, а вариабельная область легкой цепи — из двух генов —
V и J. Костномозговой предшественник B-лимфоцита содержит множество вариантов этих генов. В процессе созревания B-лимфоцита они случайным образом комбинируются друг с другом, образуя единый комплекс, состоящий из одного гена V, одного гена D и одного гена J. Увеличению разнообразия антител
способствуют также мутации этих генов.
Функции отдельных участков молекулы иммуноглобулина можно изучать,
расщепляя ее на фрагменты. Для расщепления молекулы иммуноглобулина используются ферменты (папаин и пепсин), кислоты и мочевина. При обработке
папаином молекула иммуноглобулина распадается на три части — два Fabфрагмента и один Fc-фрагмент, тогда как пепсин расщепляет молекулу на один
F(ab')2-фрагмент и два Fc-фрагмента .
а. Fab-фрагмент представлен N-концевым участком тяжелой цепи и легкой
цепью молекулы иммуноглобулина, каждая цепь Fab-фрагмента содержит одну
вариабельную и одну константную область. Специфичность антител определяется аминокислотной последовательностью вариабельных областей.
б. Fc-фрагмент представляет собой C-концевые участки тяжелых цепей и состоит только из константных областей. От строения Fc-фрагмента зависит способность иммуноглобулина проникать через плаценту, связывать комплемент и
присоединяться к разным типам клеток — макрофагам, тромбоцитам, тучным
клеткам. Кроме того, от строения этого фрагмента зависит скорость синтеза и
распада молекулы иммуноглобулина. В распознавании антигена Fc-фрагмент
участия не принимает.
81
Классы иммуноглобулинов
Иммуноглобулины G, IgG- это мономеры, включающие 4 субкласса
(IgGl - 77%; IgG2 - 11%; IgG3 - 9%; IgG4 - 3%), которые отличаются друг от
друга по аминокислотному составу и антигенным свойствам. Их содержание в
сыворотке крови колеблется от 8 до 16,8 мг/мл, период полураспада составляет
20-28 дней, а синтезируется в течение суток от 13 до 30 мг/кг. На их долю приходится 80% от общего содержания ИГ. Они защищают организм от инфекций.
Антитела субклассов IgGl и IgG4 специфически связываются через Fcфрагменты с возбудителем (иммунное опсонирование), а благодаря Fcфрагментам взаимодействуют с Fc-рецепторами фагоцитов (макрофагов, полиморфноядерных лейкоцитов), способствуя тем самым фагоцитозу возбудителя.
IgG4 участвует в аллергических реакциях и не способен фиксировать комплемент.
Антитела класса IgG играют основополагающую роль в гуморальном иммунитете при инфекционных заболеваниях, вызывая гибель возбудителя с участием комплемента и опсонизируя фагоцитарные клетки. Они проникают через
плаценту и формируют антиинфекционный иммунитет у новорожденных. Они
способны нейтрализвать бактериальные экзотоксины, связывать комплемент,
участвовать в реакции преципитации.
Иммуноглобулины М, IgM - это наиболее "ранние" из всех классов ИГ,
включающие 2 субкласса: IgMl (65%) и IgM2 (35%). Их концентрация в сыворотке крови колеблется от 0,5 до 1,9 г/л или 6% от общего содержания ИГ. За
сутки синтезируется 3-17 мг/кг, а период их полураспада составляет 4-8 суток.
Они не проникают через плаценту. IgM появляется у плода и участвует в антиинфекционной защите. Они способны агглютинировать бактерий, нейтрализовать вирусы, активировать комплемент. IgM играют важную роль в элиминации
возбудителя из кровеносного русла, в активации фагоцитоза.
Значительное повышение концентрации IgM в крови наблюдается при
ряде инфекций (малярия,трипаносомозе) как у взрослых, так и у новорожденных. Это показатель внутриутробного заражения возбудителя краснухи, сифилиса, токсоплазмоза, цитомегалии. IgM - это антитела, образующиеся на ранних
сроках инфекционного процесса. Они отличаются высокой активностью в реакциях агглютинации, лизиса и связывания эндотоксинов грамотрицательных
бактерий.
Иммуноглобулины A, IgA - это секреторные ИГ, включающие 2 субкласса: IgAl (90%) и IgA2 (10%). Содержание IgA в сыворотке крови колеблется
от 1,4 до 4,2 г/л или 13%) от общего количества ИГ; ежедневно синтезируется
от 3 до 50 мкг/кг. Период полураспада антител составляет 4-5 суток. IgA содержится в молоке, молозиве, слюне, в слезном, бронхиальном и желудочнокишечном секрете, желчи, моче. В состав IgA входит секреторный компонент,
состоящий из нескольких полипептидов, который повышает устойчивость IgA
к действию ферментов. Это основной вид ИГ, участвующих в местном иммунитете. Они препятствуют прикреплению бактерий к слизистой, нейтрализуют эн-
82
тероток-син, активируют фагоцитоз и комплемент. IgA не определяется у новорожденных. В слюне он появляется у детей в возрасте 2 месяца., причем первым обнаруживается секреторный компонент SC, и только позднее полная молекула SIgA. Возраст 3 мес. многими авторами определяется как критический
период; этот период особенно важен для диагностики врожденной или транзиторной недостаточности местного иммунитета.
Иммуноглобулины Е, IgE - это мономеры, содержание которых в сыворотке крови ничтожно мало - 0,00005-0,0003 г/л или 0,002% от общего количества ИГ. За сутки синтезируется 0,02мг/кг, а период их полураспада в сыворотке крови составляет 2-3 дня, а в коже - 9-14 дней.
К классу IgE относится основная масса аллергических антител -реагинов.
Уровень IgE значительно повышается у людей, страдающих аллергией и зараженных гельминтами. IgE связывается с Fc-рецепторами тучных клеток и базофилов. При контакте с аллергеном образуются мостики "IgE-антиген-IgE", что
сопровождается поступлением ионов кальция в клетку-мишень, активацией в
ней биохимических процессов и выделением БАВ, вызывающих аллергические
реакции немедленного типа.
Эозинофильный хемотаксический фактор, выделяемый тучными клетками, способствует аккумуляции эозинофилов и деструкции гельминтов. Предполагается также, что IgE, покрывая паразита, аккумулирует макрофаги благодаря
Fc-рецепторам этих клеток.
Иммуноглобулины D, IgD - это мономеры; их содержание в крови составляет 0,03-0,04 г/л или 1%) от общего количества ИГ; в сутки их синтезируется от 1до 5 мг/кг, а период полураспада колеблется в пределах 2-8 дней. IgD
участвуют в развитии местного иммунитета, обладают антивирусной активностью, в редких случаях активируют комплемент.
Плазматические клетки, секретирующие IgD, локализуются преимущественно в миндалинах и аденоидной ткани. IgD выявляются на В-клетках и отсутствуют на моноцитах, нейтрофилах и Т-лимфоцитах. Полагают, что IgD
участвуют в дифференцировке В-клеток, способствуют развитию антиидиотипического ответа, участвуют в аутоиммунных процессах.
Основная масса IgM и IgD находится в плазме, a IgG и IgA распределяются примерно в одинаковых соотношениях между плазмой и межсосудистой тканью. IgG проходит через плаценту к плоду, и к моменту родов их концентрацш
достигает максимума, но однако быстро снижается и в этой связи ребенок в 34-месячном возрасте наименее устойчив к инфекционным заболеваниям.
Иммунный ответ
Иммунный ответ возможен в результате активации клонов лимфоцитов и
состоит из двух фаз. В первой фазе АГ активирует те лимфоциты, которые его
распознают. Во второй (эффекторной) фазе эти лимфоциты координируют иммунный ответ, направленный на устранение АГ.
Гуморальный иммунный ответ
В гуморальном иммунном ответе эффекторными клетками являются Влимфоциты. Регуляцию антителообразования осуществляют Т-хелперы и Т-
83
супрессоры.Вторгшийся в организм АГ поглощается макрофагом и подвергается процессингу - расщеплению на фрагменты. Фрагменты АГ выставляются на
поверхности клетки вместе с молекулой MHC. Комплекс «АГ–молекула MHC
класса II» предъявляется T-хелперу При помощи рецептора Т-лимфоцита
Т-клетка распознает АГ, но только находящийся в комплексе с молекулой
MHC.
T-хелпер распознаёт комплекс «АГ–молекула MHC класса II» на поверхности антигенпредставляющей клетки. Для активации Т-хелпера специфическое узнавание Т-хелпером фрагмента АГ на поверхности антигенпредставляющей клетки оказывается недостаточным. Активацию Т-хелперов обеспечивает
взаимодействие молекулы В7 (расположена на поверхности антигенпредставляющей клетки) с молекулой CD28 на поверхности Т-хелпера. Узнавание
Т-хелпером нужных молекул на поверхности антигенпредставляющей клетки
стимулирует секрецию ИЛ1 . Активированный ИЛ1 T-хелпер синтезирует ИЛ2
и рецепторы ИЛ2, через которые стимулирует пролиферацию Т-хелперов и цитотоксических T-лимфоцитов. Таким образом, после взаимодействия с антигенпредставляющей клеткой T-хелпер приобретает способность отвечать на
действие ИЛ2 всплеском пролиферации. Биологический смысл этого процесса
состоит в накоплении такого количества Т-хелперов, которое обеспечит образование в лимфоидных органах необходимого количества плазматических клеток, способных вырабатывать АТ против данного АГ.
ИЛ2 стимулирует пролиферацию Т-хелперов и активирует цитотоксические T-лимфоциты. Отбор В-лимфоцитов производится при взаимодействии АГ
с Fab-фрагментами IgM на поверхности этих клеток. Эпитоп этого АГ в комплексе с молекулой MHC класса II узнаёт рецептор Т-хелпера, после чего из
T-лимфоцита секретируются цитокины, стимулирующие пролиферацию
В-лимфоцитов и их дифференцировку в плазматические клетки, синтезирующие АТ против данного АГ. Рецептор цитотоксических T-лимфоцитов связывается с антигенной детерминантой в комплексе с молекулой MHC класса I на
поверхности вирус-инфицированной или опухолевой клетки. В молекулярном
взаимодействии участвует дифференцировочный АГ цитотоксического
T-лимфоцита CD8. После связывания молекул взаимодействующих клеток цитотоксический T-лимфоцит убивает клетку-мишень.
Активация B-лимфоцита возможна при прямом взаимодействии АГ с Ig
на поверхности B-клетки. В этом случае сам B-лимфоцит процессирует АГ и
представляет его фрагмент в комплексе с молекулой MHC II на своей поверхности. Этот комплекс распознаёт T-хелпер, отобранный при помощи того же
АГ. В активации В-клетки участвуют две пары молекул: с одной стороны, специфическое взаимодействие АГ с рецептором (IgM) на поверхности
В-лимфоцита, а с другой стороны, молекула CD40 на поверхности В-клетки
взаимодействует с молекулой CD40L на поверхности Т-хелпера, активирующего В-клетку. Узнавание рецептором T-хелпера комплекса «Аг–молекула MHC
класса II» на поверхности B-лимфоцита приводит к секреции из Т-хелпера ИЛ2,
ИЛ4, ИЛ5 и ɤ-ИФН. Под их действием B-клетка активируется и пролиферирует,
образуя клон. Активированный B-лимфоцит дифференцируется в плазматиче-
84
скую клетку: увеличивается количество рибосом, гранулярная эндоплазматическая сеть и комплекс Гольджи становятся более выраженными.
Плазматические клетки.Плазматическая клетка синтезирует Ig. ИЛ6, выделяемый активированными Т-хелперами, стимулирует секрецию Ig. Часть зрелых В-лимфоцитов после АГ-зависимой дифференцировки циркулирует в организме как клетки памяти.
Клеточный иммунный ответ
В клеточном иммунном ответе эффекторными клетками являются цитотоксические Т-лимфоциты, активность которых регулируют Т-хелперы и Тсупрессоры.
Реакции клеточно-опосредованного цитолиза
Эффекторные клетки при помощи своих рецепторов распознают клеткумишень и уничтожают её. За клеточно-опосредованный цитолиз отвечают не
только Т-лимфоциты, но и другие субпопуляции лимфоидных клеток, а в некоторых случаях миелоидные клетки. В процессе узнавания участвуют различные
молекулы, выставленные на поверхности взаимодействующих клеточных партнеров:
- специфические АГ (например, вирусные пептиды на поверхности инфицированных клеток) в комплексе с молекулой MHC распознаются рецепторами цитотоксических Т-клеток, преимущественно CD8+-клеток и некоторыми субпопуляциями CD4+-клеток;
- антигенные детерминанты опухолевых клеток распознаются NK-клетками
без участия молекулы MHC класса I;
- связанные с АГ АТ на поверхности клеток–мишеней, распознаются рецепторами Fc-фрагментов NK-клеток (феномен АТ-зависимой цитотоксичности).
Цитотоксические T-лимфоциты
Предъявленный на поверхности клетки–мишени АГ в комплексе с молекулой
MHC класса I связывается с рецептором цитотоксического T-лимфоцита . В
этом процессе участвует молекула CD8 клеточной мембраны Tk. Секретируемый T-хелперами ИЛ2 стимулирует пролиферацию цитотоксических
T-лимфоцитов.
Цитотоксический T-лимфоцит раcпознаёт клетку-мишень и прикрепляетcя к ней. В цитоплазме активированного цитотоксического T-лимфоцита присутствуют мелкие тёмные органеллы, напоминающие запаcающие гранулы
cекреторных клеток. Гранулы концентрируютcя в той чаcти T-киллера, которая
расположена ближе к меcту контакта c клеткой-мишенью. Параллельно
проиcходят переориентация цитоcкелета и cмещение в эту облаcть комплекса
Гольджи, в котором и формируютcя гранулы. В них содержится цитолитический белок перфорин. Выделяемые T-киллером молекулы перфорина полимеризуютcя в мембране клетки-мишени в приcутcтвии Ca2+. Сформированные в
плазматической мембране клетки-мишени перфориновые поры пропуcкают воду и cоли, но не молекулы белка. Еcли полимеризация перфорина произойдет
во внеклеточном проcтранcтве или в крови, где в избытке имеетcя кальций, то
полимер не cможет проникнуть в мембрану и уничтожить клетку. Cпецифическое дейcтвие T-киллера проявляется только как результат тесного контакта
85
между ним и клеткой-мишенью, который доcтигаетcя за cчёт взаимодейcтвия
АГ на поверхноcти жертвы c рецепторами T-киллера. Cам T-киллер защищён от
цитотокcичеcкого дейcтвия перфорина.
ИММУНОПАТОЛОГИЧЕСКИЕ СОСТОЯНИЯ И РЕАКЦИИ
Расстройства механизмов иммуно-биологического надзора проявляются разнообразными иммунопатологическими состояниями и реакциями,которые являются следствием дефекта или нарушения деятельности одного или нескольких
звеньев иммунной системы , обеспечивающих в норме эффективный иммунный
ответ.К ним относятся: иммунодефицитные состояния, патологическая толерантность, реакции «трансплантат против хозяина»
Иммунодефициты и иммунодефицитные состояния
В основе развития иммунодефицитных состояний и иммунодефицитов,
как правило, находятся отсутствие или дефицит клеток иммунной системы
и/или расстройства их функций. Это обусловливает высокую частоту развития
при иммунодефицитах различных инфекционных, паразитарных, опухолевых и
аллергических заболеваний. С другой стороны, при истощающих заболеваниях
часто развиваются иммунодефицитные состояния.
Иммунодефицитные состояния и иммунодефициты - типовые формы патологии системы ИБН, характеризующиеся снижением эффективности или неспособностью иммунной системы организма к осуществлению реакций деструкции и элиминации чужеродного антигена.
Частота иммунодефицитов и иммунодефицитных состояний
Один из 500 новорожденных рождается с дефектом иммунной системы.Значительно большее количество лиц приобретают преходящий или постоянный иммунодефицит в течение жизни.
Факторы риска:
- отягощённый семейный анамнез;
- практически все вредные привычки;
- старение.
Иммунодефициты - самостоятельные заболевания (нозологические формы) и сопутствующие синдромы, характеризующиеся недостаточностью иммунной системы.
Виды
• Первичные - наследуемые и врождённые (генетические) дефекты иммунной
системы.
• Вторичные - иммунная недостаточность развивается вследствие эндо- и экзогенных воздействий на нормальную иммунную систему (например, около 90%
всех вирусных инфекций сопровождается транзиторной иммунодепрессией).
Первичные иммунодефициты проявляются развитием инфекционных поражений организма вскоре после рождения, но могут не иметь клинических
проявлений и до более позднего возраста.
Причины:
86
Генные и хромосомные дефекты (они приводят к многочисленным иммунодефицитам разных классов)
В зависимости от уровня нарушений и локализации дефекта выделяют следующие виды первичных иммунодефицитов: 1) гуморальные, или Вклеточные иммунодефицита; 2) клеточные, или Т-клеточные иммунодефициты; 3) комбинированные иммунодефициты.
Первичные В-иммунодефициты возникают в результате нарушения процессов образования и дифференцировки В-лимфоцитов. К этой группе относятся:
а) агаммаглобулинемия Брутпона. Наследственный дефект передается сцепленно с Х-хромосомой, поэтому проявляется практически только у мальчиков. Нарушается дифференцировка клеток-предшественников В-лимфоцитов. Поэтому в организме отсутствуют В-лимфоциты, плазматические клетки
и иммуноглобулины. Т-система лимфоцитов не нарушена;
б) общий вариабельный иммунодефицит. Включает в себя очень много форм.
Общим их проявлением является гипогаммаглобулинемия, возникающая достаточно поздно — в возрасте 25-30 лет. Разнообразные генетические дефекты
нарушают дифференцировку В-лимфоцитов на разных уровнях созревания.
в) селективный дефицит иммуноглобулинов. Нарушается образование одного
или нескольких классов иммуноглобулинов. Образование же других классов
антител может быть не нарушено, или даже увеличено.
Первичные Т-иммунодефициты возникают в результате нарушения процессов образования и дифференцировки Т-лимфоцитов. К ним, в частности, относятся:
а) синдром Ди Джорджи — врожденная аплазия вилочковой железы. Является
следствием пороков эмбрионального развития и часто сочетается с "волчьей
пастью", аномалиями дуги аорты, аплазией паращитовидных желез.Нарушается
дифференцировка клеток-предшественников Т-лимфоцитов в Т0-лимфоциты.
Иммунный ответ клеточного типа не возможен. Гуморальный ответ на тимуснезависимые антигены сохраняется;
б) синдром Незелофа — алимфоцитоз. Генетический дефект передается по
аутосомно-рецессивному типу. Нарушается превращение Т0-лимфоцитов в Т,лимфоциты, вследствие чего не могут осуществляться клеточные меха-низмы
иммунного ответа.
Комбинированные иммунодефициты возникают в результате ряда нарушений превращения стволовой клетки в клетку- предшественницу лимфоцитопоэза или в результате сочетания дефектов B и Т-линий лимфоцитов.
К этой группе относятся:
а) тяжелый комбинированный иммунодефицит (швейцарский тип). Генетический дефект передается аутосомно-рецессивно или сцепленно
с
Ххромосомой. Нарушается образование В- и Т- лимфоцитов, синтез иммуноглобулинов. Больные дети редко достигают 2-летнего возраста;
б) синдром Луи-Бар. Характеризуется сочетанием иммунологической недостаточности с атаксией (нарушениями координации движений) и теле-
87
ангиэктазией (поражениями мелких сосудов). Продолжительность жизни больных редко достигает 20-30 лет;
в) синдром Вискотта-Олдрича. Иммунологическая недостаточность сопровождается развитием экземы (поражений кожи) и тромбоцитопении. Как
и в предыдущих случаях страдают гуморальные и клеточные механизмы иммунного ответа. Продолжительность жизни больных не превышает 10 лет.
Вторичные иммунодефициты, или иммунодефицитные состояния
Причины иммунодефицитных состояний многообразны:
- Лекарственные средства с иммуносупрессивным действием (включая фенитоин [дифенин], пеницилламины, глюкокортикоиды).
- Недостаточность питания, полостного и мембранного пищеварения, а
также кишечного всасывания.
- Наркотики и токсические вещества.
- Лучевые воздействия, химиопрепараты.
- Рост злокачественных опухолей.
- Вирусы (например, ВИЧ).
- Состояния, приводящие к потере белка (например, нефротический синдром).
- Гипоксия.
- Гипотиреоз.
- Уремия.
- Отсутствие селезенки (аспления).
ВИЧ-инфекция и СПИД
ВИЧ-инфекция - инфекция, вызываемая вирусами иммунодефицита человека (ВИЧ), поражающими лимфоциты, макрофаги, нервные и многие другие
клетки. Проявляется медленно прогрессирующим иммунодефицитом: от бессимптомного носительства до тяжёлых и смертельных заболеваний.
Синдром приобретённого иммунодефицита (СПИД) — вторичный иммунодефицитный синдром, развивающийся в результате ВИЧ-инфекции. СПИД
является одним из наиболее клинически значимых иммунодефицитов. Этот
синдром был описан в научной литературе в 1981 г. американскими исследователями. Однако ретроспективный анализ свидетельствует о том, что СПИД поражал людей и ранее. Первые случаи синдрома официально были зарегистрированы в США, Африке и на Гаити. В последние годы, когда налажены методы
диагностики СПИДа, выяснилось, что каждые 12–14 мес число зарегистрированных случаев синдрома удваивается. Правда, соотношение инфицированных
лиц (положительный тест на появление АТ к вирусу СПИД) к заболевшим колеблется от 50:1 до 100:1.
Этиология
Возбудители-вирусы иммунодефицита человека [ВИЧ] рода Retrovirus
подсемейства Lentivirinae семейства Retroviridae) ВИЧ погибают при температуре 56 °С в течение 30 мин, но устойчивы к низким температурам; быстро раз-
88
рушаются под действием этанола, эфира, ацетона и дезинфицирующих средств.
В крови и других биологических средах при обычных условиях сохраняют
жизнеспособность в течение нескольких суток.
Известно два типа вируса.
• ВИЧ-1 (HIV-1) - основной возбудитель ВИЧ-инфекции и СПИДа (ранее был
известен как HTLV-III или LAV) в Северной и Южной Америке, Европе, Азии,
Центральной, Южной и Восточной Африке.
• ВИЧ-2 (HIV-2) - менее вирулентный вирус; редко вызывает типичные проявления СПИДа; основной возбудитель СПИДа в Западной Африке.
Наибольшее распространение СПИД имеет среди четырех групп риска:
гомо- и бисексуальных мужчин; наркоманов, вводящих наркотики в/в и пользующихся коллективными шприцами; лиц, которым часто переливают кровь
(больные анемиями); детей родителей, больных СПИДом.
Источник инфекции -человек в любой стадии инфекционного процесса.
Вирус выделяют из крови, спермы, влагалищного секрета, материнского молока (эти жидкости определяют пути передачи вируса), слюны. Пути передачи половой, парентеральный, трансплацентарный, через материнское молоко.
Группы риска
Гомосексуальные и бисексуальные мужчины (43%), наркоманы, использующие
наркотики в/в (31%), гетеросексуалы (10%), реципиенты крови и её компонентов, трансплантируемых органов (2%), больные гемофилией (1%).
Патогенез
ВИЧ поражает в основном активированные СD4+-клетки (моноциты, макрофаги и родственные клетки, экспрессирующие СD4-подобные молекулы),
используя молекулу СD4 в качестве рецептора.
В течение различных периодов времени (до 10–15 лет) у ВИЧинфицированных симптомы болезни отсутствуют. В этот период защитные системы организма эффективно сдерживают репродукцию возбудителя.Гуморальные иммунные реакции -синтез АТ различных типов, не способных
оказывать протективный эффект и не предохраняющих от дальнейшего развития инфекции.Клеточные иммунные реакции способны либо блокировать репродукцию возбудителя, либо предотвращать проявления инфекции. Вероятно,
цитотоксические реакции доминируют у ВИЧ-инфицированных с длительным
отсутствием клинических проявлений.
Центральным звеном патогенеза СПИД`а является иммуносупрессия. Она
обусловлена, в основном, уменьшением количества циркулирующих СD4+клеток.Уменьшение количества циркулирующих СD4+ T-клеток создаёт условия для репликации интегрированного ВИЧ. Появление вирусных гликопротеинов в мембране заражённых Т-клеток - пусковой механизм для запуска иммунных процессов, направленных против подобных клеток.
Механизмы реализации -активация цитотоксических Т-клеток и реакция
АТ-зависимой цитотоксичности.Аккумуляция вирусной ДНК в цитоплазме инфицированных клеток обусловливает бурную репликацию ВИЧ и гибель клеток.ВИЧ инфицирует клетки-предшественники в тимусе и костном мозге, что
приводит к отсутствию регенерации и уменьшению пула СD4+-лимфоцитов.
89
Активность цитотоксических Т-клеток и естественных киллеров также уменьшается. Это связано с дефицитом Т-хелперов. Ответ В-клеток тоже ослабевает
по мере численного сокращения ТН2-субпопуляции.
Дефект регуляторных механизмов приводит к продукции В-клетками АТ
с низкой специфичностью к АГ ВИЧ,а также синтезу иммуноглоблинов перекрёстно реагирующих с ядерными, тромбоцитарными и лимфоцитарными аутоантигенами (что обусловливает развитие цитопенических реакций тромбоцитопений и лейкопений).
О переходе бессимптомной ВИЧ-инфекции в заболевание с неспецифическими симптомами свидетельствуют лихорадка, повышенное ночное потоотделение, слабость, хроническая диарея, рассеянная лимфаденопатия и головная
боль при отсутствии какой-либо специфической или оппортунистической инфекции. При этом могут выявляться сопутствующие инфекции: саркома Капоши, кандидоз ротовой полости, лейкоплакия слизистой оболочки полости рта
(часто бессимптомная), инфекции верхних и нижних дыхательных путей и заболевания периодонта.
Патологическая толерантность
Иммунологическая толерантность — состояние, характеризующееся
«терпимостью» иммунной системы по отношению к чужеродным для неё АГ.
Иммунологическую толерантность подразделяют на физиологическую,
патологическую и индуцированную.
Физиологическая толерантность подразумевает «терпимость» системы
ИБН к собственным АГ.
Механизмы развития физиологической толерантности:
1.Элиминация в антенатальном периоде (когда иммунная система ещё недостаточно созрела) тех клонов лимфоцитов, которые подверглись антигенной
перегрузке - массированному воздействию собственных АГ. В лабораторных
условиях этот феномен воспроизводится путём подсадки эмбриону и плоду животного ткани или органа другого животного того же вида (аллотрансплантата).
Повторная трансплантация взрослому животному такого же трансплантата не
приводит к его отторжению — развивается толерантность к нему. Таких животных (в организме которых имеется генетически и антигенно чужеродная
ткань или орган), называют химерами. Подобный химеризм развивается и у
двуяйцевых близнецов, которые во время пренатального периода обмениваются
разногруппной кровью. Во взрослом состоянии им можно беспрепятственно
переливать кровь обеих групп.
2.Изоляция АГ ряда органов от контакта с иммуноцитами структурно-физиологическими барьерами. К таким органам относятся мозг, глаза, семенники, щитовидная железа, которые отделены от внутренней среды организма
гемато-тканевыми
барьерами
(гемато-энцефалическим,
гемато-офтальмическим, гемато-тиреоидным). Эту разновидность толерантности
называют изоляционной.
3.Подавление пролиферации и дифференцировки аутоагрессивных (действующих против собственных клеток) T-лимфоцитов в центральном органе иммун-
90
ной системы - тимусе. Этот феномен называют центральной селекцией и ликвидацией аутоцитотоксических лимфоцитов.
4.Гибель (апоптоз) клонов лимфоцитов, активирующихся аутоантигенами.
Патологическая толерантность
В этом случае речь идет о «терпимости» системой ИБН чужеродных АГ, чаще
всего - бактерий, вирусов, паразитов, клеток злокачественных опухолей или
трансплантата.
Механизмы развития патологической толерантности:
- Иммунодефицитные состояния и иммунодефициты.
- Чрезмерное повышение активности T-супрессоров. Последнее характеризуется торможением созревания эффекторных клеток иммунной системы:
T-киллеров, естественных киллеров, плазматических клеток.
- Ингибирование или блокада цитотоксических реакций клеточного иммунитета на соответствующий АГ (чаще всего клеток опухоли, трансплантата или
вируссодержащих клеток) в результате экранирования антигенов антителами.
- Перегрузка иммуноцитов избытком образующихся в организме или вводимых в него извне чужеродных АГ. Это может наблюдаться при синтезе аномальных белков в печени, амилоидозе, денатурации белковых молекул при
массированных ожогах, введении большого количества белоксодержащих растворов (цельной крови, плазмы).
- Гибель цитотоксических T-лимфоцитов с развитием T-клеточного иммунодефицита. Это наблюдается при экспрессии другими клетками (например, опухолевыми) Fas–лигандов. Последние, взаимодействуя с Fas-рецепторами цитотоксических T-лимфоцитов, активируют программу их апоптоза
Индуцированная толерантность
Индуцированную (искусственную, медицинскую) толерантность воспроизводят путем целенаправленного подавления активности иммунной системы.
Обычно с этой целью применяют ионизирующее излучение, высокие дозы цитостатиков и иммунодепресантов. Для создания состояния искусственной толерантности применяют также специальные (непроницаемые для иммуноцитов)
камеры, имплантируемые под кожу, слизистую оболочку, в мышцы или полости тела. В камеру помещают гомогенат или фрагменты чужеродной ткани
(например, эндокринной железы для устранения недостатка эндогенного гормона).
Состояние индуцированной толерантности применяют для повышения успеха трансплантации органов и тканей, лечения аллергии, болезней
иммунной аутоагрессии, эндокринной недостаточности и некоторых других состояний.
Трансплантационный иммунитет
Новейшие достижения в области трансплантологии позволяют все шире
использовать трансплантацию органов и тканей для лечения разных заболеваний. В последнее время наряду с трансплантацией костного мозга, почки, печени и сердца стали применять трансплантацию тонкой кишки, доли и сегментов
печени, легкого, костей, поджелудочной железы и клеток панкреатических островков, а также других органов и тканей. Для трансплантации используются
как трупные, так и полученные от живых доноров органы и ткани. Чаще доно-
91
рами служат родственники реципиента. После трансплантации в организме реципиента развивается иммунный ответ на многочисленные антигены трансплантата. Исключение составляют случаи, когда донор и реципиент - однояйцовые близнецы. Наиболее изученные антигены человека, с которым связан
иммунный ответ на трансплантат, -это антигены HLA (иногда их называют
трансплантационными антигенами).
Главный комплекс гистосовместимости человека был открыт в 1952 г.
при изучении антигенов лейкоцитов. Антигены HLA представляют собой гликопротеиды, находящиеся на поверхности клеток и кодируемые группой тесно
сцепленных генов 6-й хромосомы. Гены HLA обладают высоким полиморфизмом. Серологическими методами определено более 100 антигенов HLA С помощью молекулярно-генетических методов ежегодно открываются новые аллели генов HLA. Антигены HLA играют важнейшую роль в регуляции иммунного
ответа на чужеродные антигены и сами являются сильными антигенами.
Механизмы трансплантационного иммунитета
Иммунный ответ на трансплантат обусловлен в первую очередь распознаванием антигенов HLA донора лимфоцитами реципиента. Это вызывает активацию T-хелперов, которые, в свою очередь, стимулируют пролиферацию Bлимфоцитов и цитотоксических T-лимфоцитов.
Антитела к чужеродным антигенам HLA могут присутствовать в сыворотке реципиента и до трансплантации. Их выявление свидетельствует о предшествующей иммунизации антигенами HLA. Она возможна при переливании
цельной крови и во время беременности. Выявление в сыворотке реципиента
антител к антигенам HLA донора свидетельствует о высоком риске сверхострого отторжения трансплантата. Оно обусловлено образованием комплексов, состоящих из антигенов трансплантата и антител реципиента, которые активируют свертывание крови и приводят к тромбозу сосудов трансплантата.
Поскольку отторжение трансплантата вызывают чужеродные антигены
HLA, лучший способ его профилактики подбор донора, совместимого с реципиентом по антигенам HLA. Если реципиент уже иммунизирован антигенами
HLA, донор должен быть полностью совместим с реципиентом.
Подобрать донора, полностью совместимого с реципиентом по антигенам
HLA, очень сложно, поскольку число комбинаций, составленных более чем из
100 антигенов этого семейства, чрезвычайно велико. Вероятность найти полностью совместимого донора составляет от 1:1000 до 1:1 000 000 в зависимости от
распространенности того или иного антигена HLA. Вероятность подбора полностью совместимого донора среди родных братьев и сестер составляет 1:4, так
как гены HLA наследуются по законам Менделя.
Реакция «трансплантат против хозяина»
Реакция «трансплантат против хозяина» развивается при трансплантации реципиенту («хозяину») тканей донора, содержащих иммуноциты (например, костного мозга, селезёнки, лейкоцитарной массы).
Условия развития реакций «трансплантат против хозяина»:
- генетическая чужеродность донора и реципиента;
- наличие в трансплантате большого числа лимфоцитов;
92
- неспособность реципиента уничтожить или отторгнуть этот трансплантат.
Реакция «трансплантат против хозяина» характеризуется поражением
тканей и органов иммунной системы реципиента и развитием в связи с этим
иммунодефицита. Помимо тканей, содержащих иммуноциты, всегда повреждаются и другие ткани и органы: кожа, мышцы, ЖКТ, печень, почки. Эти повреждения проявляются некротическими и дистрофическими изменениями,
развитием недостаточности функций, лимфопенией, анемией, тромбоцитопенией, диспептическими расстройствами (тошнотой, рвотой, диареей).
У взрослых реакция «трансплантат против хозяина» проявляется трансплантационной болезнью (син.:гомологичной болезнью; термин происходит от
слова «гомотрансплантат»).У детей развивается рант-болезнь - болезнь малого
роста (от англ. runt, наименьшая особь). Последнее связано с нарушением физического развития ребёнка, сочетающегося с полиорганной недостаточностью,
склонностью к развитию инфБ и новообразований.
АЛЛЕРГИЯ
Аллергия (от греч. allos - иной, ergon - действую) — иммунная реакция
организма на какие-либо вещества антигенной природы,развивающаяся после
повторного контакта с ними и сопровождающаяся повреждением структуры и
функции клеток, тканей и органов.
Понятие “аллергия” было предложено в 1906 г. австрийским патологом и
педиатром Клемансом Пирке для определения состояния измененной реактивности, которое он наблюдал у детей при сывороточной болезни и инфекционных заболеваниях. Говоря об аллергическом состоянии организма, часто употребляют термины гиперчувствительность, или повышенная чувствительность, подразумевая способность организма болезненно реагировать на безвредные для большинства индивидов вещества (пыльца трав и деревьев, цитрусовые и др.).
Общими особенностями, объединяющими все аллергические болезни, являются:
1) этиологическая роль различных аллергенов;
2) иммунологический механизм развития;
3) повреждающее действие комплекса АГ-АТ или АГ-сенсибилизированных
лимфоцитов на клетки и ткани организма. Важно подчеркнуть, что сама сенсибилизация (иммунизация) заболевания не вызывает -лишь повторный контакт с
тем же антигеном может привести к нежелательному эффекту. В конечном счете развивается не защита от антигена (долгое время иммунный ответ считали
только защитным механизмом), а, напротив, повреждение; вместо защитной реакции возникает какая-то другая, извращенная реакция -аллергия.
93
Этиология аллергических реакций
Причиной аллергических заболеваний является аллерген, а их возникновения - определенные особенности внешней среды и состояние реактивности
организма.
Аллергенами называют вещества, вызывающие развитие аллергической
реакции. Они могут быть антигенами, с многочисленными антигенными детерминантами, и биологически активными веществами, представляющими смесь
антигенов (пыльца трав, частицы эпидермиса). Аллергены обладают чужеродностью и часто - макромолекулярностью, хотя гаптены тоже могут обладать аллергенными свойствами, становясь антигенами только после соединения с белками тканей организма (метаболиты лекарств, простые химические вещества йод, бром, хром, никель). При этом образуются так называемые комплексные
антигены, специфичность которых определяется специфичностью гаптена. По
химической структуре аллергены являются белками, белково-полисахаридными
комплексами (сывороточные, тканевые, бактериальные аллергены) или могут
быть полисахаридами или соединениями полисахаридов с липоидами (аллерген
домашней пыли, бактериальные аллергены).
Классификация аллергенов. Различают аллергены:
- бытовые (неорганические и органические вещества микробного, растительного и животного происхождения: домашняя пыль, шерсть и перхоть домашних
животных, пух домашних птиц, постельные клещи, моющие средства);
- грибковые (микроаллергены: кандиды, трихофиты, эпидермофиты, актиномицеты);
-животного происхождения (эпидермальные, яды перепончатокрылых, клещи,
материалы из шерсти животных, корм для рыб);
- лекарственные (вакцины, сыворотки, инсулин, препараты мышьяка, йода,
ртути, антибиотики, сульфаниламиды, витамины);
- микробные (возбудители туберкулеза, токсоплазмоза, бруцеллеза, вирусы
кори, гриппа, герпеса, инфекционного гепатита);
- пищевые (коровье молоко, белки куриных яиц, мясо, рыба, ракообразные,
цитрусовые, кофе, орехи, мед);
- растительные (пыльца, сок растений).
Возможность возникновения аллергического заболевания у данного индивидуума определяется характером, свойствами и количеством антигена, путем его поступления в организм, а также особенностями реактивности организма. Например, аскаридозный антиген стимулирует образование антител IgEкласса, т. е. аллергических антител. Антигены, являющиеся слабыми иммуногенами и находящиеся в окружающей среде в небольших количествах (пыльца
растений, домашняя пыль), попадая в организм, также могут вызвать развитие
аллергического заболевания.
С другой стороны, у многих людей, получавших пенициллин, обнаруживаются антитела различных классов иммуноглобулинов к пенициллину, однако
аллергические реакции на пенициллин развиваются только у некоторых из них.
Указанные факторы связывают с особенностями реактивности организма.
94
Аллергическая реактивность (аллергическая конституция) в значительной мере определяется наследственными особенностями организма. Свыше
80 % детей с аллергией в возрасте до 10 лет имеют отягощенный семейный
анамнез. В родословных больных бронхиальной астмой (наиболее тяжелое аллергическое заболевание) наследственное предрасположение к аллергии составляет более 50 %. Однако аллергические заболевания не относятся к группе
наследственных болезней, передаваемых непосредственно от родителей к
потомству; передается только предрасположенность к аллергии. Исследования
аллергических проявлений у однояйцевых близнецов показали, что реализация
аллергической предрасположенности в заболевание строго зависит от факторов
внешней среды и их природы. Особенно чувствителен к развитию аллергического процесса организм в детском возрасте.
К факторам внешней среды (или социальным факторам), способствующим аллергизации населения, относятся:
1) широкая обязательная вакцинация населения против многих инфекционных
заболеваний (оспа, дифтерия, коклюш и т. д.). Известно, что коклюшная вакцина повышает чувствительность тканей к гистамину, вызывает блокаду бетаадренергических рецепторов в бронхиальной ткани, играет роль адъюванта для
синтеза аллергических антител;
2) широкое применение сывороток в лечебных целях, которые сами могут являться аллергенами;
3) значительный рост простых и сложных химических веществ, потенциальных
аллергенов, окружающих человека. Это — лекарства, препараты бытовой химии, пестициды и гербициды в сельском хозяйстве, воздух и вода, загрязненные промышленными отходами. Считается, что в среднем аллергические заболевания охватывают около 10 % населения земного шара.
Не всегда понятно, через какие конкретные механизмы реализуются в болезнь наследственная предрасположенность к аллергии и социальные факторы.
Существенными являются следующие: 1) повышенная проницаемость кожных
или слизистых барьеров, ведущая к поступлению в организм антигенов, которые в обычных условиях либо не поступают, либо поступление их ограничено.
В формировании этого механизма значительна роль воспалительных процессов
верхних дыхательных путей, воспалительных заболеваний кишечника, дисбактериоза кишечника; 2) особый характер иммунного ответа на антиген, ведущий
к выработке преимущественно аллергических антител.
Классификация аллергических реакций
Среди многочисленных классификаций аллергических реакций, возникновение которых было связано с многообразием проявлений аллергии у человека и животных, наибольшее распространение получила классификация, предложенная Р. Куком в 1930 г., согласно которой все аллергические реакции разделяются на две большие группы:реакции немедленного и реакции замедленного
типа. В основу классификации положено время появления реакции после повторного контакта с аллергеном. Реакции немедленного типа развиваются через
15— 20 мин, замедленного — через 24—48 ч. К реакциям немедленного типа
95
относятся анафилактический шок, атопическая форма бронхиальной астмы,
поллинозы, отек Квинке, аллергическая крапивница, сывороточная болезнь,
феномен Овери и др. К реакциям замедленного типа относятся аллергический
контактный дерматит, реакция отторжения трансплантата, поствакцинальный
энцефаломиелит, тиреоидит Хашимото. Гиперчувствительность замедленного
типа сопровождает туберкулез, бруцеллез, сифилис, грибковые заболевания,
протозойные инфекции и др. Понятие об аллергических реакциях немедленного
и замедленного типа, возникшее в клинике, не отражает всего разнообразия
проявлений и механизмов развития аллергии.
В настоящее время широкое распространение получила классификация,
предложенная П. Джеллом и Р. Кумбсом в 1969 г. и основанная на патогенетическом принципе. Согласно этой классификации в зависимости от механизма
иммунной реакции выделяют четыре основных типа аллергических реакций
I тип, к которому относятся аллергические реакции немедленного типа, включает 2 подвида:
1) реагиновый, связанный с выработкой антител IgE-класса и лежащий в основе атопических заболеваний;
2) анафилактический, обусловленный в основном IgG4-антителами и наблюдающийся при анафилактическом шоке.
II тип - цитотоксический, связан с образованием IgG (кроме IgG4)- и IgMантител к антигенам, имеющимся на собственных клетках. По этому типу протекают некоторые гематологические заболевания, например аутоиммунная гемолитическая анемия, миастения и др.
III тип -иммунокомплексный, связан с образованием комплексов аллергенов
и аутоаллергенов с IgG- или IgM-антителами и с повреждающим действием
этих комплексов на ткани организма. По этому типу развиваются, например,
сывороточная болезнь, анафилактический шок и др.
IV тип - клеточно-опосредованный (другое название — гиперчувствительность замедленного типа, ГЗТ), связан с образованием сенсибилизированных лимфоцитов (Т-эффекторов ГЗТ). По этому типу развиваются: аллергический контактный дерматит, реакция отторжения трансплантата и др.
Этот механизм участвует как компонент и в инфекционно-аллергических заболеваниях, таких, как туберкулез, проказа, бруцеллез, сифилис и др.
При многих аллергических заболеваниях возможно одновременно обнаружить патогенетические механизмы различных типов аллергии. Например,
при атопической бронхиальной астме и анафилактическом шоке участвуют механизмы I и III типов, при аутоиммунных заболеваниях - реакции II и IV типов
и т. д. Однако для патогенетически обоснованной терапии всегда важно установить ведущий механизм.
Общий патогенез аллергических реакций
Независимо от того, к какому типу относится аллергическая реакция, в ее
развитии можно выделить три стадии.
I. Иммунологическая стадия. Иммунная стадия начинается при первой
встрече организма с аллергеном и заканчивается взаимодействием антитела с ан-
96
тигеном. В этот период происходит сенсибилизация организма, т.е. повышение
чувствительности и приобретение способности реагировать на повторное введение
антигена аллергической реакцией. Первое введение аллергена называется сенсибилизирующим, повторное же, которое непосредственно вызывает проявление аллергии, разрешающим.
Сенсибилизация бывает активной и пассивной. Активная сенсибилизация
развивается при иммунизации антигеном, когда в ответ включается собственная
иммунная система. Механизмы активной сенсибилизации следущие:
-распознавание антигена, кооперация макрофагов с Т- и В-лимфоцитами, выработка плазматическими клетками гуморальных антител (иммуноглобулинов) или
образование сенсибилизированныхлимфоцитов (Т-эффекторов ГЗТ) и размножение лимфоцитов всех популяций;
- распределение антител (IgE, IgG4) в организме и фиксация ихна клетках мишенях, которые сами антител не вырабатывают, в частности, на тканевых базофилах (тучных клетках), базофильных гранулоцитах, моноцитах, эозинофилах,
а также на тромбоцитах, или взаимодействие иммуноглобулинов (IgG, IgM, lgA)
либо Т-эффекторов с антигенами, если к моменту развития сенсибилизации они
еще присутствуют в организме.
На 7-14-й день после введения аллергена в сенсибилизирующей дозе организм приобретает к нему повышенную чувствительность.
Пассивная сенсибилизация осуществляется в неиммунизированном организме
при введении ему сыворотки крови, содержащей антитела, или клеточной взвеси с
сенсибилизированными лимфоцитами, полученными от активно сенсибилизированного данным антигеном донора. При этом состояние повышенной чувствительности развивается через I2—24 ч. Это время необходимо для распределения антител в организме и фиксации их на клетках.
II. Патохимическая стадия. Суть ее состоит в выделении готовых и образовании новых биологически активных веществ (медиаторов аллергии) в результате сложных биохимических процессов, запускаемых комплексами АГ-АТ
или АГ-сенсибилизированным лимфоцитом.
III. Патофизиологическая стадия. Представляет ответную реакцию
клеток, органов и тканей организма на образовавшиеся в предыдущей стадии
медиаторы.
Аллергические реакции I типа (реагиновый тип аллергии)
В основе аллергических реакций I типа лежит выработка в организме IgEантител, т. е. IgE-ответ — главное звено развития аллергической реакции 1
типа.
IgE-антитела значительно отличаются по своим свойствам от других антител . Прежде всего они обладают цитотропностью (цитофильностью). Им
присуще свойство прикрепляться к клеткам и фиксироваться в тканях. Концентрация IgE-антител в сыворотке крови потому и низка, что синтезируемые в региональных лимфоузлах молекулы IgE в меньшей степени попадают в кровоток, так как в основном фиксируются в окружающих тканях.
97
Фиксация антител клетками происходит при помощи рецептора, встроенного в мембрану клеток. Самой высокой способностью связывать IgE-антитела
обладают рецепторы для IgE, найденные на тучных клетках и базофилах крови,
поэтому эти клетки получили название клетки-мишени I порядка. На одном базофиле может фиксироваться от 3000 до 300000 молекул IgE. Рецептор для IgE
обнаружен также на макрофагах, моноцитах, эозинофилах, тромбоцитах и
лимфоцитах, однако их связывающая способность ниже. Эти клетки получили
название клетки-мишени II порядка.
Связывание IgE на клетках-зависимый от времени процесс. Оптимальная
сенсибилизация может наступить через 24—48 ч. Фиксированные антитела могут долго находиться на клетках, поэтому аллергическая реакция может быть
вызвана спустя неделю и больше. Особенностью IgE-антител является также
трудность их обнаружения, так как они не участвуют в серологических реакциях.
При развитии реакций гиперчувствительности типа I (атопические реакции немедленного типа, реагиновые, анафилактические) происходит взаимодействие Аг с АТ (IgE и IgG), приводящее к высвобождению БАВ -медиаторов
аллергии (главным образом, гистамина) из тучных клеток и базофилов.
Причиной аллергических реакций типа I чаще всего являются экзогенные
агенты (компоненты пыльцы растений, трав, цветов, деревьев, животные и растительные белки, некоторые ЛС, органические и неорганические химические
вещества).
I. Иммунологическая стадия. Как уже было сказано выше, IgE-ответ является главным звеном развития аллергической реакции I типа.
Первичное попадание аллергена в организм запускает через кооперацию
макрофагов, Т- и В-лимфоцитов сложные и до конца не ясные механизмы синтеза IgE-антител, фиксирующихся на клетках-мишенях. Повторная встреча организма с этим аллергеном приводит к образованию комплекса АГ-АТ, причем
через фиксированные молекулы IgE и сам комплекс тоже окажется фиксированным на клетках. Если аллерген оказался связанным хотя бы с двумя соседними молекулами IgE , то этого оказывается достаточным для нарушения
структуры мембран клеток-мишеней и их активации. Начинается II стадия аллергической реакции.
II. Патохимическая стадия. При повторном попадании аллергена в организм происходит его взаимодействие с фиксированными на поверхности клеток–мишеней первого порядка (тучных клеток и базофильных лейкоцитов) молекулами IgE, что сопровождается немедленным выбросом содержимого гранул этих клеток в межклеточное пространство (дегрануляция). Дегрануляция
тучных клеток и базофилов, как минимум, имеет два важных последствия:
-во-первых, во внутреннюю среду организма попадает большое количество
разнообразных БАВ - медиаторов аллергии, оказывающих самые различные
эффекты на разные эффекторные клетки (в особенности на сократительные и
секреторные);
-во-вторых, многие БАВ, высвободившиеся при дегрануляции клеток–мишеней
первого порядка, активируют клетки–мишени второго порядка (нейтрофилы,
98
эозинофилы, лимфоциты, тромбоциты, моноциты и макрофаги), которые в
свою очередь секретируют различные медиаторы аллергии.
III. Патофизиологическая стадия. Секреция клетками медиаторов аллергии и реализация их эффектов обусловливает развитие стереотипных реакций:
• повышение проницаемости стенок микрососудов и развитие отёка тканей;
• нарушения кровообращения;
• сужение просвета бронхиол, спазм кишечника;
• гиперсекрецию слизи;
• прямое повреждение клеток и неклеточных структур:
Определённая комбинация указанных выше и других эффектов и создаёт
своеобразие клинической картины отдельных форм аллергии. Чаще всего по
описанному механизму развиваются поллинозы, аллергические формы бронхиальной астмы, аллергические конъюнктивит, дерматит, гастроэнтероколит, а
также анафилактический шок.
Псевдоаллергические реакции
Сходные с описанными выше патобиохимические изменения при аллергических реакциях типа I наблюдаются и при так называемых псевдоаллергических реакциях.Последние развиваются после энтерального или парентерального попадания в организм различных агентов:продуктов питания (шоколада,
яичного белка, рыбы, молока, цитрусовых, некоторых ягод и др.),ЛС, гербицидов, пестицидов и др. Одна из таких форм патологически повышенной чувствительности (часто наследуемая как предрасположенность) к отдельным продуктам питания и ЛС получила специальное название «идиосинкразия».
Важной особенностью псевдоаллергических реакций является их развитие без видимого периода сенсибилизации. Существенно так же, что они чаще
выявляются у пациентов либо с тотальной печёночной недостаточностью (перенесших вирусные гепатиты, малярию; подвергавшихся хроническим воздействиям гепатотропных ядов), либо с избирательно нарушенной функцией печени по инактивации биогенных аминов (в частности, гистамина) и других вазоактивных веществ.Быстрое и значительное нарастание содержания этих веществ в крови после попадания в организм и приводит к проявлениям псевдоаллергических реакций: крапивницы, высыпаний различного вида, локального зуда, покраснения кожи, отёка Квинке, диареи, приступов удушья и даже состояний, напоминающих анафилактический шок.
Аллергические реакции II типа (цитотоксический тип аллергии)
При немедленных аллергических реакциях типа II (цитотоксических, цитотолитических) иммуноглобулины (обычно IgG или IgM) связываются с Аг на
поверхности клеток.Это приводит к их фагоцитозу, разрушению клетками киллерами, лизису клеток, опосредованному системой комплемента или иммуноглобулинами. Прототипом аллергии типа II является цитотоксические (цитолитические) реакции иммунной системы, направленные на уничтожение отдельных чужеродных клеток - микробных, грибковых, опухолевых, вирусин-
99
фицированных, трансплантированных. Однако, в отличие от них, при аллергических реакциях типа II повреждаются собственные клетки организма и, в связи с образованием избытка цитотропных медиаторов аллергии, это повреждение клеток нередко приобретает генерализованный характер.
Причиной аллергических реакций типа II наиболее часто являются химические вещества со сравнительно небольшой молекулярной массой (в том числе
лекарственные средства, содержащие золото, цинк, никель, медь, а также сульфаниламиды, антибиотики и гипотензивные средства) и гидролитические ферменты, в избытке накапливающиеся в межклеточной жидкости (например,
ферменты лизосом клеток или микроорганизмов при их массированном разрушении), а также активные формы кислорода, свободные радикалы, перекиси
органических и неорганических веществ.
Указанные агенты обусловливают единый общий результат - они изменяют антигенный профиль отдельных клеток и внеклеточных структур. В результате образуются две категории аллергенов:
- изменённые белковые компоненты клеточной мембраны (клеток крови, почек, печени, сердца, мозга, селезёнки. эндокринных желёз и др.);
- изменённые внеклеточные антигенные структуры (например, печени, миелина, базальной мембраны клубочков почек, коллагена и др.);
- так называемые "забарьерные" АГ, не соприкасающиеся с иммунной системой организма, такие как АГ хрусталика и прозрачных сред глаза (при "симпатической офтальмии") или АГ мозга (при демиелинизирующих болезнях).
Вовлечение в аллергические реакции внеклеточных структур сопровождается повреждением и нередко лизисом близлежащих клеток. Развитие аллергической реакции приводит к повреждению большого числа клеток, в том числе антегенно неизмененных. Кроме того, картина усугубляется развитием в регионе аллергической реакции воспаления и появленим повреждённых при нем
клеток.
Иммунологическая стадия
Коммитированные антигеном (при участии антигенпредставляющих клеток системы мононуклеарных фагоцитов) B-лимфоциты трансформируются в
плазматические клетки, синтезирующие IgG подклассов 1, 2 и 3, а также IgМ.
Указанные классы иммуноглобулинов могут связываться с компонентами комплемента.Иммуноглобулины специфически взаимодействуют с изменёнными
антигенными детерминантами на поверхности клеток и неклеточных структур
организма. При этом реализуются комплемент - и антителозависимые иммунные механизмы цитотоксичности и цитолиза:
- комплементзависимого разрушения мембраны антигенно чужеродной клетки;
- антителозависимого клеточного повреждения и лизиса носителя чужеродного Аг.
Патохимическая стадия
Комплементзависимые реакции. Цитотоксичность и цитолиз реализуются путём нарушения целостности цитолеммы клетки-мишени и её опсонизации.
100
Нарушение целостности мембраны клетки-мишени достигается благодаря активации под действием комплекса «АТ+Аг» системы комплемента.
Последовательная активация компонентов комплемента С5678 обусловливает относительно медленное повреждение мембраны клетки, С56789 — более быстрое. Ещё более эффективен комплекс C3b56789. Указанные комплексы
получили название мембраноатакующих. В результате в цитолемме образуются
поры 5–20 нм в диаметре. Через них в клетку пассивно поступают Na+, Ca2+ и
другие ионы. В связи с этим быстро и значительно повышается внутриклеточное осмотическое давление. Клетка гипергидратируется, цитолемма её перерастягивается и разрывается — наступает «осмотический взрыв» клетки-мишени.
Цитолиз осуществляется благодаря опсонизации клеток–мишеней при
помощи факторов комплемента, а также IgG и IgМ. Под влиянием комплекса
антиген-антитело активируются главным образом (хотя и не только) факторы
C4b2а3b. Наличие их стимулирует адгезию к клетке-мишени фагоцитов, высвобождение из них и последующую активацию ферментов их лизосом, генерацию
ими активных форм кислорода, свободных радикалов, других агентов, которые
лизируют антигенно чужеродную клетку.
Аналогичным образом могут повреждаться внеклеточные структуры и
базальные мембраны, на которых фиксирован чужеродный Аг. Активированные компоненты системы комплемента, находящиеся в жидких средах организма — крови, межклеточной жидкости и других, могут расширить масштаб
повреждения, воздействуя не только на антигенно чужеродные структуры, но и
на клетки и внеклеточные образования, не имеющие такого Аг. Кроме того, генерализация повреждения достигается за счёт альтерации структур организма
ферментами лизосом, активными формами кислорода, свободными радикалами,
высвобождающимися из фагоцитов и других клеток в зоне аллергической реакции.
Антителозависимый клеточный цитолиз осуществляется без непосредственного участия факторов комплемента.Прямой цитотоксический и цитолитический эффект оказывают клетки, обладающие киллерным действием: макрофаги, моноциты, гранулоциты (главным образом нейтрофилы), естественные
киллеры, T-киллеры. Все эти клетки не сенсибилизированы Аг. Киллерное действие они осуществляют путём контакта с IgG в области Fс-фрагмента АТ. При
этом Fав–фрагмент IgG взаимодействует с антигенной детерминантой на клетке-мишени.
Цитолитический эффект клетки-киллеры реализуют путём cекреции гидролитических ферментов, генерации активных форм кислорода и свободных
радикалов. Эти агенты достигают поверхности клетки-мишени, повреждают и
лизируют её.
Наряду с антигенно изменёнными клетками в ходе реакций могут повреждаться и нормальные клетки. Это связано с тем, что цитолитические агенты
(ферменты, свободные радикалы и др.) секретируются киллерами в межклеточную жидкость , где находятся и другие - антигенно неизменённые клетки.
101
Последнее является одним из признаков, отличающих данный тип аллергической реакции от иммунного - прицельного цитолиза.
Патофизиологическую стадия
Характер патологических явлений, наблюдающихся при рассматриваемом типе аллергий, во многом определяется особенностями мишеней, поражаемых цитолитическим процессом.
Если аллерген локализован на мембранах взвешенных в крови, свободно
плавающих клеток, то под влиянием мембраноатакующего компонента комплемента происходит повреждение этих клеток, приводящих к их гибели.
Таков патогенез:
-анемий (обусловленных резус-конфликтом или лекарственных);
-тромбоцитопений (чаще всего лекарственных);
-агранулоцитарных лейкопений (тоже чаще всего лекарственных).
Если поражаются мембраны, входящие в состав органов и тканей, то развиваются хронические воспалительные процессы, обостряющиеся или дающие
ремиссии в зависимости от состояния иммунной системы больного. Таковы:
-гломерулонефриты (по цитотоксическому типу развиваются приблизительно
20% этих заболеваний). Как правило, цитотоксические гломерулонефриты протекают более жестко и быстротечно, чем иммуно-комплексные;
-пульмониты (болезнь Гудпасчера);
-тиреоидит Хашимото;
-некоторые энцефалиты и невриты;
-симпатическая офтальмия.
При вирусных заболеваниях могут накапливаться AT против пораженных
вирусом клеток, т.к. вирусные АГ обязательно появляются на клеточных мембранах. Например, при вирусном гепатите присоединение аутоиммунного механизма может резко обострить течение процесса, вплоть до развития острой
печеночной недостаточности, т.к. в отличие от вирусов, которые постепенно
перемещаются с одного гепатоцита на другой, АГ атакуют практически одновременно все клетки, имеющие соответствующую АГ структуру.
Точно также аутоаллергические проявления часто осложняют вирусные
заболевания ЦНС.
Шоковые реакции, напоминающие анафилаксию, могут иметь место при
массивном цитолизе форменных элементов крови, сопровождающимся освобождением больших количеств гидрологических ферментов и других БАВ.
Аллергические реакции III типа (реакции иммунных комплексов)
Повреждение при этом типе аллергической реакции вызывается иммунными комплексами АГ-АТ. Вследствие постоянного контакта человека с какими-либо антигенами происходят иммунные реакции с образованием комплексов АГ-АТ, так как в его организме постоянно происходят реакции с образованием комплекса АГ-АТ. Эти реакции являются выражением защитной функции
иммунитета и не сопровождаются повреждением. Однако при определенных
условиях комплекс АГ-АТ может вызывать повреждение и развитие заболевания. Концепция о том, что иммунные комплексы (ИК) могут играть роль в па-
102
тологии, была высказана еще в 1905 г. Пирке и Шиком. С тех пор группа заболеваний, в развитии которых основная роль принадлежит ИК, получила название болезни иммунных комплексов.
Причиной иммунокомплексных заболеваний являются экзо- и эндоантигены и аллергены. Среди них: лекарственные препараты (пенициллин, сульфаниламиды и др.),антитоксические сыворотки, гомологичные гамма-глобулины,
пищевые продукты (молоко, яичные белки и др.), ингаляционные аллергены
(домашняя пыль, грибы и др.),бактериальные и вирусные антигены, ДНК, антигены клеточных мембран и др. Важно, что антиген имеет растворимую форму.
Иммунологическая стадия
В основе патогенеза этих реакций, как и при первом типе, лежит накопление AT к аллергену.
Но, в отличие от анафилаксии и атопии, они представлены Ig М и Ig G,
циркулирующими в крови больного.
Если в кровь попадает аллерген, то образуются ИК, которые свободно
циркулируют в сосудистой системе. Подобные явления постоянно наблюдаются и в здоровом организме.
Патологические процессы возникают, если количество ИК в крови велико
и циркуляция их продолжается достаточно длительное время.
Длительная циркуляпия больших количеств ИК приводит к их накоплению в капиллярах некоторых сосудистых областей,например, в почечных клубочках (около 80% всех гломерулонефритов имеют иммунокомплексный патогенез), суставов (при ревматоидном артрите),коже и других органах при СКВ и
т.д.
Патохимическая стадия
Поскольку скапливающиеся в капиллярах ИК при III типе имеют в своем
составе Ig G и Ig М, в месте их скопления, т.е. на внутренней поверхности капилляров и между эндотелием и базальной мембраной, начинается активация
системы комплемента и фагоцитов.
Происходит накопление активированных компонентов комплемента и
мощных лизосомальных гидролаз, освобождающихся из фагоцитов в результате экзоцитоза.
Протеазы включают системы ограниченного протеолиза, например калликреин-кининовую систему, систему гемокоагуляции и др., что приводит к
накоплению кининов, тромбообравованию и т.д.
Фосфолипазы активируют систему эйкозаноидов с последующим накоплением простагландинов, тромбоксанов и др.
Перечисленные вещества, так же как компоненты комплемента - это классические медиаторы воспаления.
Если концентрация ИК очень велика, как, например, при феномене Артюса,
на первый план выходит активация тромбоцитов и система гемокоагуляции и
накопление факторов свертывания крови, что приводит к тромбозу микрососудов и последующему некрозу тканей.
103
Патофизиологическая фаза
К патологическим процессам, которые формируются под действием ИК
можно причислить:
-хроническое рецидивирующее воспаление;
-тромбоз;
-ишемию;
-некроз.
К болезням иммунных комплексов относят:
-системную красную волчанку;
-васкулиты (узелковый периартериит и др.);
-сывороточную болезнь;
-иммунокомплексные синдромы при инфекционных заболеваниях (например,
разнообразные поражения при лепроматозной форме лепры, некрозы кишечника при брюшном тифе);
-некоторые формы геморрагических диатезов (например, болезнь ШенлейнаГеноха);
-некоторые формы инфекционной токсической бронхиальной астмы;
-некоторые варианты анафилактического шока.
Аллергические реакции IV типа (опосредованные Т-клетками)
Эта форма реактивности сформировалась на поздних этапах эволюции на
основе иммунологических реакций и воспаления. Она направлена на распознавание и ограничение действия аллергена. Гиперчувствительность IV типа лежит
в основе многих аллергических и инфекционных заболеваний, аутоиммунных
болезней, отторжения трансплантата (трансплантационный иммунитет), контактного дерматита (контактная аллергия), противоопухолевого иммунитета.
Самым ярким ее проявлением является туберкулиновая реакция, которая в клинической практике используется в виде реакции Манту. Относительно позднее
проявление этой реакции (не ранее чем через 6- 8 ч в месте введения возникает
покраснение, в дальнейшем эритема увеличивается и достигает расцвета через
24—48 ч после введения антигена) позволило также назвать ее гиперчувствительностью замедленного типа (ГЗТ).
Этиология и особенности антигенной стимуляции при ГЗТ
Антигены, индуцирующие ГЗТ, могут иметь различное происхождение:
микробы (например, возбудители туберкулеза, бруцеллеза, сальмонеллеза, дифтерии, стрептококки, стафилококки),вирусы коровьей оспы, герпеса, кори, грибы, тканевые белки (например, коллаген),антигенные полимеры аминокислот,
низкомолекулярные органические соединения.
По химической природе антигены, которые способны вызвать ГЗТ, относятся чаще к белковым соединениям.Белки, вызывающие ГЗТ, отличаются низкой молекулярной массой и “слабыми” иммуногенными свойствами. Поэтому
они не способны в достаточной мере стимулировать антителообразование. При
этом иммунологическая реакция при ГЗТ обладает рядом отличительных особенностей. Иммунный ответ направлен не только по отношению к гаптену, как
это имеет место при реакциях немедленного типа, но и к белку-носителю, при-
104
чем специфичность в отношении антигена при ГЗТ выражена гораздо сильнее,
чем при реакциях немедленного типа. Разрешающий антиген при ГЗТ должен
быть обязательно представлен комплексом антигена и белка-носителя, а при
реакциях немедленного типа в этой роли может выступать один гаптен.
На формирование ГЗТ может оказать влияние не только качество, но и
количество поступающего в организм антигена. Как правило, для воспроизведения ГЗТ требуется небольшое количество антигена (микрограммы).
Патогенез аллергической реакции IV типа
Условно в развитии ГЗТ, как и в аллергических реакциях I, II, III типов,
можно выделить три периода.
I. Иммунологическая стадия. Поступающий в организм антиген чаще всего встречается с макрофагом, обрабатывается им, а затем в переработанном виде передается Т-лимфоцитам-индукторам, имеющим на своей поверхности рецепторы для антигена. Клетки-индукторы распознают антиген, а затем с помощью интерлейкинов (веществ-посредников, выделяемых макрофагами и лимфоцитами) запускают пролиферацию антигенраспознающих клеток — Тэффекторов (Т-киллеров), а также клеток памяти. Последнее немаловажно.
Клетки памяти позволяют сформировать быстрый иммунный ответ при повторном попадании антигена в организм.
Осуществляющие ГЗТ иммунные лимфоциты захватывают антиген, повидимому, в непосредственной близости от места его введения. Необходимым
условием активации лимфоцитов является одновременное связывание Т-клетки
как с антигеном, так и с молекулами главного комплекса гистосовместимости
(ГКГ). В результате одновременного “двойного распознавания” антигена и
продуктов ГКГ начинается пролиферация клеток (трансформация лимфоцитов)
и превращение их из зрелых в бласты.
II. Патохимическая стадия. Антигенная стимуляция лимфоцитов сопровождается их трансформацией, образованием и дальнейшим выделением медиаторов ГЗТ-лимфокинов. Для каждого медиатора на клетках-мишенях обнаружены рецепторы. Действие медиаторов неспецифично (для их действия не нужен антиген). Биологический эффект лимфокинов разнообразен .Они изменяют
клеточную подвижность, активируют клетки, участвующие в воспалении, способствуют пролиферации и созреванию клеток, регулируют кооперацию иммунокомпетентных клеток. Клетками-мишенями для них служат: макрофаги и
нейтрофилы, лимфоциты, фибробласты, стволовые клетки костного мозга, опухолевые клетки, остеокласты и др. Все лимфокины - белки, большинство из них
- гликопротеиды.
В зависимости от оказываемого эффекта лимфокины делят на две большие группы:
1) факторы, подавляющие функциональную активность клеток (фактор, угнетающий миграцию макрофагов или лимфоцитов;
фактор, агглютинирующий макрофаги; хемотаксические факторы; лимфотоксины);
105
2) факторы, усиливающие функциональную активность клеток (фактор переноса; факторы, активирующие макрофаги или лимфоциты; митогенный фактор и
др.).
III. Патофизиологическая стадия. Зависит от природы этиологического
фактора и той ткани, где “разыгрывается” патологический процесс. Это могут
быть процессы, протекающие в коже, суставах, внутренних органах. В воспалительном инфильтрате преобладают мононуклеарные клетки (лимфоциты, моноциты и макрофаги). Нарушение микроциркуляции в очаге повреждения объясняется повышением проницаемости сосудов под влиянием медиаторов белковой природы (кинины, гидролитические ферменты, фактор проницаемости), а
также активацией свертывающей системы крови и усилением образования
фибрина. Отсутствие значительного отека, так характерного для аллергических
поражений при реакциях немедленного типа, связано с весьма ограниченной
ролью гистамина в ГЗТ.
При ГЗТ повреждение может развиваться в результате:
1) прямого цитотоксического действия сенсибилизированных Т-лимфоцитов на
клетки-мишени, которые приобрели аутоаллергенные свойства (растворимый
лимфотоксин и комплемент не принимают участие в этом процессе);
2) цитотоксического действия лимфотоксинов (так как действие лимфотоксина
неспецифично, то повреждаться могут не только те клетки, которые вызвали
его образование, но и интактные клетки в зоне его образования);
3) выделения в процессе фагоцитоза лизосомальных ферментов, повреждающих тканевые структуры (эти ферменты выделяют в первую очередь макрофаги).
Составной частью ГЗТ является воспаление, которое подключается к иммунной реакции действием медиаторов патохимической стадии. Как и при
иммунокомплексном типе аллергических реакций,оно подключается в качестве
защитного механизма, способствующего фиксации, разрушению и элиминации
аллергена. Однако воспаление является одновременно фактором повреждения и
нарушения функции тех органов, где оно развивается, и ему принадлежит важнейшая патогенетическая роль в развитии инфекционно-аллергических, аутоиммунных и некоторых других заболеваниях.
МЕСТНЫЕ РАССТРОЙСТВА КРОВООБРАЩЕНИЯ.АРТЕРИАЛЬНАЯ И
ВЕНОЗНАЯ ГИПЕРЕМИЯ
Сердечно-сосудистую систему можно разделить на структурные элементы системного кровообращения и структурные элементы микроциркуляторного
русла.
Структурные элементы системного кровообращения:
-Сердце — насос, генератор давления.
-Сосуды высокого давления — упруго-растяжимые сосуды — аорта, крупные
артерии обеспечивают перевод толчкообразного движения крови в плавно поступательное.
106
Структурные элементы микроциркуляторного русла:
-Артериолы - резистивные сосуды. Стенка артериолы содержит слой гладкой
мускулатуры, её сокращение и расслабление может значительно изменять просвет сосуда, а, следовательно, и сопротивление
-Прекапиллярные артериолы (прекапилляры, прекапиллярные жомы). Они регулируют количество функционирующих в микрорегионе капилляров.
-Капилляры - обменные сосуды. Основная функция капилляров - транскапиллярный обмен. Именно проходя через капилляры кровь выполняет свою "главную работу" в организме по снабжению, дренажу и гуморальной регуляции
тканей.
-Посткапиллярные венулы (посткапилляры, посткапиллярные жомы)
-Венулы - емкостные сосуды выполняют депонирующую функцию
-Артериально-венозные анастомозы. Артерио-венозные анастомоз — самый
короткий путь между артериями и венами, который снабжен сфинктерами. В
нормальном состоянии анастомозы закрыты, и кровь происходит через сеть капилляров. Если они открываются, кровь поступает в вены, минуя капилляры.
Резистивные сосуды, создавая периферическое сопротивление, как бы
"замыкают" зону высокого давления, препятствуя переходу крови из этой зоны
на периферию.
Периферическое сопротивление (показатель, характеризующий сопротивление, возникающее при движении крови по сосудам) формирует в зоне высокого давления условия, близкие к условиям замкнутого пространства. Тем
самым резистивные сосуды определяют уровень системного артериального
давления. Если все резистивные сосуды или большинство из них расширяются,
системное периферическое сопротивление снижается, кровь свободнее перемещается на периферию, покидая зону высокого давления. Это приводит и падению системного артериального давления. Если все резистивные сосуды или
большинство из них суживаются, системное периферическое сопротивление
растет, перемещение крови на периферию затрудняется, количество крови в
зоне высокого давления увеличивается. Системное артериальное давление при
этом повышается.
На преодоление сопротивления в резистивных сосуда уходит большая
часть энергии, сообщаемой работой сердечной мышцы. До артериол давление
даже в небольших артериях достаточно высокое, после артериол (резистивных
сосудов) в капиллярах давление крови сразу в несколько раз снижается.
Прекапиллярные сфинктеры регулируют количество функционирующих в микрорегионе капилляров.
То обстоятельство, что резистивные сосуды определяют уровень давления крови в микрорегионе, позволяет им регулировать кровоток в микроциркуляторном русле.
Если артериолы микрорегиона расширяются, сопротивление в них снижается, и уровень давления при прохождении крови по таким расширенным артериолам снижается незначительно. В артериальные концы капилляров кровь попадает под весьма высоким давлением и кровоток в таком микрорегионе резко
нарастает - развивается артериальная гиперемия.
107
Это не противоречит выше сказанному о том, что при системном снижении периферического сопротивления системное артериальное давление падает,
так как рассматриваются изменения просвета артериол на местном уровне.
Расширение артериол в одном микрорегионе компенсируется сужением в другом и системные показатели периферического сопротивления и артериальное
давление остаются при этом в пределах нормы.
Если резистивные сосуды микрорегиона суживаются, периферическое
сопротивление увеличивается, и редукция давления при прохождении крови
через такие сосуды будет еще больше, чем при обычном кровотоке. Кровь будет подаваться в артериальные концы капилляров под очень низким давлением,
и кровоток в микрорегионе будет резко сокращаться - разовьется ишемия.
Таким образом, резистивные сосуды регулируют важнейшие параметры
кровообращения,на системном уровне (системное периферическое сопротивление, системное артериальное давление), и на местном (интенсивность кровотока в микроциркуляторных регионах, перераспределение крови в организме в зависимости от особенностей жизнедеятельности).
Емкостные сосуды.Эти сосуды получили такое название, потому что их
суммарная емкость в 3-5 и более раз превосходит емкость резистивных сосудов.
Очень важно, кроме того, что емкость венул может изменяться в весьма широких пределах в зависимости от их функционального состояния.
При расширении этих сосудов, в них накапливается ("депонируется")
большое количество крови. При этом количество крови, которое поступает из
магистральных вен в правое предсердие - венозный возврат - уменьшается.
Уменьшение венозного возврата влечет за собой снижение сердечного выброса.
Снижается ОЦК. Развивается гак называемое депонирование крови.
Если емкостные сосуды суживается, кровь на них выталкивается в магистральные вены и поступает в правое предсердие - венозный возврат увеличивается. При нормальной сердечной деятельности это приводит к увеличению
сердечного выброса и ОЦК. Происходит мобилизация крови.
Таким образом, от функционального состояния емкостных сосудов во
многом зависят такие жизненно важные показатели, как ОЦК, венозный возврат, сердечный выброс, давление в магистральных венах.
Обменные сосуды.Основная функция капилляров - транскапиллярный
обмен. Именно проходя через капилляры кровь выполняет свою "главную работу" в организме по снабжению, дренажу и гуморальной регуляции тканей.
Транскапиллярный обмен складывается из процессов фильтрации,
диффузии и микровезикулярного транспорта (это несколько упрощенное представление).
Фильтрация
Объем профильтровавшейся жидкости и скорость фильтрации зависят от:
- фильтрационного давления
- фильтрационной поверхности
- проницаемости капилляров (т.е. от их анатомического строения, функционального состояния эндотелиоцитов).
108
Фильтрационное давление это разность между гидростатическим давлением крови (сила, способствующая выходу жидкости из капилляра) и онкотическим (коллоидно-осмотическое давление белков плазмы, сила, которая удерживает жидкость в капилляре, притягивает жидкость из межклеточных пространств в капилляры).
Если принять гидростатическое давление в артериальном конце капилляра за 35 мм рт. ст. (средняя величина, этот показатель в капиллярах различных
органов может сильно отличаться в ту или иную сторону) и онкотическое давление белков крови за 25 мм рт. ст - то фильтрационное давление составит 10
мм рт. ст. Именно с такой силой жидкая часть крови будет выталкиваться из
сосудов в ткани.
По мере продвижения крови по капиллярам, гидростатическое давление
будет падать и в венозном конце может составить 15 мм рт. ст. (средняя величина). Поскольку онкотическое давление при движении крови по капиллярам
не меняется, то разность также будет равна 10 мм рт. ст. Но направление этой
силы изменяется на противоположное. Это будет сила, притягивающая жидкость из межклеточных пространств и возвращающая ее обратно в сосудистое
русло.
Гидростатическое и онкотическое давление межклеточной жидкости
обычно не принимается во внимание, так как эти показатели невелики, по сравнению с таковыми в сосудах.
Фильтрационная поверхность определяется количеством функционирующих в данный момент капилляров.
У человека через большой круг кровообращения в сутки проходи около 89 тыс. литров крови и только 20 литров из нее фильтруется через капилляры в
ткани (в процессе фильтрации в почках в первичную мочу выходит 150-180
литров жидкости). Такой уровень фильтрации обусловлен постоянным уровнем
гидростатического давления в различных микрорайонах. В капиллярах мышц,
кожи - 30 мм рт. ст., почек - 70 мм рт. ст., легких - 10 мм рт. ст., печени - 6-7 мм
рт. ст. Уровень давления зависит от функциональных особенностей органов и
тканей. (Так при снижении фильтрационного давления в почках ниже 60-50 мм
рт. ст. приостанавливается процесс мочеобразования).
Из 20 литров фильтрующейся жидкости 16-18 литров реабсорбируется за
счет разности гидросатического давления в артериальном (30 мм рт. ст.) и венозном (15 мм рт. ст.) концах капилляров, а остальная жидкость выводится из
тканей за счет лимфооттока, таким образом, ткани как бы промывается жидкостью - этот процесс называется дренажем ткани.
"Проницаемость" капиллярной стенки требует несколько более детального рассмотрения.
Проницаемость капилляров зависит от
- размеров межэндотелиальных щелей,
- фенестрации эндотелиоцитов (образованием фенестр - ситуацией, когда
внешняя и внутренняя клеточные мембраны эндотелиоцита как бы приходят в
непосредственное соприкосновение),
-состояния базальной мембраны и др.
109
Капилляры в спокойном состоянии проницаемы практически для всех
компонентов жидкой части крови, кроме макромолекул - белков кровяной
плазмы. Тканевая жидкость в нормальных условиях отличается от кровяной
плазмы только белее низким содержанием белка (в крови 7-8%, в тканевой
жидкости 0,2-0,3-0,5%).
Таким образом, поддерживается водный баланс в тканях и их дренаж вымывание шлаков, образующихся в результате жизнедеятельности клеток.
Повышение проницаемости капилляров определяется функциональным состоянием эндотелиоцитов, их активным сокращением. В результате их плоская
форма меняется на округлую и межклеточные щели увеличиваются.
Сосуд становится проницаем не только для воды и обычных молекул: Nа+, глюкозы, аминокислот, но и для макромолекул - белков. Белки, в первую очередь
альбумины, поступают в межклеточную жидкость. Онкотичеcкое давление последней растет и приближается к онкотическому давлению крови.
Таким образом, поступившие в межклеточную жидкость белки, увеличивают ее онкотичеекое давление, препятствуют возвращению воды обратно в сосудистое русло через венозный конец капилляра. Движение жидкости приобретает односторонний характер: из сосудов в ткани. Дренаж тканей нарушается.
Диффузия
Интенсивность диффузии определяется градиентом концентраций соответствующих молекул.
Поскольку при обмене через капиллярную стенку диффузия определяет в
основном газообмен (O2, и СО2), то решающую роль играет:
а) разность парциальных давлений газов в крови и в межклеточной жидкости
б) суммарная диффузионная поверхность(определяется количеством
функционирующих капилляров)
Микровезикулярный транспорт
Эндотелиальные клетки способны активно путем пиноцитоза захватывать
капли жидкой части крови, содержащие разные молекулы. Затем такой пузырек
продвигается в цитоплазме и выталкивается с противоположной стороны клетки в межклеточную жидкость.
Таким путем эндотелиоциты избирательно транспортируют за пределы
сосуда некоторые макромолекулы, которые не могли бы пройти через межэндотелиальные щели, например, фибриноген.
Интенсивность микровезикулярного транспорта зависит от функционального состояния клеток.
«Гиперемия» или полнокровие это процессы, при которых происходит
увеличение объёма крови в каком — либо участке сосудистой сети. Данный
процесс может развиться в результате увеличения количества крови, протекающей через орган или ткань в единицу времени, а также при застойном увеличении кровенаполнения органа или ткани.
110
Артериальная гиперемия
Артериальная гиперемия — увеличение кровенаполнения органа или
ткани в результате увеличения притока крови через его сосуды, сопровождающееся увеличением линейной и объемной скорости кровотока и дренажа ткани.
По значению для организма артериальную гиперемию можно разделить на:
- физиологическую
- патологическую
Физиологическая гиперемия развивается при повышенном функционировании органа или ткани, например, в работающих мышцах, беременной матке, кишечнике во время пищеварения, секретирующей железе и т.д. При этом в ткани
поступает значительно больше кислорода и питательных веществ, а продукты их
распада быстрее выводятся.
Патологическая артериальная гиперемия сопровождается нарушениями кровоснабжения, микроциркуляции, транскапиллярного обмена наблюдается например при:
- воспалении,
- аллергии.
Причины артериальной гиперемии:
-повышение функциональной активности ткани.
-физические факторы (температура, атмосферное давление).
-химические факторы.
-нарушение сосудодвигательной иннервации (нейротоническая, нейропаралитическая). Пример: опыт К. Бернара (перерезка симпатического нерва у
основания уха кролика), Рене Ляриш - перивазальная симпатэктомия.
-нарушение тканевого обмена
-воспалительный процесс на ранних этапах развития
-аллергические реакции.
Механизмы развития артериальной гиперемии:
1. Миопаралитический механизм. Под влиянием различных метаболитов (пуринов, лактата, двуокиси углерода, калия и др.), медиаторов воспаления, увеличения концентрации внеклеточного калия, водорода и других ионов, уменьшения содержания кислорода происходит снижение миогенного тонуса сосудов и
увеличение числа функционирующих капилляров.
2. Нейропаралитический механизм заключается в уменьшении нейрогенного
вазоконстрикторного действия на сосуды и снижении нейрогенного тонуса.
Данный вид гиперемии может развиться, например, при перерезке, параличе
или повреждении нервных волокон, а также при повреждении нервных центров.
3. Нейротонический механизм заключается в повышении нейрогенной сосудорасширяющей активности или понижении тонуса вазоконстрикторов в результате истинного рефлекса, либо аксонрефлекса. Под действием симпатических вазодилятаторов артериальная гиперемия по данному типу развивается в
поджелудочной и слюнных железах, языке, кавернозных телах.
111
Внешние признаки артериальной гиперемии:
- покраснение за счет расширения сосудов, увеличения количества функционирующих капилляров и заполнения их алой (артериальной) кровью,
- повышение температуры тканей за счет активация метаболизма и прилива теплой крови от внутренних органов,
- небольшое увеличение объема,
- повышение упругости (тургора) за счет увеличения функционирующих
капилляров, усиление лимфообращения,
- пульсация - за счет снижения периферического сопротивления, повышения гидростатического давления в расширенных артериолах,
- видимое невооруженным глазом или под микроскопом расширение сосудов.
Микроскопические признаки артериальной гиперемии:
Состояние микрососудов:
- расширение артерий и артериол;
- увеличение количества функционирующих капилляров
(иногда включение в кровоток всех капилляров данного региона);
- расширение венул.
Данные изменения микрососудов происходят в результате активации того
или иного патогенетического механизма развития артериальной гиперемии.
Основные изменения гемодинамики:
- увеличение линейной скорости кровотока
- увеличение объемной скорости кровотока. Сосудистая сеть в участке артериальной гиперемий промывается очень большим количеством крови
под большим давлением и с большей скоростью.
Факторы, обусловливающие изменения гемодинамики
- уменьшение сопротивления в резистивных сосудах,
- повышение гидростатического давления в артериолах,
- повышение давления в капиллярах, особенно в их артериальных концах,
- незначительное или отсутствие повышения давления в венулах,
- увеличение артерио-венозного градиента гидростатических давлений.
Состояние транскапиллярного обмена
Фильтрация
Факторы, обусловливающие изменение баланса жидкости между сосудами и тканями:
- повышение фильтрационного давления в капиллярах - увеличение градиента гидростатических давлений между капиллярами и тканевой жидкостью,
- увеличение фильтрационной поверхности за счет увеличения количества
функционирующих капилляров,
- повышение проницаемости капиллярной мембрана.
Изменение баланса жидкости в системе: кровеносные сосуды - ткань- лимфатические сосуды.
- увеличение лимфопродукции,
112
- пропорциональное увеличение оттока лимфы по лимфатическим сосудам,
- относительно необходимое увеличение жидкости заполняющей в каждый
конкретный момент ткань и межтканевое пространство,
- усиленный "дренаж" ткани. Ткань промывается большим количеством
лимфы.
Диффузия
Изменение процессов диффузии на примере диффузии газов – газообмене
В норме парциальное давление в мм рт. столба в венозной
крови составляет: O2 - 37, CO2 - 46;
в артериальной:
О2 - 100, СО2 - 40.
Факторы, обусловливающие изменение диффузии кислорода:
- увеличение объема крови, проходящей через гнперемированный орган в
единицу времени. Кровь не успевает отдать за время прохождения через
капилляры имеющийся в ней кислород, даже если количество кислорода,
диффундирующего в тканевую жидкость и потребляемого клетками в несколько раз больше нормального. Кровь остается алой в венозном конце
капилляра.
- повышение парциального давления кислорода на протяжении
всего капилляра.
- увеличение градиента парциального давления кислорода между кровью и
тканевой жидкостью.
- увеличение количества функционирующих капилляров (через которые
идет диффузия кислорода в насыщение клеток кислородом.
Таким образом обеспечивается:
- ускорение диффузии кислорода
- увеличение общего количества диффундирующего кислорода
- оптимальное насыщение клеток (тканей) кислородом. Оптимальные
условия для обмена.
Факторы, обусловливающие изменение диффузии углекислого газа:
- увеличение объема циркулирующей крови в единицу времени через гиперемированный орган. Образующийся в тканях СО2 не может в достаточной мере насытить весь этот объем.
- понижение парциального давления углекислого газа в капиллярах, даже в
их венозном конце.
- увеличение градиента парциального давления CО2 между кровью и тканевой жидкостью.
- увеличение количества работающих капилляров (через стенки которых
идет диффузия углекислого газа, усилен дренаж ткани в отношении CО2).
Характер изменения диффузии углекислого газа:
- увеличение общего количества диффундирующего углекислого газа, даже
если его образование происходит быстрее, чем в обычных условиях.
- оптимальный дренаж тканей в отношении CO2. По аналогии с диффузией
газов происходит изменение динамики других веществ, поступающих в
ткани или попадающих в тканевую жидкость путем диффузии через капиллярную мембрану.
113
Последствия артериальной гиперемии
Положительные:
- активация обмена веществ
- оптимальные условия жизнедеятельности клетки
- обеспечение условий высокой функциональной активности
- гипертрофия ткани при длительной, постоянно повторяющейся артериальной гиперемии.
Отрицательные:
- разрыв патологически измененных сосудов
- системные расстройства гемодинамики в случае обширной артериальной
гиперемии в связи с падением периферического сопротивления.
Венозная гиперемия
Венозная гиперемия - это такое состояние микроциркуляции, при котором имеет увеличение кровенаполнения органа и ткани развивается в результате нарушения венозного оттока и сопровождается снижением линейной и объёмной скорости кровотока и нарушением дренажа ткани. Этот процесс называют также пассивной гиперемией или венозным застоем.
Причины венозной гиперемии:
- закупорка вен - тромбоз
- сдавление вен извне (опухоль, жгут, беременность и т.д.)
- структурная или функциональная неполноценность вен (варикозные расширения)
- вынужденное положение тела или его частей (в венах задних отделов лёгких — у ослабленных постельных больных, в геморроидальных венах
при сидячем положении, при продолжительном зависании вниз головой).
- гиподинамия – снижение функциональной активности поперечнополосатых мышц и снижение при этом оттока крови по венозным стволам.
- повышение давления в магистральных венах большего круга при правожелудочковой недостаточности, в малом круге при левожелудочковой
недостаточности, в бассейне воротной вены при циррозе печени.
Внешние признаки венозной гиперемии:
- "цианоз" за счет расширения и увеличения капилляров, заполненных застойной кровью с очень низким содержанием оксигемоглобина ("венозной кровью"),
- расширение вен, неравномерность их диаметра ("вздутия", "узлы") по ходу венозных сосудов. Сосуды приобретают извитой характер. Увеличение
количества венозных разветвлений, видимых невооруженным глазом
(выявляется густая венозная сеть).
- понижение температуры ткани за счет снижения интенсивности окислительных процессов, а так же вследствие уменьшения объёмной скорости
кровотока. Меньше теплой крови проходит за единицу времени через сосуды застойного участка.
- увеличение объема органа и ткани за счет избыточного скопления жидкости.
114
- пастозность ("тестоватость") за счет избыточного количества жидкости в
тканях.
Состояние микрососудов
- рефлекторное сужение приводящих артериол, прекапилляров (компенсаторное).
- расширение венул,
- увеличение числа функционирующих капилляров,
Факторы, обусловливавшие нарушение гемодинамики:
- в связи с уменьшением оттока крови при сокращении её притока повышается гидростатическое давление в венах и капиллярах.
- снижение гидростатического давления в соответствующих артериолах изза их спазма и возрастающего таким образом сопротивления кровотоку.
- уменьшение артерио-венозного градиента давления.
Основные изменения гемодинамики:
- уменьшение объёмной и линейной скорости кровотока
- кровоток становится неравномерным
- толчкообразное и маятникообразное движение крови,
- стаз (полная остановка кровотока).
Состояние транскапиллярного обмена
Фильтрация
Факторы, обусловливающие изменения процессов фильтрации (на примере
изменения баланса жидкости между сосудами и тканью):
- повышение фильтрационного давления по всей площади капилляров и
посткапилляров. Увеличение градиента гидростатических давлений между капиллярами и тканевой жидкостью,
- увеличение фильтрационной поверхности за счет увеличения количества
капилляров, участвующих в кровотоке и увеличения гидростатического
давления на всем участке капилляров,
- повышение проницаемости капиллярной мембраны за счет метаболитов,
накапливающихся в результате гипоксии.
Характер изменений баланса жидкости в системе: кровеносные сосуды - ткань лимфатические сосуды:
- многократное увеличение лимфопродукции,
- практически полное выключение возврата жидкости в кровеносное русло
через венозные концы капилляров,
- рефлекторный спазм лимфатических сосудов (когда в застой вовлечены
магистральные венозные стволы),
- повышение давления в лимфатических сосудах - снижение градиента
давления между лимфатическими сосудами и тканевой жидкостью,
- накопление тканевой жидкости – отек,
- недостаточный дренаж ткани.
Диффузия
Изменения процессов диффузии на примере диффузии газов - газообмена.
Факторы, обусловливающие изменение диффузии кислорода:
115
- резкое замедление линейной скорости кровотока, несмотря на расширение сосудов, сопровождается многократным уменьшением объема крови,
проходящей через застойный участок органа или тканей. Ткани максимально отбирают содержащийся в крови кислород. Кровь становится "венозной" уже начале артериального конца капилляров;
- снижение парциального давления кислорода в капиллярах;
- снижение градиента парциального давления кислорода между кровью и
тканевой жидкостью.
Характер изменения диффузии кислорода
В связи с уменьшением притока крови уменьшается диффузия кислорода,
что ведет снижению общего количества диффундирующего кислорода. Развивается гипоксия ткани.
Факторы, обусловливающие изменение диффузии углекислого газа
- уменьшение объема крови, проходящей за единицу времени через застойный орган: ткани сразу же практически в момент поступления крови
в капилляр насыщают ее СО2. Кровь, полностью насыщенная СО2 при
отсутствии ее значительного движения не способна обеспечить "вымывание" СО2 из тканей.
- повышение содержания парциального давления СО2 в капиллярах на
всем их протяжении.
- снижение градиента парциального давления СО2 между кровью и тканевой жидкостью.
Характер изменения диффузии углекислого газа
- замедление диффузии СО2 из ткани в кровь во времени;
- снижение общего количества диффундирующего СО2;
- тканевая гиперкапния.
По аналогии с диффузией газов происходит изменение динамики диффузии
других веществ, поступающих в ткани или покидающих ткань путем диффузии
через капиллярную мембрану.
К компенсаторным явлениям со стороны сосудистой системы на уровне микроциркуляции при венозной гиперемии можно отнести:
- отток по венозным коллатералям (которые практически хорошо развиты
во всех органах и тканях). Эти сосуды увеличиваются в объеме, приобретают более мощные сосудистые стенки, если венозный застой продолжается длительное время;
- спазм приводящих артерий и артериол, при этом увеличивается сопротивление кровотоку "закрываются краны", что препятствует нарастанию
гидростатического давления в застойной области и притоку туда избыточного количества крови.
Последствия венозной гиперемии:
Положтельные:
Замедление кровотока препятствует:
- распространению медиаторов воспаления;
- распространению патогенов из очага воспаления;
- облегчает эмиграцию лейкоцитов;
116
Отрицательные:
- гипоксия и гиперкапния тканей, активация гликолиза и лактоацидоза,
- снижение функциональной активности застойных органов и тканей,
- отеки,
- атрофия паренхиматозных, функционально активных элементов ткани
при длительном венозном застое,
- разрастание соединительной ткани - склероз органа при длительном венозном застое,
- острая сосудистая недостаточность - "коллапс" - при остром депонировании большого количества крови в расширенных емкостных сосудах при
обширном венозном застое.
МЕСТНЫЕ РАССТРОЙСТВА КРОВООБРАЩЕНИЯ. ИШЕМИЯ.
ЭМБОЛИЯ.ТРОМБОЗ
ИШЕМИЯ
Ишемия - это такое нарушение микроциркуляции, которое развивается в
результате уменьшение кровенаполнения органа или ткани вследствие уменьшения или полного прекращения притока крови в его сосудистую сеть.
Ишемия, по своей гемодинамике, состояние, обратное артериальной гиперемии.
Причины ишемии.
Причины, вызывающие ишемию делятся на:
1.Обтурационные - эмболия, тромбоз, обтурация сосудов в результате повреждения интимы;
2.Ангиоспастические – гиперкатехоламинемия, физические факторы (холод,
травма, механическое раздражение); химические агенты, биологические раздражители (токсины бактерий и т.д.);
3.Компрессионные - сдавление сосудов извне (позиционное сдавление, кровеостанавливающий жгут, гипсовая повязка, необоснованная, ошибочная перевязка сосудов, без учета наличия коллатералей, сжатие их извне опухолью и др).
Признаки ишемии:
- бледность тканей,
- снижение количества видимых сосудов и утрата ими извитой формы
- ослабление пульсации артерий,
- снижение температуры.
- снижение объема органа,
- уменьшение функциональной активности органа или ткани.
- чувство боли.
- снижение тургора ткани;
- дистрофические изменения.
Микроскопические признаки ишемии:
Состояние микрососудов:
- сужение артерий и артериол,
- «запустевание» капилляров,
117
- уменьшение количества функционирующих капилляров,
- сужение вен и венул.
Основные изменения гемодинамики:
Факторы, обусловливающие изменения гемодинамики
- снижение гидростатического давления в ишемизированном участке
вследствие увеличения сопротивления в спазмированных резистивных
сосудах
- снижение давления в капиллярах как следствие снижения давления в
приводящих сосудах
- снижение давления в венах (по тем же причинам)
- уменьшение артерио-венозного градиента давления (гидростатического).
Характер изменения гемодинамики
- снижение линейной скорости кровотока,
- резкое уменьшение объемной скорости кровотока,
- стаз.
Транскалиллярный обмен
Фильтрация
- понижение фильтрационного давления в капиллярах - снижение градиента гидростатического давления между капиллярами и тканевой жидкостью.
- сокращение фильтрационной поверхности за счет уменьшения количества принимающих участие в кровотоке капилляров.
- снижение лимфопродукции.
- сокращение количества жидкости, оттекающей по лимфатическим сосудам от ишемизированного органа
- снижение дренажа ткани.
Такое состояние водного баланса в ишемизированном участке ткани характерно только для начальных этапов процесса. В тех случаях, когда ишемия продолжается длительное время, происходят существенные изменения проницаемости капилляров за счет действия накопившихся в тканях продуктов метаболизма.
Диффузия
Факторы, обусловливающие изменение диффузии кислорода:
- уменьшение количества крови, проходящей за единицу времени через
ишемизированный орган,
- снижение парциального давления кислорода в капиллярах,
- снижение градиента парциального давления кислорода.
Характер изменения диффузии кислорода
уменьшение диффузии за счет уменьшения притока крови, что суммарно
выражается в снижении общего количества диффундируемого кислорода,
то есть развивается гипоксия ткани циркуляторного генеза.
Факторы, обусловливающие изменение диффузии углекислого газа:
- уменьшение объема крови, проходящей за единицу времени через ишемизированный орган,
118
- повышение парциального давления CO2 в капиллярах, даже в их артериальных концах,
- снижение градиента парциального давления CO2 между кровью и тканевой жидкостью.
Характер изменения диффузии углекислого газа:
- замедление диффузии CO2
- уменьшение общего количества диффундирующей CO2.
В ткани возникает гипоксия и гиперкапния, формируется местный ацидоз.
По аналогии с диффузией газов происходит изменение динамики других веществ, поступающих в ткань или попадающих в тканевую жидкость путем
диффузии через капиллярную мембрану. Главными компенсаторными механизмами со стороны сосудистой системы на местном уровне при ишемии является усиление кровотока по артериальным коллатералям. Такая перифокальная
артериальная гиперемия служит причиной выраженного отека ишемизированного участка.
Последствия ишемии
- главное последствие ишемии – гипоксия, которая обуславливает все основные
проявления.
При остром течении, особенно в работающих органах с недостаточно развитыми коллатералями развивается некроз ткани – инфаркт (легкого, сердца и
т.д.), инсульт (мозга).
При хроническом процессе:
- снижение активности обменных процессов
- снижение функциональной активности
- атрофия органа или тканей
- болевой синдром.
Изменения в организме
Изменения проявляются в постишемический период после реваскуляризации
ранее ишемизированных тканей. В период ишемии в ишемизированном участке
накапливается большое количество биологически активных веществ (продуктов
распада ткани, кининов, простагландинов, продуктов перекисного окисления
липидов), которые увеличивают проницаемость обменных сосудов, расширяют
резистивные сосуды, открываются пре- и посткапиллярные сфинктры, что приводит к резкому отеку раннее ишемизированного участка, развитию болевого
синдрома, развитию постишемического шока.
ТРОМБОЗ
Тромбоз - прижизненное внутрисосудистое свертывание крови, приводящее к образованию конгломератов, состоящих из фибриновых сгустков и форменных элементов крови.
Тромбоз — защитный физиологический ответ тканей на повреждение. Если
процесс тромбоза избыточен или недостаточен — он становится источником
тяжелой патологии
119
Тромбоз это работа всей системы гемостаза и антигемостаза (совокупность
механизмов, обеспечивающих остановку кровотечения). Основная цель системы гемостаза — остановка кровотечения и восстановление целостности сосудистой стенки.
Выделяют три основных звена гемостаза:
1. Сосудистое звено осуществляет спазм поврежденного сосуда и активирует
процессы тромбообразования и свёртывания.
2. Клеточное (тромбоцитарно-лейкоцитарное) звено, его основная задача которого — формирование белого тромба
3. Фибриновое звено — активация системы свертывания крови, формирование
красного тромба.
Все три звена гемостаза запускаются одновременно в момент повреждения сосуда.
В патогенезе тромбоза Р. Вирхов выделял 3 фактора ("триада Вирхова"):
1. повреждение сосудистой стенки;
2. замедление кровотока;
3. повышение свертываемости крови.
Первый фактор рассматривают как производящую причину, второй и третий как предрасполагающие.
Повреждение сосудистой стенки:
а) способствует активации XII фактора и тромбоцитов (обнаженный коллаген, ферменты протеолиза, тромбоксаны, АДФ и др. БАВ);
б) выключает антисвертывающую функцию эндотелиоцитов (поврежденные эндотелиоциты перестают выделять простациклины);
в) сопровождается местным спазмом сосудов и нарушениями кровотока
(турбулентностями, завихрениями).
Замедление кровотока способствует тромбообразованию.
Замедление кровотока способствует повышению концентрации БАВ и активированных факторов свертывания крови, позволяет достичь критических концентраций, необходимых для свертывания крови.
Последствия тромбоза
Положительные:
а) тромб механически укрепляет неполноценную, патологически истонченную стенку сосуда, например, при аневризмах.
б) тромб, временно, способствует прекращению кровотечения (при повреждении сосудов крупного калибра, при повышении давления в результате
улучшения состояния больного, тромб, может быть "вытолкнут" и кровотечение может возобновиться)
Отрицательные:
а) венозная, застойная гиперемия при перекрытии тромбом венозного сосуда;
б) ишемия при тромбозе артерии.
120
Профилактика и лечение тромбозов включает мероприятия направленные
на предупреждение повреждения сосудистой стенки и повышение антикоагуляционных свойств крови.
При наличии тромба проводится антикоагуляционная и фибринолитическая терапия.
ЭМБОЛИЯ
Эмболия – перенесение стоком крови и закупорка сосудов инородными
телами. Эти тела называются эмболами.
По происхождению эмболы могут быть:
- экзогенными (пузырьки воздуха, личинки гельминтов, микробы, различные
другие инородные частицы и т.д.);
- эндогенными (тромбы, жировые клетки, клетки паренхиматозных органов и
т.д.).
Эмболии делятся на:
1. Эндогенные:
- Жировая эмболия – закупорка сосудов эндогенными липопротеидными частицами
- Тканевая эмболия – закупорка сосудов амниотической жидкостью, клетками
опухоли, жировой тканью
- Газовая эмболия – закупорка эндогенными пузырьками азота при резком понижении их растворимости в крови, например при кессонной и высотной болезни.
- Тромбоэмболия - закупорка сосуда оторвавшимися тромбами или их частицами
- Опухолевая эмболия – закупорка небольшими фрагментами распадающейся
опухолевой ткани.
2. Экзогенные
- Микробная и паразитарная эмболия – наблюдается при сепсисе, паразитарных
инвазиях, бактериемии.
- Воздушная эмболия – закупорка сосудов пузырьками атмосферного воздуха
при ранении легкого и развитии пневмоторакса, искусственном кровообращении, ранении крупных вен
- Эмболия инородными телами – закупорка сосудов инородными телами при
ранениях и при медицинских инвазивных манипуляциях.
По локализации различают эмболию
- большого круга кровообращения
- малого круга кровообращения и
- системы воротной вены.
Эмболия большого круга кровообращения. Эмболы из вен попадают в
полость работающего правого сердца и блокируют сосуды малого круга кровообращения. При эмболии большого круга кровообращения попадание эмболов
в сосуды мозга и сердца возникают участки ишемии, ведущие к гибели клеточных элементов, нарушению функции.
Эмболия малого круга кровообращения.
121
Источник эмбола – глубокие вены нижних конечностей из них эмбол попадет в общую подвздошную вену и далее в нижнюю полую вену, откуда путь в правое предсердие, правый желудочек и, наконец, в легочный ствол и легочные артерии. Очень важно, каков по размерам эмбол. Закупорка самых крупных сосудов приводит к внезапной смерти вследствие нарушения кровотока через малый круг кровообращения и резкого падения кровяного давления в магистральных сосудах большого круга кровообращения. Если же нарушается кровообращение в сегменте легкого, то это не нарушает общего кровообращения, а
приведет к ишемии участка легкого, то есть местному малокровию, и омертвению части легочной ткани, что называется инфарктом легкого.
Эмболии воротной вены развивается в результате патологических изменений в бассейне воротной вены. Источник эмбола – органы брюшной полости,
которые ведут к закупорке v. Porta в результате происходит сброс венозной
крови, оттекающей из кишечника в обход печени через порто-кавальные анастомозы.
ПАТОФИЗИОЛОГИЯ КЛЕТКИ
Клетка является элементарной саморегулирующейся структурнофункциональной единицей тканей и органов. В ней протекают процессы , лежащие в основе энергетического и пластического обеспечения изменяющихся
структур и уровня функционирования тканей и органов.
Главной функцией клетки является осуществление обмена со средой информацией, веществом и энергией, что подчинено в конечном счете задаче сохранения клетки как целого при изменении условий существования.
.
Структурно- функциональная организация клетки:
1. Ядро- содержит генетический материал клетки.
2. Мембрана - обеспечивает целостность структур клетки.
3. Лизосомы - содержат широкий спектр гидролитических ферментов.
4. Митохондри и - обеспечивают энергетические потребности клетки.
5. Рибосомы - обеспечивают синтез белков.
6. Эндоплазматическая сеть - мембранная структура содержащая рибосомы
и детоксицирующие ферменты.
7. Аппарат Гольджи - накапливает и распределяет белки, необходимые для
построения различных структурных элементов клетки.
Под повреждением клетки понимают такие изменения ее, структуры,
функций, метаболизма, физико-химических свойств, которые ведут к нарушению ее жизнедеятельности.
Причины повреждения клеток
Нарушения внутриклеточного гомеостаза, составляющие сущность повреждения клетки, могут возникать как в результате непосредственного воздействия на клетку патогенного агента, так и опосредованно, вследствие
нарушений постоянства внутренней среды самого организма.
122
Непосредственное (первичное) повреждение. В зависимости от происхождения все факторы, способные при взаимодействии с клеткой вызвать
ее повреждение, можно разделить на 3 группы:
1. Физические факторы:
-механические воздействия: удары, растяжения, сдавление, гравитационные
перегрузки, и др.;
-колебания температуры;
-изменения осмотического давления в клетке (накопление продуктов неполного
окисления или избытка ионов );
-воздействие ионизирующей радиации (образование свободных радикалов и активацией перикисных липопероксидных процессов, продукты которых повреждают мембраны и денатурируют ферменты клеток);
2.Факторы химического происхождения.
Повреждение клетки могут вызвать неорганические вещества (кислоты, щелочи, соли тяжелых металлов), низкомолекулярные органические соединения (кислоты, щелочи,фенолы, альдегиды, галогеновые производные), высокомолекулярные соединения(гидролитические ферменты, основные катионные белки, иммуноглобулины, комплексы антиген-антитело, комплемент).В настоящее
время описано более 20 000 химических соединений, оказывающих повреждающее действие.
3.Факторы биологической природы.
К ним относятся вирусы, риккетсии, микроорганизмы, паразиты, грибки;
продукты их жизнедеятельности или деградации вызывают расстройства функции клеток, нарушение в них метаболических процессов ( проницаемость и целостность мембран, активность клеточных ферментов); сходство антигенов;
ферменты, белки, липиды, чрезмерная физическая нагрузка.
Опосредованное (вторичное) повреждение. Возникает как следствие
первичных нарушений постоянства внутренней среды организма. К повреждению
клетки приводят гипоксия, гипо- и гипертермия, ацидоз и алкалоз, гипер- и гипоосмия, гипогликемия, гиповитаминозы, повышение содержания в организме конечных продуктов метаболизма, оказывающих токсическое действие (аммиак, билирубин и др.).
Классификация патологических процессов в клетке.
Современное представление о природе болезней базируется на двух основных аспектах – материально-энергетическом и информационном, поскольку
болезнь может быть связана как повреждением исполнительного клеточного
аппарата ( включая ДНК- материальный носитель клеточных программ), так и с
нарушением информационных процессов – сигнализации, рецепции и т.п.
Исходя из этого выделяют следующие виды клеточных повреждений:
Информационные повреждения:
-патология сигнализации;
-патология рецепции;
-нарушения пострецепторных посредников;
-дефекты клеточных программ.
123
Повреждения исполнительного аппарата клетки:
-типовые молекулярные механизмы повреждений исполнительного аппарата;
-повреждения ядра;
-повреждения мембран;
- повреждения митохондрий и нарушения процессов энергообеспечения клетки;
-повреждения лизосом и активация процессов ограниченного протеолиза.
Интегральные механизмы повреждения клетки:
-адаптация клетки (гипертрофия, гиперплазия, гиперфункция);
- механизмы гибели клеток:
1) апоптоз;
2) некробиоз и некроз.
ИНФОРМАЦИОННЫЕ ПОВРЕЖДЕНИЯ КЛЕТКИ
Патология сигнализации
Клеточные адаптационные программы включаются в ответ на определенные входные сигналы. В большинстве случаев клетки в организме управляются
химическими регуляторными сигналами. Кажется, что разнообразие таких сигналов бесконечно. На самом деле, все они принадлежат к одной из пяти возможных категорий. Это могут быть:
1. Гормоны
2. Медиаторы
3. Антитела
4. Субстраты
5. Ионы.
Недостаток или отсутствие того или иного сигнала может воспрепятствовать включению тех или иных адаптивных программ, что приводит к определенным патологическим последствиям.
Избыток того или иного сигнала заставляет адаптивные программы,
включаемые данным регулятором функционировать излишне интенсивно или
ненормально долго, что также патогенно.
Особый случай представляет достаточно распространенная ситуация, когда клетка ошибочно принимает один сигнал за другой — так называемая мимикрия биорегуляторов, приводящая к серьезным регуляторным расстройствам.
Таблица1 содержит некоторые примеры, иллюстрирующие разнообразие
патологии сигнализации в организме.
Таб.1. Виды информационных нарушений в клетке
Биорегулятор Избыток сиг- Дефицит сигна- Мимикрия сигнала
нала
ла
Гормон
Синдром
Инсулинзависи- Болезнь фон Базедова (ми-
124
Нейромедиатор
Антитело
Субстрат
Ион
ИценкоКушинга
Отравление
цикутой
Аллергический
энцефаломиелит
Ожирение
мый
сахарный микрия антителами)
диабет
Паркинсонизм
Тяжелая миастения (мимикрия антителами)
В-клеточный им- Астматический статус (имимунодефицит
тация антителами бронхолитика)
Квашиоркор
Действие некоторых
цитостатиковантиметаболитов
(6-меркаптопурин)
Гиперкалиемия Гипокальциемия Отравление тетраэтиламмонием (имитация катиона калия)
Заболевания, вызванные неправильным использованием клетками своего
программного аппарата вследствие неправильной сигнализации весьма разнообразны. Так, при дефиците инсулина отсутствие входного сигнала не дает
возможности использовать программы синтеза инсулинозависимых белковтранспортеров глюкозы, что приводит к нарушению утилизации этого субстрата инсулинзависимыми тканями.
Разрушение или блокада функций дофаминэргических нейронов среднего
мозга при паркинсонизме приводит к дефициту дофамина в подкорковых ядрах и инактивации дофаминзависимых автоматизмов регуляции движений.
Нехватка или отсутствие иммуноглобулинов сопровождает В-клеточные
и смешанные иммунодефициты.
Примером болезни, вызванной дефицитом субстратов, может служить
квашиоркор. Дефицит кальция приводит к нарушению работы многих внутриклеточных систем, включая сократительные белки, а также межклеточные медиаторные системы, функция которых связана с каскадным ограниченным протеолизом (например, система фибринолиза).
Избыток глюкокортикоидов при синдроме Иценко-Кушинга заставляет
клетки избрать неадекватные программы метаболической регуляции, что приводит к усилению липогенеза и глюконеогенеза, отрицательному азотистому
балансу, метаболическому алкалозу и даже вызывает запрограммированную
клеточную гибель посредством апоптоза (в частности, в лимфоидных органах).
Избыточный холинэргический нейромедиаторный сигнал при отравлении
аконитом (цикутой) приводит к расстройству вегетативной, регуляции жизненно важных функций.
Избыточная продукция аутоантител приводит к аутоаллергическим заболеваниям, хотя в небольших титрах аутоантитела присутствуют у абсолютно
здоровых людей и участвуют в регуляции клеточного роста и функций.
125
Избыток ионов калия, создающийся при массивном цитолизе или длительной анурии нарушает программные автоматизмы, связанные с работой
проводящей системы сердца и ведет к аритмии.
Особый интерес представляет мимикрия клеточных сигналов, когда рецептор, контролирующий включение тех или иных программ, стимулируется
или блокируется нештатным сигналом, ошибочно принятым клеткой за гормональный или медиаторный стимул.
Ситуации, связанные с мимикрией сигналов, как правило, вовлекают иммунную систему и чаще всего речь идет о выработке аутоантител, иммунологически копирующих те или иные гормоны или медиаторы, способных связываться с соответствующими рецепторами.
Так, при болезни фон Базедова (диффузный токсический зоб) клетки щитовидной железы усиленно растут и размножаются (гиперплазия) и усиленно
синтезируют тироксин и трийодтиронин (гиперфункция), несмотря на то, что
содержание естественного стимулятора тиреоцитов — тиреотропного гормона
гипофиза — у подавляющего большинства пациентов понижено или нормально. В организме больных вырабатываются антитела против мембранных рецепторов к тиреотропину.Как известно, секреция тиреотропина при наличии в
крови достаточных количеств тироксина подавляется, и щитовидная железа перестает на какое-то время вырабатывать тироксин. Что касается антител, то их
накопление от содержания тиреоидных гормонов практически не зависит, и избыточная стимуляция железы происходит непрерывно.
При печеночной коме, вследствие нарушения метаболизма индоловых
соединений в печени, образуется ложный нейромедиатор - октопамин, нарушающий, в качестве фальшивого сигнала, работу мозга, что ведет к извращению сна и бодрствования, хлопающему тремору и другим нарушениям.
Нарушения рецепции сигналов
Даже при адекватной сигнализации клетка не в состоянии ответить должным образом при отсутствии или дефиците рецепторов, соответствующих какому-либо биорегулятору. Избыточная активность (чувствительность) тех или
иных рецепторов также способна привести к патологическим последствиям.
Так,например, патогенез семейной наследственной гиперхолестеринемии,
этого распространенного и тяжелого нарушения связан с дефектом белкарецептора, ответственного за распознавание клетками сосудистой стенки и некоторых других тканей и органов белкового компонента липопротеидов низкой
и очень низкой плотности — апопротеина В. Вследствие этого формируется
атеросклеротическое поражение.
Избыточная активность тех или иных рецепторов также патогенна.
При развитии инсульта гибель нейронов от гипоксии сопряжена с избыточной стимуляцией рецепторов глютаминовой кислоты. Адаптационный
смысл этого состоит в усилении генерации окиси азота (N0) — сильного сосудорасширяющего медиатора, регулятора адгезии нейтрофилов и активности
циклооксигеназы, повышающего резистентность нейронов к свободно-радикальному повреждению. Однако, рецепторы, находясь в состоянии длительной
126
активации, обуславливают избыточный входной ток кальция в клетку, и это ведет к повреждению и некробиозу нейронов.
Наследственный дефект рецепторов к антидиуретическому гормону обуславливает развитие периферического нефрогенного несахарного диабета.
Нарушения функционирования пострецепторных посредников
Быстрый, непосредственный ответ клеток на химические сигналы, который часто играет решающую роль в адаптации, для биорегуляторов с любой
химической структурой опосредован поверхностными рецепторами и пострецепторными передаточными механизмами.
Дефекты в функционировании этих внутриклеточных посредников приводят к разнообразным повреждениям клеток.
Большинство химических сигналов в клетке опосредуется с участием так
называемых гуанозинтрифосфатсвязывающих белков (или G-белков). Эти передатчики занимают ключевое положение в обмене информацией между поверхностным и регуляторным аппаратом клеток, потому что они интегрируют
сигналы, воспринимаемые несколькими различными рецепторами, и в ответ на
определенный рецепторно-опосредованный сигнал могут включать множество
разных эффекторных программ, вводя в действие сеть различных внутриклеточных модуляторов.
Среди этих взаимодействующих эффекторов главные роли играют аденилатциклазы, ионные каналы и фосфолипазы.
Передавая рецепторный сигнал внутрь клетки, G-белки усиливают активность аденилатциклаз, образующаяся цикло-АМФ может напрямую открывать
ионные натриевые каналы, а также запускает каталитическую субъединицу
протеинкиназ А. Протеинкиназы А расщепляют АТФ и фосфорилируют по
остаткам серина или треонина различные каталитические и распознающие белки клетки. Этот процесс для одних белков приводит к активации, для других к
ингибированию. Ингибирующие сигналы запускаются ц-АМФ и при посредстве кальция и внутриклеточного кальцийсвязывающего белка кальмодулина.
Ряд G-белков способны активировать семейство ферментов-инозитолфосфатаз, условно названных «фосфолипаза С». Этот эффект приводит к расщеплению фосфатидилинозитол-4,5-дифосфата на инозитолтрифосфат (ИТФ) и диацилглицерол (ДАГ).ИТФ вызывает освобождение кальция из внутриклеточных резервуаров в цитоплазму. Кальций влияет на активность различных клеточных белков через кальмодулин и тропонин С, а также непосредственно.
ДАГ действует, как вторичный посредник в активации протеинкиназ С,
повышая их чувствительность к кальциевой стимуляции. Протеинкиназы класса С фосфорилируют ряд белков, влияя на их активность. Эта сеть имеет общие
субстраты фосфорилирования с протеинкиназами А. ДАГ-липаза расщепляет
данный посредник и терминирует передачу сигнала, но при этом образуется
арахидоновая кислота.
Кроме фосфолипазы С и ДАГ-липазы, продукция арахидоновой кислоты
из материала плазматической мембраны осуществляется, практически во всех
127
ядерных клетках организма и в ряде безъядерных постклеточных структур
(кроме эритроцитов), с помощью семейства кальцийзависимых изоферментов,
традиционно обозначаемого как фосфолипаза А2.
Образуемые из арахидоновой кислоты эйкозаноиды являются как внутриклеточными посредниками сигналов (например, влияя на проницаемость ионных каналов клеток), так и координаторами совместного ответа соседних клеток на повреждение.
Пострецепторные поломы лежат в основе многих заболеваний в , которые
издавна известны врачам.Так, понос и водно-электролитные нарушения при холере вызваны действием холерного токсина. Данный экзотоксин вызывает
АДФ-рибозилирование а-субъединиц G -белка в клетках кишечного эпителия,
что ведет к его активации и избыточной продукции цикло-АМФ. В результате
избыточной продукции клеткой цикло-АМФ начинается экскреция воды и
электролитов в просвет кишечника.
При коклюше экзотоксины бордетеллы стойко связывают а-субъединицы
Gi/Go белков в бронхах, а также действуют сами в качестве кальмодулинзависимой аденилатциклазы. Вследствие этого, содержание цикло-АМФ в клетках
растет, что обусловливает ряд симптомов, включая снижение бактерицидной
активности лейкоцитов и кашель.
Дефекты клеточных программ
При многих болезнях клеточные адаптационные программы нормально
востребуются информационными системами клетки, однако, сама программа
содержит технические или технологические дефекты. Из-за этого она либо не
реализуется, либо дает неадекватный или несоответствующий ситуации результат.
Кратко остановимся на патологических процессах, порождаемых дефектами клеточных программ или мутациями.
Под наследственными понимают заболевания с первичными техническими дефектами в программном аппарате клеток, передаваемые по наследству через гаметы. Конечно, только мутации половых клеток передаются из поколения
в поколение.
Соматические мутации искажают запись генетической программы в неполовых клетках и, в результате митоза, передаются всему потомству данной
клетки-то есть клону. Соматические мутации, хотя и не передаются потомству
организма, имеют большое медицинское значение, так как порождают клональные заболевания. При клональных болезнях в организме живет самоподдерживающаяся линия клеток с ненормальной программой.
К клональным относятся доброкачественные и злокачественные опухоли,
клональную природу имеют лейкозы и другие гемобластозы, а также некоторые
болезни тех тканей и органов, где клеточный пул быстро обновляется (например, пароксизмальная ночная гемоглобинурия, макроглобулинемия Вальденстрема и другие болезни крови и иммунной системы).
Некоторые приобретенные до рождения заболевания похожи по клинической картине на наследственные врожденные синдромы. Такие болезни
128
именуются фенокопиями. Так, патологическая желтуха у новорожденного
может быть обусловлена наследственными нарушениями захвата, конъюгации
или экскреции билирубина (например, синдромами Жильбера, КриглераНаджара,Дабина-Джонсона и Ротора), но может развиться и как фенокопия
вследствие внутриутробного гепатита или ненаследственной атрезии желчных
ходов.
ПОВРЕЖДЕНИЕ ИСПОЛНИТЕЛЬНОГО АППАРАТА КЛЕТКИ
Все многообразные защитно-компенсаторные реакции клетки в ответ
на ее повреждение можно условно разделить на 2 группы:
1) направленные на восстановление нарушенного внутриклеточного гомеостаза;
2) направленные на создание функционального покоя поврежденной клетки.
Первая группа включает в себя активацию механизмов активного транспорта ионов, репаративный синтез поврежденных компонентов клетки, усиленную регенерацию антиоксидантных систем и др. Непременным условием
реализации этих механизмов является достаточное энергетическое обеспечение
клетки. Это достигается, с одной стороны, повышением интенсивности энергетического обмена (активация гликолиза, клеточного дыхания, пентозного
цикла), а с другой - перераспределением имеющихся в клетке энергетических ресурсов.
Вторая группа реакций направлена на то, чтобы устранить возможные дополнительные сдвиги внутриклеточного гомеостаза при действии физиологических нервных и гуморальных возмущающих факторов (стабилизация повреждения)
и свести к минимуму энергетические траты на выполнение специфических функций клетки, обеспечив таким образом энергетические ресурсы для восстановления
нарушенного гомеостаза
Можно выделить 6 групп молекулярных механизмов, имеющих важное значение в патогенезе повреждения клетки: липидные, кальциевые, электролитно-осмотические, ацидотические, протеиновые и нуклеиновые.
1.Липидные механизмы повреждения клетки включают в себя:
- перекисное окисление липидов;
- активацию мембранных фосфолипаз;
- детергентное действие свободных жирных кислот.
Перекисным окислением липидов (ПОЛ) называется свободнорадикальное
окисление ненасыщенных жирных кислот, входящих в состав фосфолипидов
клеточных мембран. Инициаторами ПОЛ являются свободные радикалы, среди
которых наибольшее значение имеют активные кислородсодержащие радикалы
(АКР): супероксидный анион-радикал, гидроксильный радикал, водородный
радикал, синглетный (возбужденный) кислород, у которого один из электронов перешел на более высокий энергетический уровень.
В процессе повреждения клетки возможны 2 механизма активации ПОЛ.
Первый механизм — избыточное образование первичных свободных радикалов. В такой ситуации имеющиеся в клетке антиоксидантные системы не
в состоянии «потушить» реакции ПОЛ. По данному механизму происходит ак-
129
тивация ПОЛ в случае повреждаюшего воздействия на клетку ультрафиолетовых лучей, ионизирующей радиации,гипероксии,четыреххлористого углерода; в условиях сильного стресса (образование свободных радикалов из
катехоламинов); при гипервитаминозе Д (образование свободных радикалов в результате процессов аутоокисления эргокальциферола).
Второй механизм активации ПОЛ — нарушение функционирования
антиоксидантных систем клетки. В этом случае инициаторами ПОЛ являются первичные свободные радикалы, образующиеся в процессе естественно протекающего обмена вешеств.
Антиоксиднты — это молекулы, обладающие лабильным водородным
атомом с неспаренным электроном:
LOO. + АН => LOOH + А.
А.+ А. => А-А
где АН — антиоксидант, А-А его стабильный несвободнорадикальный продукт.
Множество антиоксидантов, вырабатываемых клетками и поглощаемых извне в
качестве полностью, либо частично незаменимых соединений сдерживают клеточное «атомное оружие», препятствуя длительному существованию высоких
концентраций АКР. Антиоксиданты — не просто набор веществ. Они способны восстанавливать друг друга и представляют собой антиоксидантные системы клеток.
Обычно выделяют три класса антиоксидантов :
Каталаза и глютатионпероксидаза — это энзимы предупредительного действия поскольку они восстанавливают АКР (перекись водорода), провоцирующую цепной свободно-радикальный процесс, до неактивного состояния.
Супероксиддисмутаза — фермент-прерыватель цепной реакции. Она превращает при наличии восстановительных эквивалентов, супероксидный анион,
способный, формировать наиболее активные АКР, в менее активную перекись
водорода, разрушаемую каталазой. Субстратами — прерывателями цепной реакции служат фенолы (например, токоферол) и амины (например, цистамин).
Третья разновидность антиоксидантов — хелатирующие агенты, способные
связывать железо и другие металлы-катализаторы и разветвители цепных свободнорадикальных реакций (например, десферол и унитиол)
Все упомянутые энзимы и их изоэнзимы являются металлоферментами. В
состав их активных центров входят микроэлементы.|Глутатионпероксидаза и
фосфолипид-глутатионпероксидаза - селеносодержащие ферменты. Различные
тканевые изоферменты супероксиддисмутазы содержат цинк, или марганец и
медь. Митохондриальная изоформа использует марганец, а цитозольные —
цинк и медь.Калалаза является пероксисомальным железо-зависимым металлоферментом.
Главные антиоксидантные субстраты клеток — это тиоловые соединения.К ним относятся глютатион, циствин, Д-пеницилламин. Глютатион —
важнейший компонент антиоксидантных систем печени, сердца, мозга, легких
и клеток крови.Глютатион обладает радиопротекторными свойствами.
Другая группа веществ, используемых клетками нашего организма для
защиты от окислительного стресса — это витамины.Особенно тесная взаимо-
130
зависимость существует между селеном и витамином Е, которые оба служат
для инактивации липоперекисей. Витамин Е является сильнейшим антиоксидантом, так как ловит свободный электрон и не участвует в дальнейшей цепи.
Протективное действие токоферола особенно выражено в отношении клеточных мембран. Активность токоферолов восстанавливается витамином С, как и
активность системы глютатиона.
Таким образом, в системе глутатиона взаимодействуют витамины, микроэлементы и серосодержащие аминокислоты. Упомянутые витамины и микроэлементы,а также полифенолы (биофлавоноиды),липоевая кислота и каротин
действуют в комплексе и составляют антиокислительный резерв клеток, определяющий их резистентность к свободно радикальному повреждению.
Многие пищевые продукты содержат значительные количества этих ингредиентов и способны насыщать ими организм.Поэтому, питание, оптимально
обеспечивающее потребности клеток в антиоксидантах, является в настоящее
время предметом наиболее интенсивных разработок в диетологии.
Особую роль в антиоксидантной защите играет трансферрин — отрицательный глобулин острой фазы, содержание которого в крови при воспалениях
и инфекциях снижается. Он захватывает трехвалентное железо и может переносить его в клетки.
Антиоксидантная недостаточность может быть обусловлена
наследственными и приобретенными нарушениями синтеза антиоксидантных ферментов (супероксиддисмутазы, каталазы, глутатионпероксидазы, глутатионредуктазы); дефицитом железа,меди,селена,необходимых для
функционирования этих ферментов; гиповитаминозами Е,С; нарушениями
пентозного цикла и цикла Кребса ,в реакциях которых образуются НАДФН
и НАДН,обеспечивающие восстановление истинных и вспомогательных антиоксидантов и, наконец, действием детергентов, вследствие чего нарушается строение липидного бислоя мембран и открывается доступ свободных
радикалов к обычно скрытым в гидрофобном слое ненасыщенным жирным
кислотам. Независимо от механизма активации ПОЛ в клетке развиваются
тяжелые изменения, связанные с нарушениями барьерной и матричной
функций клеточных мембран.
2. Активация мембранных фосфолипаз. В патогенезе повреждения
клетки важное значение имеет чрезмерная активация фосфолипазы А2 фермента, осуществляющего гидролитическое отщепление ненасыщенной
жирной кислоты - одного из двух гидрофобных хвостов молекулы фосфолипида.Освободившиеся под действием фосфолипазы А2 ненасыщенные
жирные кислоты (арахидоновая, пентаноевая и др.) расходуются на образование физиологически активных соединений - простагландинов и лейкотриенов.
Оставшаяся часть молекулы фосфолипида (лизофосфолипид) имеет
лишь один жирнокислотный «хвост», вследствие чего обладает способностью к мицеллообразованию и является очень сильным детергентом. С детергентным действием лизофосфолипидов и связано повреждение клеточных мембран в условиях чрезмерной активации фосфолипазы А2. Основ-
131
ным фактором, вызывающим такую активацию, является высокая концентрация ионов Са2+ в цитоплазме клетки.
3. Детергептное действие избытка свободных жирных кислот.
Свободные жирные кислоты в больших концентрациях, так же как и
лизофосфолипиды, оказывают детергентное действие и вызывают нарушение липидного бислоя мембран. Можно выделить четыре основных механизма повышения содержания свободных жирных кислот в клетке:
I) усиленное поступление свободных жирных кислотв клетку при гиперлипоацидемии (повышении концентрации свободных жирных кислот
в крови), что наблюдается при активациилиполиза в жировой ткани, в
частности, при стрессе, сахарном диабете;
2) усиленное освобождение свободных жирных кислот в лизосомах из
триглицеридной части липопротеидов, поступающих в клетку, что имеет
место в условиях гиперлипопротеинемий, сопровождающих развитие атеросклероза;
3) усиленное освобождение свободных жирных кислот из фосфолипидов
мембран под действием уже упоминавшихся мембранных фосфолипаз;
4) нарушение использования клеткой свободных жирных кислот в качестве источника энергии, что отмечается при уменьшении активности ферментов бетта-окисления и цикла Кребса, а также при гипоксии.
Для того чтобы предотвратить повреждающее действие избытка жирных
кислот, клетка располагает системой ферментов, которые переводят свободные жирные кислоты в триглицериды. При этом наблюдается не свойственное в норме отложение последних в клетке в виде жировых капель, т.е. возникает жировая дистрофия клетки.
Описанные выше липидные механизмы повреждения приводят к
нарушению двух основных функций липидного бислоя клеточных мембран: барьерной и матричной. В основе нарушения барьерной функции
мембран лежат два основных механизма: ионофорный и механизм электрического пробоя.
Первый из них обусловлен появлением в клетке веществ, обладающих свойствами ионофоров, т.е. соединений, способных облегчать
диффузию ионов через мембрану благодаря образованию проходимых
через ее слои комплексов иона и ионофора. В процессе активации перскисного окисления липидов среди промежуточных продуктов его реакций появляются вещества - ионофоры по отношению к ионам кальция и водорода, в результате чего повышается проницаемость клеточных мембран для указанных ионов.
Второй механизм («самопробой») реализуется за счет существующей на многих мембранах (плазматической, внутренней митохондриальной) разности потенциалов. В результате появления гидрофильных продуктов перекисного окисления липидов, а также вследствие детергентного действия лизофосфолипидов и избытка свободных жирных кислот
нарушаются электроизолирующие свойства гидрофобного слоя клеточных мембран, уменьшается их электрическая стабильность, что приводит
132
к электрическому пробою мембраны, т.е. к электромеханическому ее
разрыву с образованием новых трансмембранных каналов ионной проводимости.
Сущность матричной функции липидного бислоя мембран состоит в
том, что в нем вмонтированы мембранные ферменты и некоторые специализированные белки. В процессе перекисного окисления липидов нарушается активность мембранных ферментов в связи с изменением их липидного микроокружения, во многом определяющего свойства белковых
молекул. Кроме того, в ходе реакций ПОЛ может произойти образование
«сшивок» между молекулами белков и фосфолипидов, а также окисление
сульфгидрильных групп активных центров, что приводит к необратимой
инактивации ферментов.
Кальциевые механизмы. Целый ряд важных патогенетических механизмов
повреждения клетки обусловлен повышением концентрации ионов кальция в ее
цитоплазме. В основе такого повышения могут лежать 2 механизма:
- избыточное поступление ионов Са2+ в цитоплазму;
- нарушение удаления ионов Са2+ из цитоплазмы.
Избыточное поступление ионизированного кальция в цитоплазму может
осуществляться через неповрежденную плазматическую мембрану в случае повышения градиента его концентрации, например при гиперкальциемии. Однако
гораздо чаще поступление кальция в цитоплазму усиливается в результате
нарушения барьерной функции мембран, как это имеет место в условиях активации уже рассмотренных липидных механизмов повреждения клетки.
Удаление ионов Са2+
из цитоплазмы нарушается вследствие недостаточности трех основных кальцийтранспортируюших систем клетки:
1) Са2+ -насосов плазматической мембраны и эндоплазматическо-го ретикулума;
2) Nа+-Са2+ -обменного механизма;
3) Са2+ -аккумулируюшей функции митохондрий.
Нарушение функционирования Са2+ -насосов может быть связано с наследственно обусловленными и приобретенными дефектами белковых компонентов
Са2+ -насосов, а также с уменьшением в клетке концентрации АТФ, необходимой
для осуществления процессов активного транспорта. Дефицит АТФ в клетке, в
свою очередь, закономерно возникает в условиях нарушения энергетического обмена: при недостаточности энергетических источников в клетке, гипоксии,
уменьшении активности ферментов гликолиза и цикла Кребса, угнетении процессов клеточного дыхания и окислительного фосфорилирования. Nа+-Са2+ обменный механизм удаления ионизированного кальция из цитоплазмы обеспечивается энергией градиента концентраций ионов Na+ по обе стороны плазматической мембраны. Поэтому основной причиной нарушения Na+- Са2+-обмена является уменьшение указанного градиента, что происходит в условиях нарушения
функции Nа+-К+-насоса, создающего этот градиент.
Са2+ -аккумулирующая функция митохондрий является одним из альтернативных путей использования энергии транспорта электронов по дыхательной цепи,
когда освобождающаяся энергия идет не на синтез АТФ, а на транспорт ионов Са2+
133
из цитоплазмы в митохондрии против концентрационного градиента. С учетом
этого Са2+ -аккумулирующая функция митохондрий угнетается во всех случаях
нарушения процессов транспорта электронов по дыхательной цепи.
Стойкое повышение содержания ионов Са2+ в цитоплазме вызывает ряд важных последствий:
1) нарушение специфических функций клетки, в осуществлении которых принимают участие ионы Са2+ ; примером является развитие контрактуры миофибрилл мышечных клеток. При этом утрачивается способность таких клеток к расслаблению, а пересокращенные миофибриллы подвергаются разрушению под
действием активированных избытком кальция протеолитичсских ферментов;
2) активация фосфолипазы А2 (см. выше);
3) разобщение окисления и фосфорилирования.
В условиях повышения концентрации ионов Са2+ в цитоплазме данный
эффект возникает в результате использования энергии клеточного дыхания не
на синтез АТФ, а на транспорт кальция из цитоплазмы в митохондрии. Кроме
того, важное значение имеет повышение проницаемости внутренней митохондриальной мембраны под влиянием фосфолипазы А2, активированной избытком ионов кальция.
Электролитно-осмотические механизмы. Электролитно-осмотические
механизмы повреждения клетки обусловлены сдвигами в содержании главных
клеточных катионов: Na+ и К+.Выравнивание концентраций этих ионов по обе
стороны плазматической мембраны приводит к увеличению внутриклеточной
концентрации ионов Na+ и уменьшению концентрации ионов К+ в клетке. В
основе указанных сдвигов могут лежать два механизма, обеспечивающих поддержание концентрационных градиентов указанных ионов:
1) усиленная диффузия ионов через плазматическую мембрану;
2) нарушение механизмов активного транспорта Na+ и К+.
Усиление диффузии ионов Na+ в клетку и выход ионов К+ из клетки могут происходить как через неповрежденную плазматическую мембрану в условиях общих нарушений водно-электролитного обмена в организме (гипернатриемия, гипокалиемия), так и при нарушении барьерной функции плазматической мембраны. Перемещение ионов Na+ и К+ в этих случаях осуществляется через имеющиеся и вновь образовавшиеся каналы ионной проводимости
за счет существующих концентрационного и электрического градиентов.
Основу нарушений активного транспорта ионов Na+ и К+через плазматическую мембрану составляет недостаточность Na+ - К+-насосов. Главной причиной нарушений работы этих механизмов является дефицит АТФ, за счет
энергии которой достигается перемещение ионов Na+ и К+ против электрохимического градиента. Поскольку основным источником АТФ для Na+ — К+насосов является гликолиз, то нарушение этого процесса при недостаточном
поступлении глюкозы в клетку или уменьшении активности соответствующих
ферментов будет приводить к рассматриваемым здесь электролитным сдвигам.
Причиной нарушения функции Na+ — К+-насосов может быть также изменение
свойств липидного бислоя наружной клеточной мембраны и, в частности,
увеличение содержания в нем холестерина, что наблюдается при атероскле-
134
розе. Угнетение работы Na+ — К+-насосов вызывается и целой группой специфических ингибиторов Na+ — К+-АТФазы (строфантин, оубаин и др.).
Сдвиги электролитного состава клетки в процессе се повреждения проявляются развитием ряда изменений, среди которых наиболее важными являются:
I) потеря клеткой электрического мембранного потенциала (потенциала покоя);
2) отек клетки и
3) осмотическое растяжение мембран, приводящее к нарушению их барьерной функции.
Ацидотические механизмы. В основе этой группы механизмов повреждения лежит увеличение концентрации ионов водорода в клетке, т.е. внутриклеточный ацидоз.
Развитие внутриклеточного ацидоза может быть обусловлено следующими
механизмами:
1) избыточным поступлением ионов водорода в клетку из внеклеточной среды,
что наблюдается в условиях общих нарушений кислотно-основного гомеостаза в
организме — при декомпенсированных газовом и негазовом ацидозе;
2) избыточным образованием кислых продуктов в самой клетке, что отмечается
при активации гликолиза (молочная кислота), нарушениях цикла Кребса (три- и
дикарбоновые кислоты), гидролитическом расщеплении фосфолипидов клеточных
мембран (жирные кислоты, фосфорная кислота), усиленном распаде свободных
нуклеотидов (фосфорная кислота);
3) нарушением связывания ионов водорода в результате недостаточности буферных систем клетки;
4) нарушением выведения ионов водорода из клетки при недостаточности Na+
—Н+-обменного механизма цитоплазматической мембраны, а также в условиях
расстройства местного кровообращения в тканях.
Повышение внутриклеточной концентрации ионов водорода приводит к
развитию ряда изменений:
1) нарушению функциональных свойств белков (ферментов, сократительных и
др.) в результате изменений конформации их молекул;
2) активации лизосомальных гидролитических ферментов;
3) повышению проницаемости клеточных мембран вследствие изменения жидкостного состояния мембранных липидов.
Протеиновые механизмы включают в себя:
1) ингибирование ферментов (обратимое и необратимое);
2) денатурацию, т.е. нарушение нативного строения белковых молекул в результате изменений вторичной и третичной структуры белка, обусловленных разрывом
нековалентных связей;
3) протеолиз, осуществляющийся под действием лизосомальных гидролитических ферментов (катепсинов) и Са2+ -активируемых протеаз.
Основу нуклеиновых механизмов повреждения клеток составляют нарушения 3
процессов: репликации ДНК, транскрипции и трансляции.
135
На субклеточном уровне реализация рассмотренных выше молекулярных
механизмов повреждения клетки приводит к нарушению строения и функции
отдельных ее органелл.
Повреждения клеточного ядра
Повреждение ядерной ДНК вызывает несколько типовых защитных реакций.В поврежденных клеточных ядрах происходит включение целого ряда аварийных генетических программ, считывание которых в нормальных условиях
отсутствует или минимально.
К ним относятся:
-Гены белков теплового шока (БТШ);
-Немедленные гены предраннего ответа;
-Антионкогены;
-Гены-регуляторы программированой клеточной гибели;
-Ген маркера стареющих и поврежденных клеток (АСК).
Работа каждой из этих генетических систем сочетает защитные и вторичные повреждающие эффекты.
Белки теплового шока
Белки теплового шока (БТШ) — многофункциональные клеточные регуляторы.
Впервые они были обнаружены в клетках дрозофил, подвергавшихся тепловому воздействию.Экспрессия данных белков специфична не только для тепловой травмы, она может быть индуцирована различными воздействиями (воспаление, инфекция, гипоксия, химические повреждения клеток тяжелыми металлами, мочевиной, перекисью водорода, мышьяком и этиловым спиртом).
Вместе с тем, повышенная экспрессия БТШ увеличивает и термпературную устойчивость клеток.
Считается, что БТШ могут представлять собой систему белковых клеточных детергентов и регуляторов ограниченного протеолиза. В этом качестве они
способны поддерживать нативную конформацию синтезируемых в клетке белков, предохранять их от денатурации и солюбилизировать белки, которые при
клеточном стрессе утрачивают свою растворимость, либо способствовать протеолизу денатурированных белков.
БТШ синтезируются в небольших количествах и в норме и могут, в частности, принимать участие в регуляции клеточной пролиферации, обеспечивая
конденсацию хромосом при митозе.
БТШ — элементы наиболее древних, неспецифических механизмов клеточной реактивности. БТШ способны оказывать цитопротекторное действие
при повреждении, предохраняя клетку и от апоптоза и, в определенной степени,
на ранних стадиях (до развития тяжелой гипоксии)— также и от некроза.
Работа системы БТШ способна как предохранять клетки от преждевременной запрограммированной гибели (БТШ-70), так и индуцировать ее (убиквитины). Повреждение способно индуцировать программы, устраняющие сами
поврежденные клетки.
136
Немедленные гены предранней реакции (НГПР)
При повреждении клеток различных органов и тканей, независимо от
причин этого повреждения, происходит очень быстраят неспецифическая активация ряда генов, которые были условно объединены под названием «немедленные гены предранней реакции».
Считается, что НГПР способны активировать клеточную пролиферацию,
в том случае, если их считывание в клетках происходит на фоне достаточного
количества ростовых сигналов.Таким образом, работа НГПР может рассматриваться, как подготовка к репаративным процессам при повреждении тканей.
Вместе с тем, продукты тех же самых НГПР могут запустить процесс запрограммированной клеточной гибели, если ростостимулирующий фон недостаточен.
Антионкогены
Продукты антионкогенов это регуляторы генной стабильности, останавливающие митотический цикл в мутировавших клетках . Остановка цикла дает
мутантной клетке время для срабатывания репаразных механизмов. Если мутация не репарируется, продолжение экспрессии антионкогенов ведет к запуску
программы апоптоза клеток с поврежденной ДНК.
Активация генов, контролирующих антигены- маркеры стареющих
и повреждённых клеток.
В начале 80-х годов Маргарет Кей и соавторы обнаружили, что один из
белков поверхностной мембраны красных кровяных телец, движущийся при
электрофорезе в составе третьей полосы и выполняющий функции ионного канала, практически отсутствует или скрыт поверхностными гликопротеидами
мембраны в молодых клетках и, наоборот, активно экспрессируется в мембране
старых и поврежденных эритроцитов.
Впоследствии оказалось, что данная молекула является маркером, появляющимся на мембране после истечения онтогенетически отпущенного эритроцитам срока в 120 дней — то есть, опознавательным знаком стареющих клеток.
Таким образом, маркер или антиген стареющих клеток оказался частью
фундаментального механизма запрограммированного устранения из организма
тех клеточных элементов, которые исчерпали вследствие изнашивания или повреждений свой генетический ресурс.
Повреждение плазмолеммы
Поскольку большинство органелл относится к мембранным образованиям, то универсальным механизмом повреждения субклеточных структур является нарушение проницаемости и целостности клеточных мембран. Можно выделить 5 основных механизмов повреждения мембран: 1) перекисное
окисление липидов; 2) активация фосфолипаз; 3) осмотическое растяжение мембран; 4) адсорбция белков на мембране (например, комплексов
137
антиген-антитело); 5) изменение фазового состояния мембранных липидов (ацидоз, изменения температуры).
При повреждении создается цепь последствий первичной альтерации
плазмолеммы, которая включает:
1. Недостаточность натрий-калиевого насоса и функций ионных каналов;
2. Утрату физиологических трансмембранных ионных градиентов;
3. Избыточный входной ток натрия и воды в клетку;
4. Набухание клетки;
5. Избыточный входной ток кальция в клетку;
6. Активацию мембранных фосфолипаз;
7. Освобождение и превращения арахидоновой кислоты;
8. Нарушение локальной микроциркуляции;
9. Появление вокруг клетки липидных медиаторов воспаления.
Повреждение цитоплазматическоп мембраны может проявляться:
- нарушениями ее барьерной функции;
- расстройствами систем активного транспорта веществ (Na+ — К+- и
Са2+ -насосов, Na+ — Са2+ -и Na+ -H+-обменных механизмов и др.);
- изменениями белков, образующих специфические каналы ионной проводимости; повреждением рецепторных макромолекул, воспринимающих внешние регуляторные сигналы;
- нарушениями белковых комплексов, осуществляющих межклеточные
взаимодействия;
-изменениями гликопротеидов, определяющих антигенность клетки.
Повреждение цитоскелета
Цитоскелет — это система микротрубочек (диаметром 20-25 нм), промежуточных филаментов (диаметром 15 нм), а также тонких актиновых (6-8 нм) и
толстых миозиновых (15 нм) филаментов.Элементы цитоскелета способны к
самосборке и обратимой полимеризации и состоят из глобулярных и фибриллярных белков: тубулина, динеина и динамина (микротрубочки), актина, миозина (микрофиламенты), виментина, кератинов, десмина и других (промежуточные филаменты).
Цитоскелет ответственен за поддержание формы клеток и за все способы
их движения (деятельность ресничек, жгутиков, ундулирующих мембран и
псевдоподий). Он же обеспечивает внутриклеточное перемещение органоидов
и включений (например, гранул при дегрануляции тучных клеток или хромосом
при митозе). Прикрепление клеток к межклеточному веществу и друг к другу,
передача сигнала от рецепторов плазматической мембраны внутрь клетки также
проходит при участии этой внутриклеточной опорно-двигательной системы.
Фагоцитоз, пиноцитоз, хемотаксис также всецело связаны с функцией
цитоскелета.
При энергодефиците в клетке работа цитоскелета существенно нарушается. Например, сахарный диабет сопровождается «синдромом ленивых фагоцитов», при котором замедлен хемотаксис и снижена фагоцитарная активность
этих клеток.
138
При вирусном поражении клеток зачастую вирусы взаимодействуют
именно со структурами цитоскелета.Так как вирусы содержат специфические
рецепторы, распознающие белки цитоскелета, иммунный ответ против вирусных антигенов может приводить к появлению аутоантител, копирующих способность вируса связывать элементы цитоскелета. Поэтому многие вирусиндуцированные процессы продолжаются как аутоиммунные и сопровождаются появлением антител, поражающих цитоскелет.Характерным примером служит
агрессивный хронический гепатит. Данное аутоиммунное заболевание провоцируется вирусной инфекцией, в частности, вирусом гепатита С. Однако, его
персистирующее волнообразное течение обусловлено появлением аутоантител
против белков цитоскелета — актина и кератина.
Аутоантитела к элементам цитоскелета сперматозоидов вызывают снижение их подвижности и могут быть причиной бесплодия. Показано, что они
могут присутствовать в цервикальной слизи у женщин при бесплодном браке.
Существуют токсины, избирательно поражающие цитоскелет. Цитохалазины вызывают деполимеризацию, а токсин бледной поганки фаллоидин —
стойкую полимеризацию актина. Колхицин блокирует полимеризацию, а такбол — деполимеризацию микротрубочек.
Важным аспектом повреждения цитоскелета считается его трансформация в злокачественных клетках, под влиянием онкобелков. Один из онкобелков,
вырабатываемых злокачественными клетками, способен вызывать необратимое
фосфорилирование цитоскелетного белка винкулина, участвующего в прикреплении клеток к межклеточному веществу. Из-за этого злокачественные
клетки легко отсоединяются от межклеточного вещества и покидают свои места. Это считается важным механизмом, составляющим основу их способности
расселяться по организму —метастазировать.
Повреждения внутриклеточных мембран
Большая часть внутриклеточных мембран принадлежит к эндоплазматическому реткулуму. Различные факторы, включая отравления алкоголем, ДДТ,
четыреххлористым углеродом, инфекцию, ионизирующее излучение, гипоксию
вызывают набухание ретикулума, изменение его конфигурации появление
крупных вакуолей и петель или распад на мелкие гранулы.При повреждении
клеток в результате действия многих различных причин наступает такое типовое последствие, как отсоединение рибосом от мембран шероховатого эндоплазматического ретикулума ( ШЭР).Транспорт вновь образованных белков нарушается и избыток их оказывается в цитоплазме, где они денатурируются по
мере нарастания клеточной гипоксии и ацидоза и формируют картину «мутного набухания» или начальные стадии так называемой «зернистой дистрофии».
При повреждении клетки затрагивается и гладкий эндоплазматический
ретикулум (ГЭР).Мембраны ГЭР являются отсеком клетки, содержащим систему мощных оксидаз со смешанной функцией. Эти ферменты содержат цитохром
Р450 и являются железозависимыми. Они представляют собой дезинтоксикационную систему клетки. Оксидазы со смешанной функцией принимают участие
в биотрансформации эндогенных соединений, в частности, инактивации сиг-
139
нальных молекул, например, стероидных гормонов и обезвреживании билирубина, детоксикации ксенобиотиков, например, многих лекарств.
Избыточная акгивация оксидаз ГЭР приводит к повышению продукции
клеткой окиси азота. Избыток N0 ведет к ряду неблагоприятных результатов.
При образовании окиси азота формируются, в качестве сопутствующих продуктов, свободные кислородные радикалы. Они способны причинять вторичные повреждения клеточным мембранам, нуклеиновым кислотам и другим
компонентам клеток.
Повреждение пластинчатого комплекса
Важной составной частью исполнительного аппарата клетки служит пластинчатый комплекс (комплекс Гольджи, описанный К. Гольджи в 1898 г.).
Этот органоид также представляет собой мембранную структуру, состоящую из
стопок, содержащих каждая по 5-6 уплощенных ограниченных мембранами цистерн. В комплекс Гольджи поступают из ШЭР в специальных везикулах синтезированные там белки, снабженные углеводными остатками и предназначенные для распределения внутри клетки или экспортных нужд.Ферменты комплекса подвергают белки гликозилированию и фосфорилированию.
Аппарат Гольджи поражается при ряде приобретенных и наследственных расстройств. Так как он является местом образования и наполнения лизосом, часто
его повреждение ассоциируется с лизосомальной патологией.
Повреждение лизосом и пероксисом
При повреждении клеток лизосомы участвуют в процессе аутофагии, захватывая и переваривая своими гидролазами в составе аутофагосом остатки разрушающихся органоидов.
Процесс удержания ферментов в лизосоме еще не ясен. Ведь ферменты
весьма агрессивны. Ферменты лизосом могут действовать внутри клетки, расщепляя, растворяя внутриклеточные структуры. Они в фаголизосомах разрушают чужеродные и собственные стареющие клетки, то есть лизосомы участвуют в механизме разрушения собственных отживших клеток.
Лизосомальная мембрана весьма стабильна и не повреждается даже при
наличии в лизосоме агрессивных энзимов, радикалов и кислой среды (рН в работающей лизосоме достигает 1.5-2 единиц). При обратимых повреждениях
клетки, даже если они глубоки, пероксисомы и лизосомы не дают утечки агрессивного содержимого, несмотря на то, что в этих условиях может наблюдаться их значительное набухание.
Гипоксия, ацидоз, радиация, голодание, избыток и недостаток витаминов
А, Е и Д, ряд ядов (например, эндотоксины бактерий тифо-паратифозной группы) и многие другие факторы увеличивают проницаемость лизосомальных
мембран. При глубоком необратимом повреждении, некробиотические изменения в клетке всегда приводят к разрушению лизосом, что вызывает аутолиз
клетки, неизбежный при некрозе. Выход ферментов за пределы клетки приводит к активации функциональных биохимических систем ограниченного протеолиза, липолиза и др. с образованием биологически активных веществ.
140
При некоторых наследственных энзимопатиях лизосомы неспособны переварить те или иные субстраты (гликозаминогликаны, липиды или их комплексы, гликоген и др.). Подобные болезни характеризуются как тезаурисмозы
или «болезни накопления», поскольку непереваренные субстраты образуют в
лизосомах стойкие включения.
К лизосомальным болезням относятся болезни накопления липидов и
гликолипидов (например, наследственные ганглиозидозы — болезнь Тея-Сакса
и болезнь Зандхоффа, наследственный галактоцереброзидоз — болезнь Краббе,
наследственный сфинголипидоз — болезнь Ниманна-Пика), мукополисахаридозы (например, болезни Сан-Филипио А. В, С и О — гепарансульфатозы, болезнь Моркио —кератансульфатоз, болезнь Марото-Лами — дерматансульфатоз), гликогеноз 2 типа (болезнь Помпе или дефицит кислой мальтазы).
Клинические симптомы, как правило, касаются, главным образом, тех
тканей, где в норме должен идти наиболее интенсивный лизосомальный гидролиз того или иного субстрата.Общей чертой таких заболеваний нередко является нарушение психомоторного развития и иммунитета, потому что среди загруженных субстратами клеток оказываются нейроны и макрофаги.
Поражение ЦНС наиболее характерно для липидозов и глико (муко) липидозов, так как компоненты миелина и клеточных рецепторов — цереброзиды,
сфинголипиды и их комплексы с углеводами — наиболее широко представлены
в нервной ткани.
Пероксисомы выполняют в клетке ряд важных функций, включая образование и инактивацию перекиси водорода, окисление жирных кислот до ацетилкоэнзима А, окисление мочевой кислоты.Наследственный дефект, связанный с
отсутствием пероксисом, абсолютно смертелен и приводит к гибели новорожденных через несколько месяцев при явлениях иммунодефицита и гипоксии.
Пероксисомы обеспечивают кислородзависимый бактерицидный эффект
при фагоцитозе. Во время повреждения клетки альтерация пероксисом способствует процессам образования свободных радикалов. Нарушение утилизации
жирных кислот позволяет этим субстратам формировать в цитоплазме детергенты, что способствует омылению клетки и разрушению ее мембран.
Повреждение митохондрий
При многих клеточных повреждениях окисление в митохондриях тормозится.Если прекратится окисление в цикле трикарбоновых кислот, отсутствие
возобновления протонового градиента приведет к прекращению выработки
АТФ.Если в цепи биологического окисления образуются разрывы из-за нехватки каких-либо коферментов или ингибирования энзимов — это также остановит работу митохондриального генератора. В обоих случаях возникает невозможность утилизации кислорода, несмотря на его поставку — то есть тканевая
гипоксия.
Разрывы в цепи биологического окисления могут быть вызваны авитаминозами по витаминам В, РР, Q, входящим в митохондриальные окислительно-восстановительные комплексы. К таким же последстиям приводит дефицит
микроэлементов (в первую очередь, железа и меди). Не случайно, авитаминозы
141
по витаминам-коферментам биоокисления и дефицит указанных микроэлементов имеют ряд общих симптомов, например, генерализованное поражение слизистых и кожи, эпителий которых является в норме высокоаэробной тканью.
Отравления мочевиной, фторотаном, сероводородом и сульфитами также
протекают с ингибированием тканевого дыхания. Цианистый калий — эффективно блокирует систему цитохромов, вызывая картину острой тканевой гипоксии, прежде всего, в миокарде (что вызывает синдром стенокардии) и мозге
(что проявляется потерей сознания и остановкой дыхания).
Одним из факторов, нарушающих нормальную работу митохондрий, может служить сам дефицит кислорода. Это происходит, если кислород не поступает в клетку в течение более или менее длительного времени (3-5 минут для
нейронов, 20-60 минут для миокарда или много часов — для фибробластов).
Так как нехватка кислорода ломает митохондрии, все виды гипоксии имеют парадоксальную и опасную тенденцию — переходить в тканевую форму.
При нарушении окисления в митохондриях в цитозоле клетки растет
концентрадия ацетил-КоА, увеличивается синтез и количество триглицеридов (жировая инфильтрация клетки). Ацетил-КоА при его большой концентрации ингибирует образование ацильных эфиров СЖК, то есть включение
СЖК в энергообразование.Избыток ацетил-КоА и промежуточные продукты недоокисления СЖК подавляют тканевое дыхание. Наиболее характерными проявлениями повреждения митохондрий являются эффект
разобщения окисления и фосфорилирования и угнетение клеточного
дыхания.
Основным источником АТФ при этом становится гликолитический
путь окисления глюкозы в цитозоле. Он в 18 раз менее эффективен, чем ее
митохондриальное окисление и не может в достаточной мере компенсировать дефицит макроэргов.
Активация гликолиза в последующем приводит к его торможению в результате накопления кислых продуктов гликолиза - молочной и пировиноградной кислот, ведущих к развитию внутриклеточного ацидоза. В условиях ацидоза снижается активность ряда ферментов гликолиза: фосфофруктокиназы, фосфатдегидрогеназы, гексокиназы, фосфорилазы.
Расстройства транспорта энергии.Заключённая в макроэргических связях энергия АТФ в норме доставляется от мест ресинтеза — митохондрий и цитозоля — к эффекторным структурам (миофибриллам, мембранным ионным
насосам и др.) с помощью АДФ-АТФ-транслоказы (адениннуклеотидилтрансферазы) и КФК( креатинфосфокиназа).
Адениннуклеотидилтрансфераза обеспечивает транспорт энергии макроэргической фосфатной связи АТФ из матрикса митохондрий через их внутреннюю мембрану, а КФК переносит её далее на креатин с образованием креатинфосфата, который поступает в цитозоль. КФК эффекторных клеточных структур транспортирует фосфатную группу креатинфосфата на АДФ с образованием АТФ, который и используется в процессах жизнедеятельности клетки.
Было отмечено, что при воздействиях, повреждающих энергетику клетки,
в первую очередь снижается содержание креатининфосфата. Так при 30 сек
142
ишемии миокардиоцита снижение концентрации ATФ составляет 18%, а креатининфосфата 56%. Последнее свидетельствует о важности нарушения транспорта энергии при повреждении энергетики клетки.
Нарушение энергообразования и транспорта энергии в клетке приводит к
торможению всех энергозавиcимых процессов,страдают, в частности, ферментные системы, регулирующие ионный баланс клетки: Са2+-зависимая АТФаза,
Мg2+-зависимая АТФаза,
К+- зависимая АТФаза,
Nа+- зависимая АТФаза.
Эти ферменты называют мембранными насосами, которые сдерживают
распределение ионов по градиенту концентрации, т.е. переход Са2+, Nа+ и Мg2+
в клетку и К+ из клетки, и выводят Nа+, Мg2+, Са2+ из клетки в окружающую
среду, и К+ в клетку против градиента концентрации.
При нарушении действия этих систем возникает ионный дисбаланс и гибель клетки.
Мутации ядерных и местных генов, контролирующих митохондриальные
белки, как правило, не смертельны для клеток, которые способны выживать,
переходя к анаэробному метаболизму. Однако, в тканях с высокой потребностью в кислороде (мышечная, нервная) при этом возникают клинически значимые нарушения, кроме того, вследствие усиленного гликолиза формируется метаболический ацидоз. Эти нарушения описаны у человека, как группа «митохондриальных болезней».
Характерные повреждения митохондрий при этих болезнях придают мышечным волокнам изорванный вид. Органоиды увеличены, содержат кристаллические включения и кристы искаженной формы. При специальной гистологической окраске скопления митохондрий приобретают красный цвет, вследствие чего мышечные волокна при митохондриальных болезнях описывают в
патологической литературе, как «красные изорванные волокна». Характерно
компенсаторое увеличение количества митохондрий в клетках.Как правило,
наследственные митохондриопатии выражаются в поражении головного мозга
и мышц.
Роль митохондрий в процессе повреждения клеток огромна. Их деструкция является решающим событием при смерти клетки от гипоксии.
ИНТЕГРАЛЬНЫЕ МЕХАНИЗМЫ
ПОВРЕЖДЕНИЯ И ГИБЕЛИ
КЛЕТКИ
Интактная клетка находится в неком стабильно-динамическом состоянии
гомеостаза-гомеореза, поддерживая оптимальный метаболический уровень,
соответствующий текущим требованиям среды.При стимуляции различными
сигналами клетка может перейти на новый уровень интенсивности своего
функционирования, не снижая своей жизнеспособности. При этом ее функциональные резервы уменьшаются, но остаются достаточно существенными.
Интенсивность метаболизма и функционирования клеточных структур
увеличивается. Если это не сопровождается ограничением поставки или относительной нехваткой необходимых метаболитов, то гомеорез сохраняется на
143
новом стабильно-динамическом уровне и повреждений не возникает. В общем
виде, такой ответ может быть назван клеточной адаптацией.
Включаются новые программы и происходит учащенное считывание старых. Это приводит к увеличению синтеза и сборки органоидов. Результатом является увеличение размера клетки и числа функционирующих структур в ней
— то есть адаптация в форме гипертрофии. Примером может служить увеличение массы миокарда и диаметра кардиомиоцитов при гиперфункции сердца, сопровождаемое внутриклеточным увеличением числа миофибрилл, митохондрий и других исполнительных элементов.
В ряде случаев управляющий сигнал позволяет клетке включить программу деления. И происходит адаптация в форме пролиферации клеток с увеличением их числа — гиперплазии. Так, при дефиците йода и пониженном
синтезе гормонов щитовидной железы тиреотропный гормон гипофиза, секретируемый в ответ на гипотиреоз, стимулирует пролиферацию тиреоцитов и развитие зоба .Если адаптационные возможности недостаточны или. если применяемые для адаптации и защиты программы, вследствие своего несовершенства, сами порождают вторичные нарушения, следует повреждение клетки.
Судьба клетки, претерпевающей повреждение, может быть различной.
Процессы, предшествующие гибели клетки, и представляющие собой начальные, обратимые стадии ее повреждения именуются паранекрозом. Паранекроз
рассматривают как начальный этап повреждения клетки. Некоторые изменения,
свойственные паранекрозу, например, повышение сорбционных свойств цитоплазмы, отмечаются и при адаптации клеток к действию физиологических раздражителей.
Глубокая, частично необратимая стадия повреждения клетки, непосредственно предшествующая моменту ее смерти именуется некробиозом. Некробиоз иногда отождествляется с собственно процессом гибели клетки.
Посмертные изменения необратимого характера, заключающиеся в постепенном ферментативном разрушении клетки и денатурации ее белков, называются некрозом.
Выделяют две основные разновидности некроза — коагуляционный и
колликвационный. При коагуляционном или сухом некрозе в клетке развивается выраженный ацидоз и идет коагуляция белков и выраженное накопление
кальция с агрегацией элементов цитоскелета. Очаги такого некроза легко кальцифицируются. Ядро удаляется, а протеолиз блокируется или носит ограниченный характер, касаясь предшественников воспалительных медиаторов, вокруг
возникает воспаление с выраженным отложением фибрина и фибриноидным
набуханием межклеточного вещества. Данный тип некроза — самый частый.
Он провоцируется тяжелой гипоксией в клетках миокарда при инфаркте, моделируется при действии концентрированной кислоты и высокой температуры на
ткани. Считается, что этот тип некроза характерен для богатых белком и кальцием тканей и предусматривает ранние и серьезные повреждения митохондрий.
Разновидность бактериального некроза с преобладанием коагуляции носит
название сухой гангрены.
144
При колликвационном некрозе доминируют гидролитические процессы
лизосомального аутолиза или гетеролиза при участии фагоцитов. Ткань размягчается, процессы коагуляции и фибринообразования менее выражены, отмечается значительное накопление активных гидроксильных радикалов и эндогенное омыление клеток, разрушающее мембраны.Колликвационный некроз
моделируется действием сильных щелочей, его элементы отмечаются при отморожениях. Бактериальные очаги некроза также часто дают колликвационную
картину, так как лейкоциты вызывают выраженные цитолитические процессы.
Очаги колликвационного некроза являются основой для формирования кист и
пустот. Разновидностью бактериального некроза с преобладанием колликвационного процесса является влажная гангрена.
Творожистый (казеозный) некроз при туберкулезе некоторые авторы считают комбинацией обоих типов некроза.
Все случаи клеточной гибели могут быть отнесены к одной из двух основных категорий, обозначаемых как насильственная и естественная (программируемая) клеточная гибель(апоптоз).
Насильственная гибель возникает как следствие лишения клеток источников питания и кислорода или необратимого подавления важнейших метаболических путей химическими и физическими факторами. Принципиально важно различать гипотоксическое повреждение клетки, механизм которого запускается любыми воздействиями, вызывающими более или менее продолжительное кислородное голодание и свободно-радикальное повреждение клетки,
при котором она может подвергаться разрушению без первичной гипоксии,
иногда даже в условиях избытка кислорода.
Указанные механизмы могут комбинироваться и взаимопроникать — так
например, реперфузия или восстановление кровоснабжения ишемизированного
участка миокарда сопровождается продукцией активных кислородных радикалов в поврежденных на стадии ишемической гипоксии митохондриях. Эти радикалы вызывают деструкцию клеточных мембран и вносят вклад в некробиотические процессы, спровоцированные гипоксией.
При остром отравлении кислородом основным механизмом повреждения
считается действие активных кислородных радикалов на мембраны. Однако, в
финальной стадии повреждения деструкция митохондрий приводит к невозможности утилизовать кислород и развитию сопутствующей глубокой тканевой
гипоксии.
Физиологическая (программируемая) гибель интактной клетки имеет более сложную природу, так как она реализуется путем индукции генной активности и синтеза специфических белков (факторов), способных выступать в качестве триггеров клеточной гибели.
Механизмы апоптоза
Этот вид клеточной гибели представляет собой важнейший интегральный
компонент эмбриогенеза, морфогенеза и роста тканей, а также гормонозависимой инволюции. Он, наряду с лизосомальной аутофагией участвует в механизмах таких клеточных адаптаций, как атрофия (уменьшение размеров клеток и
145
числа функционирующих структур в них при сохранении жизнеспособности
клетки) и гипоплазия (уменьшение органа вследствие уменьшения числа клеток в нем при сохранении его жизнеспособности).
Для обозначения процесса запрограммированной клеточной гибели, морфологически и патохимически отличного от некробиоза, Н. Уолкер в 1968 году
предложил термин «апоптоз».
Выделяют следующие основные процессы, при которых доминирует гибель по типу апоптоза:
-Устранение клеток в раннем онтогенезе;
-Физиологическая инволюция и уравновешивание митозов в зрелых тканях и
клеточных популяциях;
-Реализация процессов атрофии и регрессия гиперплазии;
-Альтруистический суицид мутантных и пораженных вирусами клеток;
-Клеточная гибель после слабого воздействия агентов, вызывающих при массированном поражении некроз.
В процессе реализации апоптоза условно выделяют четыре стадии.
Стадия инициации.На этой стадии информационные сигналы рецептируются клеткой. Патогенный агент либо сам является сигналом, либо обусловливает генерацию сигнала в клетке и его проведение к внутриклеточным регуляторным структурам и молекулам
Стадия программирования.На этой стадии специализированные белки
либо реализуют сигнал к апоптозу путём активации исполнительной программы (её эффекторами являются цистеиновые протеазы — каспазы и эндонуклеазы), либо блокируют потенциально летальный сигнал.Белки-ингибиторы
апоптоза (например, продукты экспрессии антиапоптозных генов Bcl–2,
Bcl-XL) блокируют апоптоз (например, путём уменьшения проницаемости
мембран митохондрий, тем самым уменьшая вероятность выхода в цитозоль
одного из пусковых факторов апоптоза — цитохрома C).Белки-промоторы
апоптоза (например, белки, синтез которых контролируется генами Bad, Bax,
антионкогенами Rb или p53) активируют эффекторные каспазы и эндонуклеазы.
Стадия реализации программы апоптоза (исполнительная, эффекторная)
состоит в собственно гибели клетки, осуществляемой посредством активации
протеаз и эндонуклеаз .Непосредственными исполнителями процесса «умертвления» клетки являются Ca2+,Mg2+-зависимые эндонуклеазы (катализируют
распад нуклеиновых кислот) и эффекторные каспазы (подвергают протеолитическому расщеплению различные белки, в том числе белки цитоскелета, ядра,
регуляторные белки и ферменты).В результате разрушения белков и хроматина
в процессе апоптоза клетка подвергается деструкции. В ней формируются и от
неё отпочковываются фрагменты, содержащие остатки органелл, цитоплазмы,
хроматина и цитолеммы — апоптозные тельца.
Стадия удаления фрагментов погибших клеток.На поверхности апоптозных телец экспрессируются лиганды, с которыми взаимодействуют рецепторы
фагоцитирующих клеток. Фагоциты обнаруживают, поглощают и разрушают
апоптозные тельца. Благодаря этому, содержимое разрушенной клетки не попадает в межклеточное пространство, а при апоптозе отсутствует воспалительная
146
реакция. Этот признак отличает апоптоз от некроза, который сопровождается
развитием перинекротического воспаления.
Апоптоз - генетически управляемый процесс, который может быть включен различными пусковыми сигналами без какого — либо существенного предварительного повреждения исполнительного аппарата клетки, хотя может и
включиться после умеренного повреждения.
Принципиально важно, что при неспособности вступить в апоптоз возникает неограниченно пролиферирующий клон клеток, что ведет к серьезным
нарушениям в многоклеточном организме и наблюдается, например, при онкологических заболеваниях.
ГИПОКСИЯ
Гипоксия – типический патологический процесс, возникающий в результате недостаточного поступления кислорода в организм или не полной утилизации кислорода тканями.
Классификация гипоксических состояний
Гипоксические состояния классифицируют с учётом различных критериев: этиологии,распространенности, степени тяжести расстройств, скорости развития и длительности гипоксии.
Этиология
По этиологии выделяют несколько типов гипоксии , условно объединяемых в две группы: экзогенные (нормо- и гипобарическую гипоксию) и эндогенные:
1.Экзогенная (гипоксическая):
- гипобарическая
- нормобарическая
2.Эндогенная:
- респираторная (дыхательная);
- циркуляторная (сердечно-сосудистая);
- гемическая (кровяная);
- тканевая;
- гипоксия нагрузки (перегрузочная гипоксия);
- смешанная.
По распространенности
1. Местная
2. Общая
По скорости развития и длительности
По критериям скорости возникновения и длительности гипоксического
состояния выделяют несколько его разновидностей.
Молниеносная.Молниеносная (острейшая) гипоксия развивается в течение
нескольких секунд. Как правило, через несколько десятков секунд (в пределах
первой минуты) после действия причины гипоксии выявляется тяжёлое состояние пациента, нередко служащее причиной его смерти (например, при разгер-
147
метизации летательных аппаратов на большой (более 9000–11 000 м) высоте
или в результате быстрой потери большого количества крови (например, при
ранениях крупных артериальных сосудов или разрыве аневризмы их стенки).
Острая.Острая гипоксия развивается через несколько минут (как правило, в пределах первого часа) после воздействия причины гипоксии (например, в
результате острой кровопотери или острой дыхательной недостаточности).
Подострая.Подострая гипоксия формируется в течение нескольких часов
(но в пределах первых суток). Примерами такой разновидности могут быть гипоксические состояния, развивающиеся в результате попадания в организм
метгемоглобинообразователей (нитратов, окислов азота, бензола), венозной
кровопотери, медленно нарастающей дыхательной или сердечной недостаточности.
Хроническая.Хроническая гипоксия развивается и/или длится более чем
несколько суток (недели, месяцы, годы), например, при хронической анемии,
сердечной или дыхательной недостаточности.
По степени тяжести
По степени тяжести расстройств жизнедеятельности организма различают следующие виды гипоксии: лёгкую, среднюю (умеренную), тяжёлую, критическую (опасную для жизни, летальную).В качестве основных признаков тяжести гипоксии используют следующие:
• степень нарушения нервно-психической деятельности;
• выраженность расстройств функций ССС и дыхательной систем;
• величину отклонений показателей газового состава и КЩР крови, а также некоторых других показателей.
Экзогенная(гипоксическая) гипоксия
К экзогенным типам гипоксии относят нормо- и гипобарическую гипоксию. Причина их развития: уменьшение парциального давления кислорода
(pO2) в воздухе, поступающем в организм.
• При нормальном барометрическом давлении говорят о нормобарической экзогенной гипоксии.
• При снижении барометрического давления экзогенную гипоксию называют
гипобарической.
Нормобарическая экзогенная гипоксия.
Причины нормобарической экзогенной гипоксии: ограничение поступления в организм кислорода с воздухом при нормальном барометрическом давлении. Такие условия складываются при:
- нахождении людей в небольшом и/или плохо вентилируемом пространстве
(помещении, шахте, колодце, лифте);
- нарушениях регенерации воздуха и/или подачи кислородной смеси для дыхания в летательных и глубинных аппаратах, автономных костюмах (космонавтов, лётчиков, водолазов, спасателей, пожарников);
- несоблюдении методики искусственной вентиляции легких (ИВЛ).
Гипобарическая экзогенная гипоксия
148
Причины гипобарической экзогенной гипоксии: снижение барометрического давления при подъёме на высоту (более 3000–3500 м, где pO2 воздуха
снижено примерно до 100 мм рт.ст.) или в барокамере. В этих условиях возможно развитие горной, высотной или декомпрессионной болезни.
Горная болезнь наблюдается при подъёме в горы, где организм подвергается воздействию не только пониженного содержания кислорода в воздухе и
пониженного барометрического давления, но также более или менее выраженной физической нагрузки, охлажения, повышенной инсоляции и других факторов средне- и высокогорья.
Высотная болезнь развивается у людей, поднятых на большую высоту в
открытых летательных аппаратах, на креслах-подъёмниках, а также- при снижении давления в барокамере. В этих случаях на организм действуют в основном сниженные pO2 во вдыхаемом воздухе и барометрическое давление.
Декомпрессионная болезнь наблюдается при резком снижении барометрического давления (например, в результате разгерметизации летательных аппаратов на высоте более 10 000–11 000 м). При этом формируется опасное для
жизни состояние, отличающееся от горной и высотной болезни острым или даже молниеносным течением.
Патогенез экзогенных гипоксий
К основным звеньям патогенеза экзогенной гипоксии (независимо от её
причины) относятся артериальная гипоксемия, гипокапния, газовый алкалоз,
сменяющийся ацидозом; артериальная гипотензия, сочетающаяся с гипоперфузией органов и тканей.
Снижение напряжения кислорода в плазме артериальной крови (артериальная гипоксемия) - инициальное и главное звено экзогенной гипоксии. Гипоксемия ведёт к уменьшению насыщения кислородом Hb, общего содержания
кислорода в крови и как следствие — к нарушениям газообмена и метаболизма
в тканях.
Снижение напряжения в крови углекислого газа (гипокапния) возникает в
результате компенсаторной гипервентиляции лёгких (в связи с гипоксемией).Газовый алкалоз является результатом гипокапнии.
При наличии во вдыхаемом воздухе высокого содержания углекислого
газа (например, при дыхании в замкнутом пространстве или в производственных условиях) экзогенная гипоксемия может сочетаться с гиперкапнией и ацидозом. Умеренная гиперкапния (в отличие от гипокапнии) не усугубляет влияний экзогенной гипоксии, а напротив — способствует увеличению кровообращения в сосудах мозга и сердца. Однако значительное увеличение рCO2 в крови
приводит к ацидозу, дисбалансу ионов в клетках и биологических жидкостях,
гипоксемии, снижению сродства Hb к кислороду и ряду других патогенных эффектов.
Снижение системного АД (артериальная гипотензия), сочетающееся с
гипоперфузией тканей в значительной мере являются следствием гипокапнии.
CO2 относится к числу основных факторов регуляции тонуса сосудов мозга.
Выраженное снижение раCO2 является сигналом к сужению просвета артериол
мозга, сердца и уменьшения их кровоснабжения. Эти изменения служат причи-
149
ной существенных расстройств жизнедеятельности организма, включая развитие обморока и коронарной недостаточности (проявляющейся стенокардией, а
иногда - инфарктом миокарда).
Параллельно с указанными отклонениями выявляются нарушения ионного баланса как в клетках, так и в биологических жидкостях: межклеточной,
плазме крови (гипернатриемия, гипокалиемия и гипокальциемия), лимфе, ликворе. Описанные выше отклонения могут быть уменьшены или устранены путём добавления к вдыхаемому воздуху необходимого (расчётного) количества
углекислого газа.
Респираторная (дыхательная) гипоксия
Этот вид гипоксии развивается в результате недостаточного газообмена в
легких. Гиповентиляция, гиподиффузия, гипер- или гипоперфузия приводят к
нарушению оксигенации крови.
Гиповентиляция : снижение кислорода в альвеолярном воздухе в результате
нарушения проходимости дыхательных путей (бронхиты, бронхоспазм), потери
эластичности альвеол (эмфизема), параличе дыхательных мышц, нарушении
подвижности грудной клетки, пневмотораксе. Тяжелое состояние дыхательной
гипоксии возникает при частом поверхностном дыхании.
Диффузионная способность легких зависит от толщины альвеолокапиллярной мембраны и степени проницаемости ее для газов. Диффузия кислорода из альвеолярного воздуха в кровь затрудняется при саркоидозе, пневмокониозе, пневмонии, эмфиземе, отеке легкого, при деструкции обширных
участков легкого (кавернозный туберкулез, абсцесс), ателектазе, фиброзе, дистресс-синдроме и т.д.
Гипоперфузия – снижение поступления кислорода в общий кровоток в
единицу времени.
Гиперперфузия в результате открытия артерио-венозный анастомозов и
сброса крови по внутри- и внелегочным шунтам справа налево, минуя капилляры происходит быстрое прохождение крови через малый круг кровообращения,
вследствие чего нарушается ее оксигенация.
Нарушение легочного кровотока наблюдается при: воспалении легких,
эмфиземе, бронхоспазме, эмболии малого круга кровообращения, поражении
дыхательного центра, болезненном дыхании, плевритах, пневмотораксе.
Вне зависимости от происхождения дыхательной гипоксии, инициальным
патогенетическим звеном является артериальная гипоксемия, обычно сочетающаяся с гиперкапнией и ацидозом (на раннем этапе острой дыхательной недостаточности - газовый, а затем и негазовый).
Циркуляторная (сердечно-сосудистая,гемодинамическая) гипоксия
Причина циркуляторнойгипоксии: недостаточность кровоснабжения тканей и органов. Недостаточность кровоснабжения формируется на основе гиповолемии, сердечной недостаточности, снижения тонуса стенок сосудов, расстройств микроциркуляции, нарушений диффузии кислорода из капиллярной
крови к клеткам.
150
Гиповолемия - уменьшение общего объёма крови в сосудистом русле и
полостях сердца. Это один из важных механизмов развития недостаточности
кровообращения и циркуляторной гипоксии. Причины гиповолемии:
- большая кровопотеря;
- гипогидратация организма (например, при хронических поносах, ожоговой
болезни, массивном длительном потоотделении).
Сердечная недостаточность проявляется снижением выброса крови из
желудочков сердца и как следствие - уменьшением ОЦК. Причины:
- прямое повреждение миокарда (миокардиты, инфаркт, диффузный кардиосклероз);
- перегрузка миокарда (например, увеличенной массой крови или повышенным сосудистым сопротивлением её току при сердечных пороках);
- нарушение диастолического расслабления сердца (например, при его сдавлении - тампонаде экссудатом или кровью, накопившимися в полости перикарда).
Снижение тонуса стенок сосудов (как артериальных так и венозных)
приводит к увеличению ёмкости сосудистого русла и уменьшению ОЦК. Причины:
- снижение адренергических влияний на стенки сосудов (например, при
надпочечниковой недостаточности, повреждении нейронов кардиовазомоторного центра);
- доминирование холинергических воздействий (например, при невротических
состояниях, на торпидной стадии шока, при отклонениях показателей электролитного баланса и КЩР);
- дефицит минералокортикоидов в организме ( гипоальдостеронизм при
надпочечниковой недостаточности).
Гипотония стенок сосудов любого происхождения обусловливает снижение артериального и перфузионного давлений, а также объёма кровотока в сосудах тканей и органов.
Расстройства микроциркуляции развиваются при ишемия, венозной гиперемии, тромбозе, эмболии,а также избыточном артерио-венозном шунтировании крови, обусловленном спазмом прекапиллярных сфинктеров.
Нарушение диффузии кислорода через стенку микрососудов, в межклеточной жидкости, через плазмолемму и цитозоль к митохондриям приводит к
дефициту кислорода в матриксе митохондрий и, следовательно, к снижению
интенсивности тканевого дыхания. Причины:
- уплотнение стенок микрососудов (например, при дистрофиях их стенок, васкулитах, артериолосклерозе, интерстициальном отёке, микседеме);
- мембранопатии клеток (например, при активации липопероксидного процесса, клеточных дистрофиях, опухолевом росте).
Циркуляторная гипоксия часто является результатом комбинации указанных выше механизмов (например при коллапсе, шоке, надпочечниковой недостаточности и гиперкортицизме различного генеза, артериальной гипер- и гипотензии).
151
Виды циркуляторной гипоксии
Важной особенностью гипоксии циркуляторного типа является возможность
развития локальной и системной её форм.
Локальная гипоксия.Причины:
- местные расстройства кровообращения (венозная гиперемия, ишемия, стаз);
- регионарные нарушения диффузии кислорода из крови к клеткам и их митохондриям.
Системная гипоксия.Причины:
- гиповолемия;
- сердечная недостаточность;
- генерализованные формы снижения тонуса сосудов.
Изменения газового состава и рН крови:
- снижение рvО2 (венозная гипоксемия);
- нормальное (как правило) раО2( артериальная нормоксемия);
- увеличение артерио-венозной разницы по кислороду (за исключением вариантов с масштабным «сбросом» крови по артериовенозным шунтам минуя капиллярную сеть).
- негазовый ацидоз.
Гемическая гипоксия
Такой вид гипоксии развивается в результате снижения количества эритроцитов и/или гемоглобина (анемическая гипоксия), или при снижении способности гемоглобина связывать О2.
Анемическая гипоксия обусловлена уменьшением содержания эритроцитов и гемоглобина в циркулирующей крови, при этом нарушается доставка кислорода к органам и тканям. Анемии могут быть развиться при кровопотере,
угнетении костномозгового кроветворения, повреждении токсическими факторами, ионизирующей радиацией, лейкозным процессом, дефицитом компонентов, необходимых для нормального эритропоэза и синтеза гемоглобина: железа,
витаминов, эритропоэтина, избыточном разрушении эритроцитов (гемолитические).
Нарушение транспорта кислорода кровью может развиваться при качественных изменениях гемоглобина. Такая форма гемической гипоксии может
развиться при отравлении окисью углерода, метгемоглобинообразователями,
при врожденных аномалиях гемоглобина, нарушениях физико-химических
свойств внутренней среды организма, которые влияют на процессы его оксигенации в легких и дезоксигенации в тканях.
Сродство Нв с угарным газом в 300 раз выше, чем с кислородом, поэтому
его ничтожные концентрации при длительной экспозиции дают тяжелое отравление. Соединенный с СО гемоглобин (карбоксигемоглобин HbCO) не может
выполнять функцию переноса кислорода. СО тормозит диссоциацию оставшегося гемоглобина, блокирует железосодержащие дыхательные ферменты. Развитие интоксикации окисью углерода возможно в производственных условиях:
металлургических цехах, на коксохимических, кирпичных и цементных заводах, химических производствах, гаражах, на городских магистралях с интен-
152
сивным автотранспортным движением, в жилых помещениях при неисправности газовых приборов или печного отопления, при пожарах.
Теряет способность к переносу кислорода и метгемоглобин (met-Hb).
Метгемоглобин – met-Hb отличается от нормального гемоглобина тем, что железо гемма в нем находится не в виде Fe2+ , а окислено до Fe3+. К транспорту
кислорода met-Hb не способен. К основным метгемоглобинообразователям относятся:
- Нитросоединения: окислы азота, нитриты, нитраты, органические нитросоединения.
В эксперименте используют нитрат натрия для получения различной тяжести анемий в дозе 3-5-10 мг/100 подкожно в 1% растворе.
- Аминосоединения: гидроксиламин, анилин, фенилгидрозин, аминофенолы и
др.
- Окислители: хлораты, хроматы, перманганаты.
- Окислительно-восстановительные красители: метиленовая синь, нильский
голубой, бриллиантовый голубой.
- Лекарственные препараты: висмут, новокаин, анестезин, фенецетин, сульфаниламиды, барбитураты, аспирин и др.
Содержание Мет - Нв обусловливает тяжесть гипоксии:
1. бессимптомная - 5-15% Мет.-Нв,
2. легкая Мет - Нв - до 18-20%,
3. средняя - Мет - Нв до 35-36%,
4. тяжелая - Мет - Нв больше 36%,
5. смертельная
Мет - Нв 75%.
Изменения газового состава и рН крови
- снижение объёмного содержания кислорода в артериальной крови (VaO2 в
норме равно 19,5–21 объёмных %);
- нормальное (!) парциальное напряжение кислорода в артериальной крови;
- снижение рvO2 (венозная гипоксемия);
- негазовый ацидоз;
- снижение артерио-венозной разницы по кислороду.
Тканевая гипоксия (первичная тканевая гипоксия)
Тканевая гипоксия (первичная тканевая гипоксия) возникает вследствие
нарушения способности тканей использовать доставленный кислород. Причины
тканевой гипоксии: факторы, снижающие эффективность утилизации кислорода клетками тканей и/или сопряжения окисления и фосфорилирования.
Снижение эффективности усвоения кислорода клетками наиболее часто
является результатом ингибирования активности ферментов биологического
окисления, значительного изменения физико-химических параметров в тканях,
торможения синтеза ферментов биологического окисления и повреждения мембран клеток. Подавление активности ферментов биологического окисления
наблюдается при:
- специфическом ингибировании ферментов. Примером могут служить ионы
циана (CN–), препятствующие окислению цитохрома. В результате блокируется
153
восстановление железа дыхательного фермента и транспорта кислорода к цитохрому. Аналогичные последствия вызывает блокада активных центров ферментов тканевого дыхания антимицином А, соединениями, содержащими сульфид-ион S2– и некоторыми другими веществами;
- неспецифическом ингибировании ферментов биологического окисления
ионами металлов (Ag2+, Hg2+, Cu2+). При этом указанные металлы обратимо взаимодействуют с SH–группами фермента с образованием его неактивной меркаптоидной формы;
- конкурентном ингибировании ферментов биологического окисления. В роли
конкурентных ингибиторов могут выступать оксалат и малонат, блокирующие
взаимодействие сукцината с сукцинатдегидрогеназой в цикле трикарбоновых
кислот; фторлимонная кислота, конкурирующая за активный центр аконитазы с
цитратом.
Изменения физико-химических параметров в тканях (температуры, электролитного состава, рН, фазового состояния мембранных компонентов) существенно влияют на эффективность биологического окисления . Отклонение от
нормы указанных и других параметров наблюдается при многих болезнях и патологических состояниях: гипертермиях и гипотермиях, недостаточности различных органов (сердца, почек, печени), анемиях и ряде других).
Торможение синтеза ферментов биологического окисления может
наблюдаться при:
- общем или частичном (особенно белковом) голодании;
- при большинстве гипо- и авитаминозов;
- нарушении обмена минеральных веществ, необходимых для синтеза ферментов.
Повреждение мембран. В наибольшей мере это относится к мембранам
митохондрий. Важно, что выраженная гипоксия любого типа сама по себе активирует многие механизмы, приводящие к повреждению мембран и ферментов
клеток с развитием тканевой гипоксии.
Снижение степени сопряжения окисления и фосфорилирования макроэргических соединений в дыхательной цепи
Выраженной способностью разобщать процессы окисления и фосфорилирования обладают многие эндогенные агенты (например, избыток Ca2+, H+,
ВЖК, йодсодержащие гормоны щитовидной железы), а также экзогенные вещества (2,4-динитрофенол, дикумарин, пентахлорфенол, грамицидин и другие).
В этих условиях увеличиваются расход кислорода тканями и интенсивность функционирования компонентов дыхательной цепи. Однако, большая
часть энергии транспорта электронов трансформируется в тепло и не используется для ресинтеза макроэргов. Эффективность биологического окисления снижается. Клетки не получают энергетического обеспечения. В связи с этим
нарушаются их функции и нарушается жизнедеятельность организма в целом.
Изменения газового состава и рН крови
- увеличение парциального напряжения кислорода в венозной крови;
- увеличение объёмного содержания кислорода в венозной крови;
154
- уменьшение артерио-венозной разницы по кислороду (исключение — тканевая гипоксия, развившаяся при действии разобщителей окисления и фосфорилирования);
- негазовый ацидоз.
Перегрузочная гипоксия
Перегрузочная гипоксия возникает при чрезмерно высокой функциональной нагрузке какого-либо органа или ткани. Наблюдается ситуация когда
функциональные резервы систем транспорта и утилизации кислорода и субстратов оказываются недостаточными для обеспечения увеличенной потребности тканей в связи с повышенной нагрузкой. Например, при повышенных
нагрузках на мышечные органы: скелетные мышцы и миокард (при чрезмерной
нагрузке на сердце возникают относительная коронарная недостаточность,
местная гипоксия сердца и вторичная общая циркуляторная гипоксия).
В случае повышенной мышечной работы вместе с гипоксией скелетных
мышц развиваются конкурентные отношения в распределении кровотока, которые приводят к ишемии других тканей и развитию распространенной гипоксии.
Изменения газового состава и рН крови:
- снижение парциального напряжения кислорода в венозной крови (венозная
гипоксемия), оттекающей от гиперфункционирующей мышцы;
- увеличение артерио-венозной разницы по кислороду;
- увеличение парциального напряжения углекислого газа (гиперкапния) в венозной крови, что является результатом активированного метаболизма в ткани
мышцы.
Смешанный тип гипоксии
Смешанный тип гипоксии является результатом сочетания нескольких
разновидностей гипоксии.
Примером могут служить наркотические вещества, способные в высоких
дозах угнетать функцию сердца, нейронов дыхательного центра и активность
ферментов тканевого дыхания. В результате развивается смешанная гипоксия
гемодинамического, дыхательного и тканевого типов.
Острая массивная кровопотеря приводит как к снижению кислородной
ёмкости крови (в связи с уменьшением содержания Hb), так и к расстройству
кровообращения: развивается гемический и гемодинамический типы гипоксии.
Часто возникшая гипоксия любого типа, достигнув определенной степени, вызывает нарушения деятельности других органов и систем, участвующих в
обеспечении биологического окисления. Практически любая тяжелая гипоксия
носит смешанный характер
Патогенез гипоксии смешанного типа включает звенья механизмов развития разных типов гипоксии. Смешанная гипоксия часто характеризуется взаимопотенцированием отдельных её типов с развитием тяжёлых экстремальных и
даже терминальных состояний.
Изменения газового состава и рН крови при смешанной гипоксии определяются доминирующими расстройствами механизмов транспорта и утилизации
кислорода, субстратов обмена веществ, а также процессов биологического
окисления в разных тканях.
155
Адаптивные реакции организма при гипоксии
При развитии гипоксии в организме повышается функциональная активность систем, обеспечивающих достижение и поддержание оптимального
уровня биологического окисления в клетках.К ним относятся системы внешнего дыхания, сердечно- сосудистая система, кровь, системы биологического
окисления и регуляторные системы.
Адаптивные реакции принято делить на две группы: экстренной адаптации и долговременной адаптации.
Экстренная адаптация
Причина активации механизмов срочной адаптации организма к гипоксии: недостаточность биологического окисления. Как следствие, снижается
содержание АТФ в тканях, необходимого для обеспечения оптимального уровня жизнедеятельности.Ключевой фактор процесса экстренной адаптации организма к гипоксии — активация механизмов транспорта O2 и субстратов обмена
веществ к тканям и органам. Повышенное функционирование систем транспорта кислорода и субстратов метаболизма к клеткам сопровождается интенсивным расходом энергии и субстратов обмена веществ. Таким образом, эти механизмы имеют высокую «энергетическую и субстратную цену». Именно это является (или может стать) фактором, ограничивающим уровень и длительность
гиперфункционирования.
Система внешнего дыхания.
Недостаточность биологического окисления при гипоксии ведёт к гипервентиляции - возрастанию объёма альвеолярной вентиляции вследствие активация афферентной импульсации от хеморецепторов (аорты, каротидной зоны
сонных артерий, ствола мозга и других регионов организма) в ответ на изменение показателей газового состава крови (снижение раО2, увеличение раCO2 и
др.).Увеличивается частота и глубина дыхательных движений и число раскрывшихся резервных альвеол. В результате минутный объём дыхания (МОД)
может возрасти более чем на порядок: с 5–6 л в покое до 90–110 л в условиях
гипоксии.
Сердечно- сосудистая система
При острой гипоксии функция сердца значительно интенсифицируется в
результате активации симпатоадреналовой системы и высвобождения катехоламинов.Это приводит к тахикардии,увеличению ударного объема сердца.Возрастает минутный объём кровообращения (МОК),повышается линейная
и объёмная скорость кровотока в сосудах.
В условиях гипоксии развивается феномен перераспределения, или централизации кровотока.
Последствия
• Расширение артериол и увеличение кровоснабжения мозга и сердца.
• Одновременное сужение просвета артериол и уменьшение объёма кровоснабжения в других органах и тканях: мышцах, подкожной клетчатке, сосудах
брюшной полости, почках.
156
Система крови
Реакция системы крови: усиление эритропоэза, повышение сродства Нв к
кислороду.
Усилена диссоциация оксигемоглобина. Происходят качественные изменения в цепи дыхательных ферментов (цитохромоксидазы). Увеличивается количество дыхательных ферментов и количество митохондрий.
Резкое увеличение эритропоэза может ухудшить реологические свойства
крови из-за ее сгущения.
Системы биологического окисления(биохимическая адаптация)
Активация метаболизма — важное звено экстренной адаптации организма к
острой гипоксии.
Системы биологического окисления в тканях обеспечивают оптимальное
энергетическое обеспечение функционирующих структур и уровень пластических процессов в них в условиях гипоксии. Это достигается благодаря:
- активации гликолиза
- увеличению числа митохондрий и количества крист митохондрий
- увеличению числа молекул ферментов тканевого дыхания в каждой митохондрии, а также активности ферментов, особенно цитохромоксидазы.
- повышению эффективности процессов биологического окисления и сопряжения его с фосфорилированием.
- повышению эффективности механизмов анаэробного ресинтеза АТФ в
клетках.
Долговременная адаптация
Причина включения механизмов долговременной адаптации к гипоксии:
повторная или продолжающаяся недостаточность биологического окисления
умеренной выраженности.Долговременная адаптация к гипоксии реализуются
на всех уровнях жизнедеятельности: от организма в целом до клеточного метаболизма.
Системы, обеспечивающие доставку кислорода и продуктов обмена веществ к тканям (внешнего дыхания и кровообращения), при устойчивой адаптации к гипоксии также приобретают новые качества: повышенные мощность,
экономичность и надёжность функционирования.
Системы биологического окисления
Системы биологического окисления в тканях обеспечивают оптимальное
энергетическое обеспечение функционирующих структур и уровень пластических процессов в них в условиях гипоксии. Это достигается благодаря увеличению:
- числа митохондрий и количества крист митохондрий;
- числа молекул ферментов тканевого дыхания в каждой митохондрии, а также
активности ферментов, особенно – цитохромоксидазы;
- эффективности процессов биологического окисления и сопряжения его с
фосфорилированием;
- эффективности механизмов анаэробного ресинтеза АТФ в клетках.
157
Система внешнего дыхания
Система внешнего дыхания обеспечивает уровень газообмена, достаточный для оптимального течения обмена веществ и пластических процессов в
тканях. Это достигается благодаря:
- гипертрофии лёгких и увеличению в связи с этим площади альвеол, капилляров в межальвеолярных перегородках, уровня кровотока в этих капиллярах;
- увеличению диффузионной способности аэро-гематического барьера лёгких;
- гипертрофии и возрастанию мощности дыхательной мускулатуры;
- возрастанию жизненной ёмкости лёгких (ЖЁЛ).
Сердечно-сосудистая система
При долговременной адаптации к гипоксии увеличивается сила, а также
скорость процессов сокращения и расслабления миокарда. В результате происходит возрастание объёма и скорости выбрасываемой в сосудистое русло крови
-ударного и сердечного (минутного) выбросов. Эти эффекты становятся возможными благодаря:
- умеренной сбалансированной гипертрофии всех структурных элементов
сердца: миокарда, сосудистого русла, нервных волокон;
- увеличению числа функционирующих капилляров в сердце;
- увеличению числа митохондрий в кардиомиоцитах и эффективности реакций
биологического окисления. В связи с этим сердце расходует на 30–35% меньше
кислорода и субстратов обмена веществ, чем в неадаптированном к гипоксии
состоянии;
- повышению эффективности трансмембранных процессов (транспорта ионов,
субстратов и продуктов метаболизма, кислорода и др.);
- возрастанию мощности и скорости взаимодействия актина и миозина в миофибриллах кардиомиоцитов;
- повышению эффективности адрен- и холинергических систем регуляции
сердца.
В адаптированном организме сосудистая система способна обеспечивать
такой уровень перфузии тканей кровью, который необходим для осуществления их функции даже в условиях гипоксии. В основе этого лежат следующие
механизмы:
- увеличение количества функционирующих капилляров в тканях и органах;
- снижение миогенного тонуса артериол и уменьшение реактивных свойств
стенок резистивных сосудов к вазоконстрикторам: катехоламинам, АДГ, лейкотриенам, отдельным эйкозаноидам и другим.
Это создаёт условия для развития устойчивой артериальной гиперемии в
функционирующих органах и тканях.
Система крови
При устойчивой адаптации организма к гипоксии существенно возрастают кислородная ёмкость крови, скорость диссоциации HbO2, сродство дезоксигемоглобина к кислороду в капиллярах лёгких.
Увеличение кислородной ёмкости крови является результатом стимуляции эритропоэза и развития эритроцитоза в результате активации под влиянием
158
ишемии и гипоксии образования в почках эритропоэтина. Эритропоэтин стимулирует эритропоэз.
Метаболизм
Метаболические процессы в тканях при достижении состояния устойчивой адаптированности к гипоксии характеризуются:
- снижением их интенсивности;
- экономным использованием кислорода и субстратов обмена веществ в реакциях биологического окисления и пластических процессах;
- высокой эффективностью и лабильностью реакций анаэробного ресинтеза
АТФ;
- доминированием анаболических процессов в тканях по сравнению с катаболическими;
- высокой мощностью и мобильностью механизмов трансмембранного переноса ионов.Это является следствием повышения эффективности работы мембранных АТФаз, что обеспечивает регуляцию трансмембранного распределения ионов, миогенного тонуса артериол, водно-солевого обмена и др. важных
процессов.
Системы регуляции
Системы регуляции адаптированного к гипоксии организма обеспечивают достаточную эффективность, экономичность и надёжность управления его
жизнедеятельностью. Это достигается благодаря включению механизмов нервной и гуморальной регуляции функций.
Нервная регуляция
Значительные изменения как в высших отделах мозга, так и в вегетативной нервной системе адаптированного к гипоксии организма характеризуются:
- повышенной резистентностью нейронов к гипоксии и дефициту АТФ, а также некоторым другим факторам (например, токсинам, недостатку субстратов
метаболизма);
- гипертрофией нейронов и увеличением числа нервных окончаний в тканях и
органах;
- увеличенной чувствительностью рецепторных структур к нейромедиаторам;
- уменьшением синтеза и высвобождения нейромедиаторов.
Указанные изменения в нервной системе способствуют:
- быстрой выработке и сохранению новых условных рефлексов;
- переходу приобретённых навыков из кратковременных в долговременные;
- устойчивости нервной системы к патогенным воздействиям.
Гуморальная регуляция
Перестройка функционирования эндокринной системы при гипоксии
обусловливает:
- снижение степени стимуляции мозгового вещества надпочечников, гипоталамо-гипофизарно-надпочечниковой и других систем; это ограничивает активацию механизмов стресс-реакции и её возможные патогенные эффекты;
- повышение чувствительности рецепторов клеток к гормонам, что способствует уменьшению объёма их синтеза в железах внутренней секреции.
159
В целом изменения в системах регуляции потенцируют как системные,
так и органные приспособительные реакции организма, жизнедеятельность которого осуществляется в условиях гипоксии.
Расстройства жизнедеятельности при гипоксии
Характер, динамика и степень изменений жизнедеятельности организма
зависят от ряда факторов: типа гипоксии, её степени, скорости развития, а также от состояния реактивности организма.
Острейшая (молниеносная) тяжёлая гипоксия приводит к быстрой потере сознания, подавлению функций организма и его гибели. Такая картина
наблюдается, например, при вдыхании газовых смесей, не содержащих кислорода или содержащих его в малых количествах. Это может быть при авариях в
производственных условиях (например, в шахтах), в летательных аппаратах, в
подводных лодках, при поломке скафандров. Молниеносная гипоксия развивается также при фибрилляции желудочков сердца, при острой массивной (артериальной) кровопотере, отравлении цианидами и других подобных ситуациях.
Хроническая (постоянная или прерывистая) умеренная гипоксия сопровождается, как правило, адаптацией организма к гипоксии.
Расстройства обмена веществ
Расстройства обмена веществ являются одним из ранних проявлений гипоксии.
Содержание АТФ и креатинфосфата при гипоксии любого типа прогрессирующе снижаются вследствие подавления процессов биологического окисления (особенно — аэробных) и сопряжения их с фосфорилированием, содержание АДФ, АМФ и креатина нарастают вследствие нарушения их фосфорилирования,концентрация неорганического фосфата в тканях увеличивается.
Процессы тканевого дыхания в клетках подавлены вследствие дефицита
кислорода, недостатка субстратов обмена веществ, подавление активности
ферментов тканевого дыхания.
Гликолиз на начальном этапе гипоксии активируется вследствие дефицита АТФ и активации гликолитических ферментов продуктами гидролиза АТФ:
АДФ и АМФ.Снижается содержания гликогена и глюкозы в клетках, увеличивается внутриклеточное содержание молочной и пировиноградной кислот.
Содержание H+ в клетках и биологических жидкостях прогрессирующе
нарастает и развивается ацидоз вследствие торможения окисления субстратов,
особенно лактата и пирувата, кетоновых тел.
Биосинтез нуклеиновых кислот и белков подавлен вследствие дефицита
энергии, необходимой для этих процессов.Параллельно активируется протеолиз, обусловленный активацией в условиях ацидоза протеаз, а также неферментного гидролиза белков.Азотистый баланс становится отрицательным. Это
сочетается с повышением уровня остаточного азота в плазме крови и аммиака в
тканях
Жировой обмен характеризуется:
- активацией липолиза вследствие повышения активности липаз и ацидоза;
- торможением ресинтеза липидов;
160
- накоплением в результате вышеуказанных процессов избытка кетоновых тел
(ацетоуксусной, b-оксимасляной кислот, ацетона) и жирных кислот в плазме
крови, межклеточной жидкости, клетках;При этом ВЖК оказывают разобщающее влияние на процессы окисления и фосфорилирования, что усугубляет дефицит АТФ;
Обмен электролитов и жидкости в тканях существенно нарушен:
- нарушено соотношения ионов в клетках;
- увеличено в крови содержания Na+, Cl–, отдельных микроэлементов; изменения содержания разных ионов различно. Оно зависит от степени гипоксии, преимущественного повреждения того или иного органа, изменений гормонального статуса и других факторов.
- накапливается избыток жидкости в клетках (набухание клеток) в результате
увеличения осмотического давления в цитоплазме клеток( в связи с накопление
в них Na+, Ca2+ и некоторых других ионов), гидролиза крупных молекул органических веществ (например, гликогена, белка),повышения онкотического давления в клетках в результате распада полипептидов, липопротеидов и других
белоксодержащих молекул, обладающих гидрофильными свойствами.
Нарушения функций органов и тканей
При гипоксии нарушения функций органов и тканей выражены в разной
мере. Это определяется различной резистентностью органов к гипоксии, а также скоростью её развития, степенью и длительностью её воздействия на организм.
Резистентность органов к гипоксии
Наибольшая устойчивость к гипоксии у костей, хрящей, сухожилий, связок. Даже в условиях тяжёлой гипоксии в них не обнаруживается значительных
морфологических отклонений.
В скелетной мускулатуре изменения структуры миофибрилл, а также их
сократимости выявляются через 100–120 мин, а в миокарде — уже через 15–20
мин.
В почках и печени морфологические отклонения и расстройства функций
обнаруживаются обычно через 20–30 мин после начала гипоксии.
Наименьшей резистентностью к гипоксии обладает ткань нервной системы. При этом различные её структуры по-разному устойчивы к гипоксии одинаковой степени и длительности.Резистентность нервных клеток уменьшается в
следующем порядке: периферические нервные узлы - спинной мозг - продолговатый мозг - гиппокамп - мозжечок - кора больших полушарий. Прекращение
оксигенации коры мозга вызывает значительные структурные и функциональные изменения в ней уже через 2–3 мин, в продолговатом мозге через 8–12 мин,
а в ганглиях вегетативной нервной системы через 50–60 мин.
Отсюда следует, что последствия гипоксии для организма в целом определяются степенью повреждения нейронов коры больших полушарий и временем их развития.
161
Нарушения высшей нервной деятельности(ВНД)
Нарушения ВНД в условиях гипоксии выявляются наиболее рано - уже
через несколько секунд. Это проявляется:
- Снижением способности адекватно оценивать происходящие события и
окружающую обстановку.
- Ощущениями дискомфорта, тяжести в голове, головной боли.
- Дискоординацией движений.
- Замедлением логического мышления и принятия решений (в том числе простых).
- Расстройством сознания и его потерей в тяжёлых случаях.
- Нарушением бульбарных функций, что приводит к расстройствам функций
сердца и дыхания, вплоть до их прекращения.
Нарушения кровообращения
Расстройства кровообращения выражаются:
- Снижением сократительной функции миокарда, уменьшением ударного и
сердечного выбросов.
- Расстройством кровотока в сосудах сердца и развитием коронарной недостаточности, обусловливающей эпизоды стенокардии и даже инфаркт миокарда.
- Развитием аритмий сердца, включая мерцание и фибрилляцию предсердий и
желудочков.
- Гипертензивными реакциями (за исключением отдельных разновидностей
гипоксии циркуляторного типа), сменяющимися артериальной гипотензией, в
том числе -острой (коллапсом).
- Изменением объёма и реологических свойств крови. Так, при гипоксии гемического типа, вызванной острой кровопотерей, развиваются характерные
стадийные их изменения.
- Возможны расстройства микроциркуляции, проявляющиеся чрезмерным замедлением тока крови в капиллярах, турбулентным его характером, артериолярно-венулярным шунтированием, трансмуральными и экстраваскулярными
нарушениями микроциркуляции. В тяжёлых случаях эти расстройства завершаются сладжем и капилляротрофической недостаточностью.
Система внешнего дыхания
Отклонения функции системы внешнего дыхания проявляются:
- Вначале увеличением объёма альвеолярной вентиляции, а затем (при нарастании степени гипоксии и повреждения нервной системы) прогрессирующим её
снижением.
- Уменьшением общей и регионарной перфузии ткани лёгких. Это обусловлено падением сердечного выброса, а также регионарной вазоконстрикцией в
условиях гипоксии.
- Нарушением вентиляционно-перфузионного соотношения (вследствие местных расстройств перфузии и вентиляции в различных участках лёгких).
- Снижением диффузии газов через аэрогематический барьер (в связи с развитием отёка и набуханием клеток межальвеолярной перегородки).
В итоге развивается дыхательная недостаточность, усугубляющая степень
гипоксии.
162
Нарушения функций почек
Нарушения функций почек разнообразны. Они зависят от степени, длительности и типа гипоксии. Как правило, при гипоксии развиваются:
- Расстройства диуреза (от полиурии до олиго- и анурии).
- Нарушения состава мочи
- Отклонение за пределы нормального диапазона содержания глюкозы, ионов,
азотистых соединений и других веществ, имеющихся и в норме.
- Появление в моче отсутствующих в норме компонентов: эритроцитов, лейкоцитов, цилиндров, белка.
Выраженные повреждения почек при тяжёлых формах гипоксии могут
привести к развитию почечной недостаточности, уремии и комы.
Расстройства функций печени
Расстройства функций печени развиваются, как правило, при хронически
протекающей гипоксии. Их проявления зависят от особенностей патогенеза основной формы патологии (например, сердечной или дыхательной недостаточности, анемического состояния, расстройств обмена веществ, биологического
окисления и др.). В любом случае при гипоксии выявляются признаки парциального или тотального нарушения функций печени:
- Расстройства обмена веществ (углеводного, липидного, белкового, витаминов).
- Нарушения антитоксической функции.
- Угнетение образования различных веществ (например, факторов системы
гемостаза, коферментов, мочевины, жёлчных пигментов и др.).
Система пищеварения
Нарушения системы пищеварения проявляются:
-Расстройствами аппетита (как правило, его снижением).
-Нарушением моторики желудка и кишечника (обычно — снижением перистальтики, тонуса и замедлением эвакуации желудочного и/или кишечного содержимого).
-Развитием эрозий и язв (особенно при длительной тяжёлой гипоксии).
Иммунная система
При хронических и выраженных гипоксических состояниях происходит
снижение эффективности системы иммуно-биологического надзора, что проявляется:
- Низкой активностью иммунокомпетентных клеток.
- Недостаточной эффективностью факторов неспецифической защиты организма: комплемента, ИФН, мураминидазы, белков острой фазы, естественных
киллеров и др.
Указанные и некоторые другие изменения в системе ИБН при выраженной длительной гипоксии могут привести к развитию различных иммунопатологических состояний: иммунодефицитов, патологической иммунной толерантности, аллергических реакций, состояний иммунной аутоагрессии.
163
ВОСПАЛЕНИЕ
Воспаление - это типовой патологический процесс, развивающийся в васкуляризированных тканях в ответ на любое повреждение и проявляющийся в
виде поэтапных изменений микроциркуляторного русла, крови и стромы органа
или ткани в виде альтерации,экссудации и пролиферации, направленных на локализацию, разведение, изоляцию и устранение агента, вызвавшего повреждение, и на восстановление поврежденной ткани.
Воспаление относится к числу наиболее распространенных патологических процессов. Одновременно оно представляет собой важную защитноприспособительную реакцию, эволюционно сформировавшуюся как способ сохранения целого организма ценою повреждения его части. С помощью воспаления обеспечиваются локализация и элиминация воспалительного агента
(флогогена — от лат. phlogosis — воспаление, синоним термина inflammatio) и
(или) поврежденной под его воздействием ткани.
ЭТИОЛОГИЯ
Причиной воспаления может быть первичное повреждение агентом любой природы, который по силе и длительности воздействия превосходит адаптационные возможности организма. Такой агент называется флогогенным (от
греч. phlogos - гореть).
Классификация флогогенных агентов:
В зависимости от происхождения и природы флогогенные аг енты под
164
разделяются на:
1. Экзогенные
1.1. биологические (бактерии, вирусы, грибы, простейшие, гельминты)
1.2. механические (инородное тело, травма)
1.3. физические (высокая и низкая температура, радиация)
1.4. химические (кислоты и щелочи)
2. Эндогенные (опухоли, желчные, мочевые камни, тромбоз, комплексы антиген-антитело, депозиты мочевой кислоты, солей кальция).
В зависимости от участия инфекции воспаление также делят на септическое
и асептическое.
Компоненты воспаления
Для воспаления, как и для любого типического патологического процесса, характерна стереотипность развития. Закономерная динамика воспаления,
как типового патологического процесса, определяется тем, что в основе его
развития находится несколько общих и взаимосвязанных компонентов:
1. Альтерация - повреждение, которое может быть первичным (в результате
действия флогогенного агента) и вторичным (издержки защиты).
2.Экссудация - включает сосудистые реакции и изменения крово- и лимфообращения, собственно экссудацию, эмиграцию лейкоцитов и выход других
форменных элементов крови в ткань, фагоцитоз.
3. Пролиферация - завершающая стадия воспаления, в которой происходит
либо восстановление структуры и функции тканей (регенерация), либо замещение утраченных паренхиматозных элементов органа соединительной тканью
(фиброплазия, склерозирование).
Каждый из компонентов воспаления, в свою очередь, - сложный динамический комплекс взаимозависимых реакций, процессов и факторов. Как правило, по ходу воспаления, преимущественно альтеративные изменения в очаге
воспаления закономерно сменяются преимущественно экссудативными и далее
- преимущественно пролиферативными. Однако, в большинстве случаев, особенно при значительной площади воспаления и/или при его хроническом течении, даже в соседних участках очага воспаления одновременно выявляются
признаки различных компонентов воспалительной реакции -и альтерации, и
экссудации, и пролиферации ( Рис.1).
Определённая пространственная и временная мозаика этих компонентов в
очаге воспаления обусловливает, с одной стороны, закономерный характер развития и проявлений воспаления, а с другой - своеобразие его течения у каждого
конкретного пациента.
165
Рис.1. Соотношение компонентов воспаления.
Признаки воспаления
Кардинальные признаки воспаления были описаны античными врачами
Цельсом (I в н. э.), и Галеном (II в н. э.). К ним относятся:
1. Tumor (опухоль, припухлость) - развивается в результате увеличения кровенаполнения ткани и развития отека в очаге воспаления.
2. Rubor (покраснение) - возникает в результате развития артериальной гиперемии в очаге воспаления.
3. Calor (жар, повышение температуры в очаге воспаления) - является
следствием артериальной гиперемии и повышения интенсивности обмена веществ в очаге воспаления.
4. Dolor (боль) - возникает в результате воздействия на рецепторы медиаторов воспаления (гистамин, брадикинин), ацидоза, гиперосмии и отека
воспаленной ткани.
5. Functio laesa (нарушение функции) - развивается как вследствие действия
первичного флогогенного фактора, так и в результате морфо-функциональных
изменений характерных для воспалительного процесса.
Эти признаки особенно хорошо определяются, когда очаг воспаления
находится на наружных покровах. Наряду с местными воспаление может проявляться также общими признаками, выраженность которых зависит от интенсивности и распространенности процесса. Общие проявления воспаления
включают лихорадку, реакции кроветворной ткани с развитием лейкоцитоза,
повышенную скорость оседания эритроцитов, ускоренный обмен веществ, измененную иммунологическую реактивность, явления интоксикации организма.
Классификация воспаления
В зависимости от доминирующего местного процесса различают 3 вида
воспаления:
1. Альтеративное - преобладание процессов повреждения, дистрофии,
некроза.
2.Экссудативное - характеризуется выраженным нарушением кровообращения с явлениями экссудации и эмиграции лейкоцитов.
3.Пролиферативное или продуктивное - характеризуется размножением клеток гематогенного и гистогенного происхождения с развитием клеточных инфильтратов.
166
В зависимости от длительности течения различают:
1.Острое воспаление, характеризующееся интенсивным развитием и разрешением процесса в течение 1-2 недель, умеренными явлениями альтерации,
экссудации и пролиферации.
2. Хроническое воспаление, характеризующееся затяжным, вялым течением с
длительной персистенцией альтеративных и экссудативных изменений и выраженной пролиферацией.
АЛЬТЕРАЦИЯ
Комплекс изменений, вызванных непосредственным действием повреждающего агента, называют первичной альтерацией. Действие флогогенного
фактора зависит от его природы, однако можно выделить несколько базовых
механизмов повреждения, присущих многим флогогенам. Эти механизмы
включают повреждение структуры мембран или нарушение их функции, нарушение функций внутриклеточных ферментов, а также повреждение структур
межклеточного вещества. Так как воздействие этиологического фактора распределяется неравномерно, часть клеток гибнет, а часть адаптируется и играет
важную роль в последующем воспалении и репарации ткани.
Вторичная альтерация. Продукты, возникающие в результате деструкции
клеток при первичной альтерации и производимые клетками-участниками воспаления, могут вызвать вторичное самоповреждение тканей. Многие факторы
вторичной альтерации являются также важнейшими факторами антимикробной
защиты, действующими внутри фаголизосом нейтрофилов и макрофагов. Однако эти агенты не обладают специфичностью, и, попадая во внеклеточное пространство, они могут оказывать цитолитический эффект на собственные клетки
организма и разрушать структуры межклеточного вещества. Основными факторами вторичной альтерации являются:
1.Гидролитические ферменты лизосом (нейтральные и кислые протеазы,
липазы, гликозидазы, фосфатазы). При некрозе клеток целостность их мембран
нарушается, и внутриклеточное содержимое, в том числе лизосомальные ферменты, поступает в интерстициальное пространство.
Другим источником лизосомальных гидролаз в очаге воспаления являются
фагоциты, выделяющие их при гибели и дегрануляции. Нейтральные протеазы
(коллагеназа, эластаза, катепсин) разрушают коллаген, эластин и фибриллин
межклеточного вещества, базальные мембраны, а также способны прямо активировать компоненты комплемента СЗ и С5, гидролизуя их до СЗа и С5а. Кислые протеазы разрушают гликопротеины и протеогликаны, а гликозидазы (гиалуронидаза и др.) - гликозаминогликаны основного вещества соединительной
ткани и компоненты клеточных стенок бактерий. Липазы и фосфолипазы повреждают липиды клеточных мембран.
2. Активные кислородные радикалы (АКР) и оксид азота (NO). Повреждение
клеток сопровождается повреждением мембран митохондрий, пероксисом, ГЭР
в результате чего АКР, продуцируемые этими органеллами поступают в цитоплазму и далее в интерстиций. АКР также являются важными бактерицидными
факторами фагоцитов и активно продуцируются этими клетками. Наконец, N0
167
продуцируется эндотелиоцитами и макрофагами и выделяется во внеклеточное
пространство, где может трансформироваться в пероксинитрит — крайне реакционноспособный свободный радикал. Действие всех свободных радикалов заключается в перекисном окислении липидов, что нарушает структуру и функции клеточных мембран, ковалентных модификациях белков и нуклеиновых
кислот .
3. Продукты активации комплемента. Конечным этапом активации системы комплемента является образование мембраноатакующего комплекса
(МАК), который встраивается в мембрану клетки-мишени и формирует в ней
канал, что приводит к гибели клетки. Как и большинство эффекторных защитных систем, комплемент не обладает специфичностью, поэтому может атаковать собственные клетки организма.
Важную роль в развитии вторичной альтерации играют также гипоксия,
ацидоз и расстройства осмотического баланса, развивающиеся в очаге воспаления в результате изменений микроциркуляции и метаболизма в очаге воспаления.
Биохимические и физико-химические изменения в очаге воспаления
Изменения метаболизма в очаге воспаления могут быть охарактеризованы как «пожар обмена». Они развиваются в следствие действие флогогенного
фактора и вторичных расстройств в тканях, выражающихся в перестройке
местных механизмов нервной и гуморальной регуляции, микроциркуляции,
формировании физико-химических сдвигов. В очаге воспаления наблюдаются
закономерные фазные изменения обмена веществ, направленные на энергетическое и пластическое обеспечение местных адаптивных и защитных реакций.
На начальном этапе воспаления в ткани преобладают реакции катаболизма, затем - при активации процессов пролиферации, - начинают доминировать анаболические реакции.
Метаболизм углеводов в очаге воспаления характеризуется преобладанием
гликолиза. Это обусловлено повреждением мембран и снижением активности
ферментов митохондрий. При этом увеличение поглощения тканью кислорода
30-35%, характерное для начальных этапов воспаления, сопровождается снижением окисления глюкозы. Активация гликогенолиза и гликолиза сопровождается уменьшением продукции АТФ, накоплением пирувата и лактата, и развитием ацидоза.
В метаболизме липидов доминируют процессы липолиза. Это обусловлено, в
основном, интенсификацией гидролиза липидов липазами и фосфолипазами,
высвобождающимися из поврежденных клеток, а также лейкоцитов. Усиливаются и процессы перекисного окисления липидов. Следствиями подобной
направленности липидного обмена являются накопление в очаге воспаления
свободных высших жирных кислот, обладающих детергентным действием и
повреждающих мембраны; накопление токсичных кетокислот (ацетоуксусной,
β-оксимасляной, β-кетоглутаровой); накопление липоперекисей; активация
продукции метаболитов арахидоновой кислоты.
168
Белковый обмен характеризуется повышением процессов протеолиза в результате накопления в очаге воспаления большого количества протеолитических ферментов.
Физико-химические нарушения в очаге воспаления
В очаге воспаления накапливаются ионы водорода, что приводит к снижению рН в клетках и межклеточной жидкости и, следовательно, развитию
ацидоза. Причинами метаболического ацидоза являются: образование большого
количества недоокисленных продуктов в результате активации гликолиза,
нарушение дренажа тканей, в результате изменения микроциркуляции, и истощение буферных систем клеток и межклеточной жидкости (бикарбонатной,
фосфатной, белковой). Следствиями ацидоза являются: повышение проницаемости мембран, активация лизосомальных ферментов, повышение проницаемости сосудистых стенок в результате неферментативного гидролиза компонентов
базальных мембран, изменения чувствительности рецепторов (особенно адренорецепторов, что приводит к снижению вазоконстрикторных влияний).
Характерно повышение осмотического и онкотического давления в очаге
воспаления (гиперосмия, гиперонкия). Причины повышения осмотического давления: гидролиз макромолекул с образованием низкомолекулярных дериватов,
выход ионов из поврежденных и разрушенных клеток. Гиперонкия развивается
в результате увеличения концентрации белка в очаге воспаления, который поступает из разрушенных клеток, а также из крови при повышении сосудистой
проницаемости. Последствия гиперонкии и гиперосмии: гипергидратация очага
воспаления и развитие отека.
Медиаторы воспаления, их классификация
I. Провоспалительные медиаторы( медиаторы альтерации и
экссудации)
1.Активные кислородные и кислородгалогеновые радикалы, оксид азота –
мощный неселективный цитотоксический эффект.
2.Гидролитические ферменты лизосом–нейтральные и кислые протеазы, липазы, гликозидазы, фосфатазы-разрушение коллагена, эластина, базальных
мембран, гликопротеидов, компонентов межклеточного вещества, липидов и
т.д , и активация полипептидных каскадных медиаторных систем (калликреинкининовая, свертывающая система, система комплемента, система фибринолиза),активация образования эйкозаноидов и др. производных арахидонового
каскада.
3.Биогенные амины:
- Гистамин (тканевые базофилы, тромбоциты, эндотелий, гладкомышечные
клетки) –
Н 1 + Н 2 - рецепторы: боль, зуд, гиперсекреция слизи, фибрилляция сердца.
Н1 -рецепторы: бронхоспазм, активация хемотаксиса и лимфоцитотоксичности,
генерация липидных медиаторов, повышение проницаемости, подавление номотопного водителя сердечного ритма.
169
Н2- рецепторы:бронходилятация, расширение артериол, торможение хемотаксиса, дегрануляции, экзоцитоза, лимфоцитотоксичности,стимуляция супрессорного действия лимфоцитов, аритмогенный эффект.
- Серотонин (энтерохромаффинные клетки, тромбоциты, эозинофилы) - агрегация тромбоцитов, бронхоспазм, расширение артериол, повышение проницаемости, спазм поврежденных сосудов,стимуляция стероидогенеза.
- Адреналин, норадреналин (тромбоциты) – спазм сосудов, снижение проницаемости, агрегация тромбоцитов.
4. Полипептидные медиаторы:
А.Контактная полисистема плазмы крови: кининовая система, комплемент, свертывающая система, система фибринолиза.
Кинины – повышение сосудистой проницаемости (намного превышает
эффект гистамина), боль, спазм гладкомышечных клеток венул, бронхов, матки, кишечника, расширение артериол (вазодилятаторы), хемотаксис лейкоцитов, стимуляция миграции и митогенеза лимфоцитов, стимуляция фибробластов (пролиферация и коллагеногенез), торможение миграции нейтрофилов,
усиление дегрануляции тканевых базофилов (мастоцитов), усиление продукции
простациклинов эндотелиоцитами, стимуляция циклооксигеназы в различных
клетках, действие на системную гемодинимику-гипотензия, стимуляция сердечной деятельности, диуретическое действие.
Комплемент:
С5а-сверхсильный анафилотоксин, освобождает гистамин из мастоцитов и базофилов, повышение проницаемости эндотелия посткапиллярных венул, хемоаттрактант нейтрофилов, базофилов, эозинофилов, макрофагов, ингибитор миграции макрофагов, стимулятор липоксигеназы фагоцитов, спазм гладких
мышц, стимуляция лейкоцитарной адгезии, увеличивает освобождение ИЛ-1
(интерлейкина –1) и ФАТ (фактора активации тромбоцитов).
С3а-анафилотоксин средней силы, эффекты сходны с С5а, хемоаттрактантное
действие очень слабое, не активирует липооксигеназу.
С2а-вазоактивный пептид, расширяет микроциркуляторные сосуды, увеличивает сосудистую проницаемость, эффектор наследственного ангионевротического отека.
С3b,С4b-маргинация лейкоцитов, синтез простагландинов, опсонизация клеток, стимуляция экзоцитоза и фагоцитоза.
С5b67-хемоаттрактант лейкоцитов.
Б.Цитокины:
ИЛ-1(интерлейкин) (макрофаги, эндотелий, кератиноциты, микроглия,Влимфоциты, фибробласты, дендритные клетки) - провоспалительные эффекты,
индукция адгезивных молекул, эндопироген, причина продромального синдрома, триггер ответа острой фазы воспаления, главный медиатор иммунного ответа на чужеродные вещества, стимулятор стресса.
ИЛ-6 ( Т- и В- клетки, макрофаги, фибробласты, эндотелий, эпителий тимуса)провоспалительные эффекты, индуктор ответа острой фазы, эндопироген, стимулятор антителопродукции.
170
ИЛ-8 (фибробласты, моноциты, макрофаги) - инициация воспаления и ответа
острой фазы, хемоаттрактант и активатор дегрануляции гранулоцитов и Тлимфоцитов, фактор роста лимфоцитов.
ФНО (фактор некроза опухолей) альфа и бета (макрофаги, мастоциты, лимфоциты, астроциты) – эндогенный пироген, стимулятор острофазного ответа,
индуктор ИЛ-1, ИЛ-6, стимулятор цитотоксичности, гранулоцитов, апоптоза
опухолевых и других клеток, кахексия, гиперкатаболизм, контринсулярное
действие, индукция коллагеназы, прокоагулянтов, ФАТ, фиброгенеза, гранулематоза, ангиогенеза.
ИФН (интерферон) альфа, бета, гамма (макрофаги, Тh1, НК-клетки, фибробласты) - активатор макрофагов, всех видов цитотоксичности, тормозит синтез
цитокинов, пролиферацию тимоцитов, эндопироген, противовирусный, антипролиферативный, противоопухолевый эффекты.
5.Липидные медиаторы.Компоненты системы эйкозаноидов
Циклоксигеназный путь активации:
1. Простагландины (Pg Е2,PgF2 ,-различные клетки,PgD2 –тканевые базофилы,
PgF1a-эндотелий) - расширение сосудов и повышение проницаемости
(Е1,У2,D2), сужение сосудов кожи и понижение проницаемости, подавление
эмиграции (F2a,спазм гладких мышц бронхов(D2,G2,H2),потенцирование болевых эффектов кининов и гистамина (Е2),активация фагоцитов, стимуляция
адгезии и фагоцитоза, хемотаксис нейтрофилов, адгезия и реакция освобождения тромбоцитов (G2,H2), опосредуют внутриклеточное действие пирогенов в
гипоталамусе (Е1 и Е2) и цитокинов при преиммунном ответе, стимулируют
секрецию синовиальной жидкости в суставах и протеолиз в мышцах (Е2), стимуляция дегрануляции мастоцитов (D2,E2).
2. Тромбоксаны(тромбоциты, эндотелий, макрофаги) - вазоконстрикция, хемотаксис и маргинация нейтрофилов, адгезия, агрегация, реакция освобождения тромбоцитов, бронхоспазм
3. Простациклины (эндотелий) – вазодилятация, стимуляция коллатерального
кровотока, антитромботическое, антиадгезивное, антикоагуляционное действие, стимуляция фибринолиза, антиатерогенный эффект.
Липоксигеназный путь активации:
4. Лейкотриены ( различные клетки, особенно нейтрофилы, мастоциты) – вазоконстрикция и увеличение проницаемости, бронхоспазм, хемотаксис, маргинация нейтрофилов, макрофагов, хемотаксис и ингибирование пролиферации
лимфоцитов, хемотаксис эозинофилов, дегрануляция базофилов.
5. ФАТ (фактор активации тромбоцитов) – (базофилы, нейтрофилы, макрофаги, мастоциты, эозинофилы, эндотелий) – стимулятор адгезии, агрегации и
реакции освобождения тромбоцитов, активатор гранулоцитов, вазо- и бронхоконстриктор, способствует продукции эйкозаноидов, в малых концентрациях
расширяет сосуды, синергист тромбоксанов, стимулирует эмиграцию нейтрофилов и базофилов,мощный повышающий сосудистую проницаемость агент (в
10 000 раз активнее гистамина).
171
6. Гидроксиэйкозаполиеновые кислоты (нейтрофилы, другие клетки)хемотаксис, хемокинез и активация нейтрофилов, эозинофилов, макрофагов,
ингибирование пролиферации лимфоцитов.
II.Противовоспалительные медиаторы( медиаторы пролиферации)
1. Полисахаридные медиаторы (гликозамингликаны)
-Гепарин (мастоциты, эозинофилы, базофилы, макрофаги,
фибробласты).
–связывает биогенные амины, ингибирует комплемент, коагуляцию,
адгезию, агрегацию, снижает активность кининовой системы,служит структурным компонентом межклеточного вещества соединительной ткани,
участвует в процессах регенерации.
-Гепаран-сульфаты,
хондроитин-сульфаты,
дерматан-сульфаты
2.Ингибиторы протеаз– альфа-1-антитрипсин, альфа-2-макроглобулин, антиплазмин, антитромбин –III, ингибиторы комплемента и др. – подавляют
активность лизосомальных гидролаз и сторожевой контактной полисистемы
крови, уменьшают альтерацию и ликвидируют последствия экзоцитоза.
3.Антифосфолипазы -макрокортин,ренокортин, липомодулин - ингибируют
образование эйкозаноидов.
4.Антиоксиданты - церулоплазмин, гаптоглобин, гемопексин, транскобаламин, пероксидаза, супероксиддисмутаза, бета-2-микроглобулин, амилоидА, С-реактивный белок - инактивация активных кислородных радикалов и
липоперекисей.
5.Инактиваторы провоспалительных медиаторов:
-арилсульфатаза – инактивация лейкотриенов;
-гистаминаза – инактивация гистамина;
-кининаза – инактивация кининов; и др.
6. Полиамины – кадаверин, путресцин, спермин, спермидин (все клетки) –
торможение экссудации, стимуляция регенерации.
7. ИЛ-10 ( Т-лимфоциты) – ингибитор продукции цитокинов, блокирует
функции Th1-хелперов.
III.Регуляторы пролиферации
1. Факторы роста(макрофаги, лимфоциты, фибробласты, тромбоциты и др.)
–стимуляция пролиферации и ограничение апоптоза.
2. Кейлоны – гликопротеидные тканеспецифические ингибиторы роста.
3. Фибронектин-хемоаттрактант фибробластов.
4. Ламинин-главный адгезивный белок базальных мембран.
5. Синдекан-интегральный протеогликан клеточных мембран, связывает
коллаген, фибронектин и тромбоспондин.
6. Тромбоспондин – гликопротеид, образует комплексы с синдеканом, коллагеном и гепарином, играет существенную роль в сборке костной ткани.
Образование и реализация эффектов биологически активных веществ (БАВ)
- одно из ключевых звеньев воспаления. БАВ обеспечивают закономерный ха-
172
рактер развития воспаления, формирование его общих и местных проявлений, а
также исходы воспаления. Именно поэтому БАВ нередко именуют как «медиаторы воспаления».
Медиаторы воспаления - это местные химические сигналы, образующиеся, высвобождаемые либо активируемые в очаге воспаления, действующие
и разрушаемые также в пределах очага. Под медиаторами (посредниками) воспаления понимают биологически активные вещества, ответственные за возникновение или поддержание тех или иных воспалительных явлений, например
повышенной сосудистой проницаемости, эмиграции и т. д.
Это те же вещества, которые в условиях нормальной жизнедеятельности
организма, образуясь в различных органах и тканях в физиологических концентрациях, ответственны за регуляцию функций на клеточном, тканевом уровне.
При воспалении, местно высвобождаясь (вследствие активации клеток и жидких сред) в больших количествах, они приобретают новое качество — медиаторов воспаления. Практически все медиаторы являются и модуляторами воспаления, т. е. способны усиливать или ослаблять выраженность воспалительных
явлений. Это обусловлено комплексностью их влияния и взаимодействием их
как с клетками-продуцентами этих веществ, так и между собой. Соответственно
эффект медиатора может быть добавочным (аддитивным), потенцирующим
(синергистическим) и ослабляющим (антагонистическим), а взаимодействие
медиаторов возможно на уровне их синтеза, секреции или эффектов.
Медиаторное звено является основным в патогенезе воспаления. Оно координирует взаимодействие множества клеток — эффекторов воспаления, смену клеточных фаз в очаге воспаления. Соответственно патогенез воспаления
можно представить себе как цепь множественных межклеточных взаимодействий, регулируемых медиаторами-модуляторами воспаления.
Медиаторы воспаления обусловливают развитие и регуляцию процессов
альтерации (включая изменение обмена веществ, физико-химических параметров, структуры и функции), развитие сосудистых реакций, экссудации жидкости и эмиграции клеток крови, фагоцитоза, пролиферации и репаративных процессов в очаге воспаления.
Большинство медиаторов выполняют свои биологические функции специфически воздействуя на рецепторы клеток-мишеней. Однако некоторые из них
имеют прямую ферментативную или токсическую активность (например, лизосомальные гидролазы и активные кислородные радикалы). Функции каждого
медиатора регулируются соответствующими ингибиторами.
Источниками медиаторов воспаления могут служить плазма крови и клеткиучастники воспаления. В соответствии с этим выделяют 2 большие группы медиаторов воспаления: гуморальные и клеточные. Гуморальные
медиаторы в основном представлены полипептидами, которые постоянно циркулируют в крови в неактивном состоянии и синтезируются преимущественно
в печени. Эти медиаторы составляют так называемую «сторожевую полисистему плазмы крови». Клеточные медиаторы могут синтезироваться de novo
(например, метаболиты арахидоновой кислоты) или высвобождаться из клеточ-
173
ных депо (например, гистамин). Источниками клеточных медиаторов в очаге
воспаления являются, в основном, макрофаги, нейтрофилы и базофилы.
Из гуморальных медиаторов воспаления наиболее важными являются
производные комплемента. Среди почти 20 различных белков, образующихся
при активации комплемента, непосредственное отношение к воспалению имеют его фрагменты С5а, С3а, С3b и комплекс С5b—С9. При этом С5а и в меньшей степени С3а являются медиаторами острого воспаления. СЗb опсонизирует
патогенный агент и соответственно способствует иммунной адгезии и фагоцитозу. Комплекс С5b—С9 ответствен за лизис микроорганизмов и патологически
измененных клеток. Источником комплемента является плазма крови и в
меньшей мере тканевая жидкость. Усиленная поставка плазменного комплемента в ткань является одним из важных назначений экссудации. С5а, образующийся из него в плазме и тканевой жидкости под влиянием карбоксипептидазы N, C5a des Arg и С3а повышают проницаемость посткапиллярных венул.
При этом C5a и С3а, будучи анафилатоксинами (т. е. либераторами гистамина
из тучных клеток), повышают проницаемость как прямо, так и опосредованно
через гистамин Эффект C5a des Arg не связан с гистамином, но является
нейтрофилзависимым, т. е. осуществляется за счет факторов проницаемости,
вьсвобождаемых из полиморфноядерных гранулоцитов, — лизосомальных
ферментов и неферментных катионных белков, активных метаболитов кислорода. Кроме того, C5a и C5a des Arg привлекают нейтрофилы. В отличие от них
С3а практически не обладает хемотаксическими свойствами. Активные компоненты комплемента высвобождают не только гистамин и гранулоцитарные
продукты, но и интеряейкин-1, простагландины, лейкотриены, фактор, активирующий тромбоциты, и синергистически взаимодействуют с простагландинами
и веществом Р.
Кинины — вазоактивные пептиды, образующиеся из кининогенов (альфа2-глобулинов) под влиянием калликреинов в плазме (нонапептид брадикинин) и в тканевой жидкости (декапептид лизилбрадикинин, или каллидин).
Пусковым фактором активации калликреин-кининовой системы является активация при повреждении ткани фактора Хагемана (XII фактор свертывания крови), превращающего прекалликреины в калликреины.
Кинины опосредуют расширение артериол и повышение проницаемости
венул путем контракции эндотелиальных клеток. Они сокращают гладкую мускулатуру вен и повышают внутрикапиллярное и венозное давление. Кинины
угнетают эмиграцию нейтрофилов, модулируют распределение макрофагов,
стимулируют миграцию и митогенез Т-лимфоцитов и секрецию лимфокинов.
Они также усиливают пролиферацию фибробластов и синтез коллагена и, следовательно, могут иметь значение в репаративных явлениях и в патогенезе хронического воспаления.
Один из наиболее значимых эффектов кининов— активация рефлексов
путем раздражения окончаний чувствительных нервов и опосредование, таким
образом, воспалительной боли. Кинины вызывают или усиливают высвобождение гистамина из тучных клеток, синтез простагландинов многими типами клеток, поэтому некоторые из их основных эффектов— вазодилятация, сокраще-
174
ние гладкой мускулатуры, боль — связывают с высвобождением других медиаторов, особенно простагландинов.
Активация фактора Хагемана запускает не только процесс кининообразования, но и свертывания крови и фибринолиза. При этом образуются такие медиаторы, как фибринопептиды и продукты деградации фибрина, которые являются мощными хематтрактантами. Кроме того, фибринолиз и образование
тромбов в сосудах очага имеют существенное значение как в патологических,
так и в защитных явлениях воспаления.
Из клеточных медиаторов первостепенный интерес вызывают эйкозаноиды поскольку скорее всего именно они являются центральным медиаторным
звеном воспалительной реакции. В пользу этого свидетельствуют продолжительное поддержание продукции эйкозаноидов в очаге, их тесная связь с ключевым событием воспалительного процесса — лейкоцитарной инфильтрацией,
мощный противовоспалительный эффект ингибиторов их синтеза.
Основную роль в продукции эйкозаноидов в очаге воспаления играют
лейкоциты, особенно моноциты и макрофаги, хотя они образуются почти всеми
типами ядерных клеток при стимуляции последних. Преобладающими эйкозаноидами в очаге воспаления почти всегда оказываются простагландин (ПГ) Е2,
лейкотриен (ЛТ) B4 и 5-гидроксиэйкозатетраеновая кислота (5-ГЭТЕ). Образуются также, хотя и в меньшем количестве, тромбоксан (Ткс) А2, ПГF2альфа,
ПГD2, простациклин (ПГ12), ЛТС4, ЛТD4, ЛТЕ4, другие ГЭTЕ.
Главными эффектами эйкозаноидов при воспалении являются влияния на
лейкоциты. ПГ, Ткс и особенно ЛТ являются мощными хематтрактантами и играют, таким образом, важную роль в механизмах самоподдержания лейкоцитарной инфильтрации. ПГ сами не повышают сосудистую проницаемость, но,
будучи сильными вазодилятаторами, усиливают гиперемию и, следовательно,
экссудацию. ЛТС4, JITD4, ЛТЕ4 повышают проницаемость сосудов путем прямой контракции эндотелиальных клеток, а ЛТВ4 — как нейтрофилзависимый
медиатор. ПГ и ЛТ имеют значение в генезе воспалительной боли. При этом
ПГЕ2, не обладая прямой болевой активностью, повышает чувствительность
рецепторов афферентных болевых нервных окончаний к брадикинину и гистамину. ПГЕ2 является сильным жароповышающим агентом, и лихорадка при
воспалении может быть отчасти обусловлена его высвобождением. ПГ играют
ключевую роль в модуляции воспалительного процесса, осуществляя двунаправленную регуляцию экссудации, эмиграции и дегрануляции лейкоцитов, фагоцитоза. Так, например, ПГЕ способны потенцировать развитие отека, вызванного гистамином или брадикинином, а ПГF2альфа, напротив, ослаблять. Аналогичные отношения между ПГЕ и ПГF2альфа распространяются также на
эмиграцию лейкоцитов.
Особо широкий спектр взаимодействий с другими медиаторами воспаления характерен для ЛТ. Они синергистически взаимодействуют в отношении
бронхоспазма с гистамином, ацетилхолином, ПГ и Ткс, стимулируют высвобождение ПГ и Ткс. Модуляторная функция эйкозаноидов осуществляется через изменения соотношения циклических нуклеотидов в клетках.
175
Источниками гистамина являются базофилы и тучные клетки. Серотонин (нейромедиатор) у человека кроме незначительного количества в тучных
клетках содержится также в тромбоцитах и энтерохромаффинных клетках. Благодаря быстрому высвобождению при дегрануляции тучных клеток, способности изменять просвет микрососудов и вызывать непосредственную контракцию
эндотелиальных клеток венул гистамин и серотонин считаются основными медиаторами первоначальных микроциркуляторных нарушений в очаге острого
воспаления и немедленной фазы повышения проницаемости сосудов. Гистамин
играет дуалистическую роль как в отношении сосудов, так и клеток. Через Н2рецепторы он расширяет артериолы, а через H1-рецепторы суживает венулы и,
таким образом, повышает внутрикапиллярное давление. Через Hi -рецепторы
гистамин стимулирует, а через Нг-рецепторы угнетает эмиграцию и дегрануляцию лейкоцитов. При обычном течении воспаления гистамин действует преимущественно через Нг-рецепторы на нейтрофилах, ограничивая их функциональную активность, и через Hi -рецепторы на моноцитах, стимулируя их. Таким образом, наряду с провоспалительными сосудистыми эффектами он оказывает противовоспалительные клеточные. Серотонин также стимулирует моноциты в очаге воспаления. Гистамин осуществляет двунаправленную регуляцию
пролиферации, дифференцировки и функциональной активности фибробластов
и, следовательно, может иметь значение в репаративных явлениях. Модуляторные эффекты гистамина также опосредуются циклическими нуклеотидами.
Что касается взаимодействий биогенных аминов в очаге воспаления, то
известно, что гистамин через Hi-рецепторы может запускать или усиливать
синтез простагландинов, а через На -рецепторы — угнетать. Биогенные амины
взаимодействуют как между собой, так и с брадикинином, нуклеотидами и нуклеозидами, веществом Р в повышении проницаемости сосудов. Сосудорасширяющее действие гистамина усиливается в комплексе с ацетилхолином, серотонином, брадикинином.
Основным источником лизосомальных ферментов в очаге воспаления
являются фагоциты — гранулоциты и моноциты-макрофаги. Несмотря на
огромную важность в патогенезе воспаления фагоцитоза, фагоциты являются
прежде всего подвижными носителями медиаторов-модуляторов, секретируемых внеклеточно. Вывобождение лизосомального содержимого осуществляется
в ходе их хемотаксической стимуляции, миграции, фагоцитоза, повреждения,
гибели. Главными компонентами лизосом у человека являются нейтральные
протеиназы— эластаза, катепсин G и коллагеназы, содержащиеся в первичных,
азурофильных, гранулах нейтрофилов. В процессах противомикробной защиты,
в том числе при воспалении, протеиназы относятся к факторам “второй очереди” после кислородзависимых (миелопероксидаза — перекись водорода) и таких кислороднезависимых, как лактоферрин и лизоцим, механизмов. Они обеспечивают главным образом лизис уже убитых микроорганизмов. Основные же
эффекты протеиназ — медиация и модуляция воспалительных явлений, в том
числе повреждения собственных тканей. Медиаторный и модуляторный эффек-
176
ты протеиназ осуществляются в отношении сосудистой проницаемости, эмиграции, фагоцитоза.
Повышение проницаемости сосудов под влиянием лизосомальных ферментов происходит за счет лизиса субэндотелиального матрикса, истончения и
фрагментации эндотелиальных клеток и сопровождается геморрагией и тромбозом. Образуя или расщепляя важнейшие хемотаксические вещества, лизосомальные ферменты являются модуляторами лейкоцитарной инфильтрации. В
первую очередь это касается компонентов системы комплемента и калликреинкининовой.
Лизосомальные ферменты, в зависимости от концентрации, могут и сами
усиливать или угнетать миграцию нейтрофилов. В отношении фагоцитоза
нейтральные протеиназы также обладают рядом эффектов. В частности, эластаза может образовывать опсонин С3b; С3b является также важным для адгезии
частиц к поверхности нейтрофила. Следовательно, нейтрофил сам обеспечивает
себе механизм усиления фагоцитоза. Как катепсин G, так и эластаза повышают
сродство Fc-рецептора мембраны нейтрофила к комплексам иммуноглобулинов
и, соответственно, усиливают эффективность поглощения частиц.
Благодаря также способности лизосомальных ферментов активировать
системы комплемента, калликреин-кининовую, свертывания и фибринолиза,
высвобождать цитокины и лимфокины, воспаление развертывается и самоподдерживается в течение длительного времени.
Важнейшим свойством неферментных катионных белков, содержащихся как в азурофильных, так и в специфических гранулах нейтрофилов, является их высокая микробицидность. В этом отношении они находятся в синергистическом взаимодействии с системой миелопероксидаза — перекись водорода. Катионные белки адсорбируются на отрицательно заряженной мембране
бактериальной клетки путем электростатического взаимодействия. В результате этого нарушаются проницаемость и структура оболочки и наступает гибель
микроорганизма, что является предпосылкой для последующего эффективного
лизиса его лизосомальными протеиназами. Высвободившиеся внеклеточно катионные белки опосредуют повышение проницаемости сосудов (главным образом путем индукции дегрануляции тучных клеток и высвобождения гистамина), адгезию и эмиграцию лейкоцитов.
Главным источником цитокинов (монокинов) при воспалении являются
стимулированные моноциты и макрофаги. Кроме того, эти полипептиды продуцируются нейтрофилами, лимфоцитами, эндотелиальными и другими клетками. Наиболее изученными из цитокинов являются интерлейкин-1(ИЛ-1) и
фактор некроза опухоли (ФНО). Цитокины повышают сосудистую проницаемость (неитрофилзависимым путем), адгезию и эмиграцию лейкоцитов. Наряду
с провоспалительными свойствами цитокины могут иметь значение и в непосредственной защите организма, стимулируя нейтрофилы и моноциты к
умерщвлению, поглощению и перевариванию внедрившихся микроорганизмов,
а также усиливая фагоцитоз путем опсонизации патогенного агента.
Стимулируя раневое очищение, пролиферацию и дифференцировку клеток, цитокины усиливают репаративные процессы. Наряду с этим они могут
177
опосредовать тканевую деструкцию (деградацию хрящевого матрикса и резорбцию кости) и, таким образом, играть роль в патогенезе заболеваний соединительной ткани, в частности ревматоидного артрита.
Действие цитокинов вызывает также ряд метаболических эффектов, лежащих в основе общих проявлений воспаления — лихорадки, сонливости, анорексии, изменения обмена веществ, стимуляции гепатоцитов к усиленному
синтезу белков острой фазы, активации системы крови и т. д.
Цитокины взаимодействуют между собой, с простагландинами, нейропептидами и другими медиаторами.
К медиаторам воспаления относится также ряд лимфокинов — полипептидов, продуцируемых стимулированными лимфоцитами. Наиболее изученными из лимфокинов, модулирующих воспалительный ответ, являются фактор,
угнетающий макрофаги, макрофагактивирующий фактор, интерлейкин-2. Лимфокины координируют взаимодействие нейтрофилов, макрофагов и лимфоцитов, регулируя таким образом воспалительную реакцию в целом.
Активные метаболиты кислорода, прежде всего свободные радикалы
— супероксидный анион радикал, гидроксильный радикал НО, пергидроксил,
вследствие наличия на их внешней орбите одного или нескольких непарных
электронов обладают повышенной реактивностью с другими молекулами и,
следовательно, значительным деструктивным потенциалом, который имеет
значение в патогенезе воспаления . Источником свободных радикалов, а также
других кислородпроизводных медиаторов и модуляторов воспаления — перекиси водорода (Н202), синглетного кислорода (f02), гипохлорида (НОС1) служат: дыхательный взрыв фагоцитов при их стимуляции, каскад арахидоновой
кислоты в процессе образования эйкозаноидов, ферментные процессы в эндоплазматическом ретикулуме и пероксизомах, митохондриях, цитозоле, а также
самоокисление малых молекул, таких, как гидрохиноны, лейкофлавины, катехоламины и др.
Роль активных метаболитов кислорода в воспалении состоит, с одной
стороны, в повышении бактерицидной способности фагоцитов и, с другой стороны, — в их медиаторной и модуляторной функциях. Медиаторная роль активных метаболитов кислорода обусловлена их способностью вызывать перекисное окисление липидов, окисление белков, углеводов, повреждение нуклеиновых кислот. Указанные молекулярные изменения лежат в основе вызываемых активными метаболитами кислорода явлений, характерных для воспаления, — повышения проницаемости сосудов (вследствие повреждения эндотелиальных клеток), стимуляции фагоцитов.
Модуляторная ролъ, активных метаболитов кислорода может заключаться как в усилении воспалительных явлений (путем индукции высвобождения
ферментов и взаимодействия с ними в повреждении ткани; не только инициации, но и модуляции каскада арахидоновой кислоты), так и в противовоспалительных эффектах (за счет инактивации лизосомальных гидролаз и других медиаторов воспаления).
Важное значение имеют активные метаболиты кислорода в поддержании
хронического воспаления.
178
К медиаторам и модуляторам воспаления относят также нейропептиды
— вещества, высвобождаемые С-волокнами в результате активации воспалительным агентом полимодальных ноцицепторов, играющих важную роль в возникновении аксонрефлексов в конечных разветвлениях первичных афферентных (чувствительных) нейронов. Наиболее изученными являются вещество Р,
кальцитонин-генсвязанный пептид, нейрокинин А. Нейропептиды повышают
проницаемость сосудов, и эта их способность во многом опосредована медиаторами, происходящими из тучных клеток. Между немиелинными нервами и
тучными клетками имеются мембранные контакты, которые обеспечивают сообщение центральной нервной системы с очагом воспаления.
Нейропептиды синергистически взаимодействуют в повышении проницаемости сосудов как между собой, так и с гистамином, брадикинином, С5а,
фактором, активирующим тромбоциты, лейкотриеном В4; антагонистически —
с АТФ и аденозином. Они оказывают также потенцирующее воздействие на
привлечение и цитотоксическую функцию нейтрофилов, усиливают адгезию
нейтрофилов к эндотелию венул. Кроме того, нейропептиды повышают чувствительность ноцицепторов к действию различных медиаторов, в частности
простагландина Е2 и простациклина, участвуя, таким образом, в воссоздании
воспалительной боли.
Кроме вышеперечисленных веществ к медиаторам воспаления относятся
также ацетилхолив и катехоламины, высвобождающиеся при возбуждении
холин- и адренергических структур. Ацетилхолин вызывает расширение сосудов и играет роль в аксонрефлекторном механизме артериальной гиперемии
при воспалении. Норадреналин и адреналин тормозят рост сосудистой проницаемости, выступая главным образом как модуляторы воспаления.
ЭКССУДАЦИЯ
Изменения кровообращения в очаге воспаления.
При воспалении реакция сосудов стереотипна и развивается в 4 стадии:
1.Кратковременный преходящий спазм артериол и прекапилляров, сопровождающиеся развитием ишемии.
2.Расширение артериол, сопровождающееся ускорением кровотока и развитием артериальной гиперемии.
3.Дальнейшее расширение сосудов и замедление кровотока с развитием венозной гиперемии.
4.Развитие стаза и остановка кровотока.
Начальный спазм сосудов отчетливо выражен при быстро развивающемся
повреждении (ожог, травма) и менее заметен при постепенном развитии повреждения (инфекционный процесс). Причиной вазоконстрикции является выделение под влиянием повреждающего фактора сосудосуживающих БАВ: катехоламинов из симпатических нервных окончаний, тромбоксана А2 из тромбоцитов, эндотелина-1 из поврежденных эндотелиоцитов. Кратковременность ишемии обусловлена быстрой инактивацией этих эффекторов и накоплением вазодилататоров.
179
Расширение артериол, метартериол и прекапиллярных сфинктеров возникает в результате воздействия вазодилататоров: гистамина, кининов, оксида
азота и PgI2 (простациклина). В результате скорость кровотока в микроциркуляторном русле повышается, возрастает количество функционирующих
капилляров, увеличивается кровенаполнение тканей, улучшается их оксигенация - развивается артериальная гиперемия (см. главу «Нарушение периферического кровотока»). Артериальная гиперемия сохраняется недолго (обычно
10-30 мин) и сменяется венозной гиперемией.
Развитие венозной гиперемии начинается с максимального расширения
артериол и прекапиллярных сфинктеров, которые становятся резистентны к сосудосуживающим стимулам, а также с затруднения венозного оттока. Скорость
кровотока в микроциркуляторных сосудах падает. Причинами этого состояния
являются сдавление венул накапливающимся экссудатом, повышение тонуса их
стенок под влиянием гистамина, действующего на Н1-рецепторы. Немаловажную роль в развитии венозной гиперемии имеет изменение реологических
свойств крови - повышение ее вязкости в результате выхода жидкой части крови из сосудистого русла при экссудации и «сладжирования» форменных элементов.
Сладж эритроцитов - это прилипание их друг к другу вследствие адсорбции на их поверхности высокомолекулярных глобулинов (белков острой фазы
воспаления), что снижает их поверхностный потенциал. Агрегаты эритроцитов
формируют структуры напоминающие монетные столбики, которые значительно затрудняют кровоток. Накопление лейкоцитов у стенок посткапилляров и
венул в процессе маргинации также способствует замедлению тока крови.
Закономерное развитие причин венозной гиперемии, приводит к стазу и
прекращению кровотока в очаге воспаления.
Экстравазация жидкости ( экссудация )
Отличительной особенностью сосудистых изменений при воспалении является значительное повышение сосудистой проницаемости и выход богатой
белками жидкости (экссудата) в ткани. Потеря белков плазмой крови приводит
к снижению внутрисосудистого онкотического давления и повышению онкотического давления интерстициальной жидкости. Совместно с повышением гидростатического давления в расширенных сосудах, это приводит к значительному оттоку жидкости в ткани и формированию отека.
Повышение проницаемости сосудистой стенки при остром воспалении
развивается в 3 фазы, каждая из которых обусловлена различными механизмами.
1. Ранняя преходящая фаза возникает вскоре после повреждения и продолжается 15-30 минут. Механизм повышения сосудистой проницаемости заключается в сокращении эндотелиоцитов и формировании промежутков между
ними. Развивается эта реакция под действием гистамина, лейкотриенов, простагландинов и кининов в венулах и не затрагивает капилляры и артериолы.
2. Поздняя продленная фаза начинается через 2 часа после повреждения и
продолжается 24 часа и более. Повышается проницаемость капилляров и венул.
180
Изменения в микрососудах являются следствием активации эндоте-лиоцитов и
обеспечивается цитокинами (ИЛ-1, ФНО, ИФН-γ). Происходит перестройка цитоскелета эндотелиоцитов, что приводит к разрушению межклеточных контактов и втягиванию цитоплазматических отростков с образованием щелей между
соседними эндотелиоцитами. Важное значение имеет усиление транспорта через эндотелиоциты - трансцитоза за счет увеличения количества специфических
транспортных органелл.
3.Раннее стойкое повышение проницаемости возникает вследствие прямого
повреждения эндотелия, некроза эндотелиоцитов и их отделения от базальной
мембраны. Подобная ситуация часто встречается при значительной альтерации
в результате ожогов или инфекции экзотоксинпродуцирующими возбудителями (например, Corynebacterium diphteriae, Str pyogenes). Повышение сосудистой
проницаемости развивается сразу после повреждения, продолжается несколько
часов и наблюдается в артериолах, капиллярах и венулах.
Жидкость, выходящая из микрососудов при повышении их проницаемости, содержащая большое количество белка и форменные элементы крови
формирует экссудат, который накапливается в тканях и/или полостях тела при
воспалении. Экссудат следует отличать от транссудата, формирующегося, в основном, в результате повышения гидростатического давления в сосудах без повышения их проницаемости (например, при сердечной недостаточности). Для
транссудата характерны удельная плотность < 1.012, отсутствие клеток и содержание белка < 2%; для экссудата - удельная плотность > 1.015, наличие клеток-участников воспаления и содержание белка > 2%.
Клеточный и химический состав экссудата имеет диагностическое значение и зависит от причины воспаления, ткани, в которой развивается воспаление, реактивности организма и ряда других факторов.
Различают следующие типы экссудатов:
Серозный - содержит преимущественно альбумины в умеренной концентрации (3-5%) небольшое количество клеток, образуется на ранних стадиях
воспаления.
Катаральный (слизистый) - образуется при воспалении слизистых оболочек воздухоносных путей, ЖКТ. Отличается высоким содержанием мукополисахаридов и секреторных антител (IgA), содержит лизоцим.
Фибринозный экссудат отличается высоким содержанием фибриногена,
что является результатом значительного повышения проницаемости сосудов.
При контакте с поврежденными тканями фибриноген превращается в фибрин и
выпадает в виде ворсинчатых масс (на серозных оболочках) или пленки (на
слизистых), вследствие чего экссудат уплотняется. Если фибринозная пленка
расположена рыхло, поверхностно, легко отделяется без нарушения целостности слизистой, такое воспаление называется крупозным. Оно наблюдается в желудке, кишечнике, трахее, бронхах. В том случае, когда пленка плотно спаяна с
подлежащей тканью и ее удаление обнажает язвенную поверхность, речь идет о
дифтеритическом воспалении. Оно характерно для миндалин, полости рта,
пищевода. Такое различие обусловлено характером эпителия слизистой оболочки и глубиной повреждения. Фибринозные пленки могут самопроизвольно
181
отторгаться благодаря аутолизу, развертывающемуся вокруг очага, и демаркационному воспалению и выходить наружу; подвергаться ферментативному
расплавлению или организации, т. е. прорастанию соединительной тканью с
образованием соединительно-тканных сращений, или спаек. Фибринозный экссудат может наблюдаться при дифтерии, дизентерии, туберкулезе.
Гнойный - содержит большое количество лейкоцитов, фрагменты некротизированных тканей. Образуется чаще всего при инфекциях, вызываемыхпиогенными бактериями (стафилококками, стрептококками, пневмококками
и др.).
Геморрагический - содержит большое количество белка и эритроцитов. Образуется при повреждениях сосудов с разрушением базальной мембраны, характерен для сибирской язвы, гриппозной пневмонии и др.
Смешанные формы экссудата могут быть самыми разнообразными (серозно-фибринозный, гнойно-фибринозный и др.)
Биологическое значение экссудации состоит в том, что, являясь одним из
основных компонентов воспаления как, патологического процесса, она выполняет вместе с тем важную защитную роль, которая заключается прежде всего в
локализации воспалительного процесса.
Положительные последствия:
1. Экссудат разбавляет, снижает концентрацию повреждающих агентов и
тем самым ослабляет их вредное воздействие.
У новорожденных и эмбрионов, у которых экссудация практически не
развивается, воспаление принимает характер альтеративного, проявляется тканевыми дистрофиями и некрозом.
2. С экссудатом в очаг воспаления поступают защитные факторы - иммуноглобулины, факторы свертывания крови и др.
3. Экссудат обладает бактерицидными свойствами.
4. Если в экссудате содержится фибрин, он блокирует лимфатические сосуды и препятствует резорбции и генерализации повреждающих факторов.
Отрицательные последствия:
1. Экссудат может механически смещать органы и ткани, тем самым
нарушая их работу (экссудат в плевральной полости, перикарде).
2. Экссудат может сдавливать ткани, обусловливая боль и повреждения
(особенно в замкнутых полостях - при артритах, гайморитах, пульпитах и др.).
3. Если происходит потеря экссудата или его удаление при медицинских
манипуляциях - это может привести к обезвоживанию и белковому дефициту
(при диарее, потере "плазмы" с обожженной кожи, нерациональном пункгировании при плевритах).
4. Резорбция экссудата может сопровождаться интоксикацией.
Скопление в ткани экссудата обусловливает такой внешний местный признак воспаления, как припухлость. Кроме того, наряду с действием брадикинина, гистамина, простагландинов, нейропептидов давление экссудата на окончания чувствительных нервов имеет некоторое значение в возникновении воспалительной боли.
182
Эмиграция лейкоцитов
Наряду с микроциркуляторными изменениями в очаге воспаления происходят характерные изменения функций ряда клеточных элементов, обусловленные их активацией. Активируются эндотелиоциты, различные популяции
лейкоцитов, тромбоциты, клетки соединительной ткани и др. Эти клеточные
процессы определяют защитную функцию воспаления.
Для обобщающей характеристики этих процессов приняты термины: эмиграция клеточных элементов, то есть их выход за пределы микрососудов в
ткани и пролиферация.
В ходе воспалительной реакции в результате внедрения патогенных возбудителей или повреждения какой-либо другой природы имеет место активация макрофагов.
Это проявляется фагоцитозом и выделением большого количества регуляторных молекул главным образом пептидной природы - цитокинов. Цитокины в свою очередь активируют множество других клеток-мишеней. Цитокины интерлейкины (ИЛ-1, ИЛ-3, ИЛ-8) и фактор некроза опухолей (ФНО) и др. в
очаге воспаления активируют эндотелиоциты и нейтрофилы.
Активация эндотелиоцитов и нейтрофилов под влиянием ИЛ-1 и ФНО
проявляется во-первых в появлении на их мембранах особых адгезивных молекул (адгезинов), относящихся к классам селектинов и интегринов. В результате
нейтрофилы выходят из кровотока к сосудистой стенке и "прокатывается" по
эндотелию, а затем "прилипают" - фиксируются к эндотелиоцитам в местах
межэндотелиальных контактов. Потом нейтрофилы формируют псевдоподии,
проникающие между эндотелиальными клетками и как бы "переливаясь" в
псевдоподии в конце концов выходят за пределы сосудов. "Прокатывание"
осуществляется с помощью адгезинов типа селектинов, "прилипание" и прохождение в межэндотелиальные пространства с помощью интегринов.
Неактивированные нейтрофилы и эндотелиоциты взаимодействовать не
могут и эмиграция нейтрофилов не происходит.
Наследственный дефект по одному из адгезинов эндотелиоцитов (болезнь
неадгезивных лейкоцитов) снижает сопротивляемость организма к инфекционным заболеваниям.
Активация эндотелиоцитов проявляется выделением в кровь нескольких
прокоагулянтных факторов и их пролиферацией и при формировании грануляционной ткани.
Состояние активации эндотелиоцитов достигает пика за 4-6 часов после
воздействия ИЛ-1 и других активаторов. Исходный уровень активности, в том
числе освобождение мембраны от адгезинов, восстанавливается за 24-36 часов,
если нет повторных активирующих воздействий.
Мембрана нейтрофила быстро теряет адгезивные молекулы после прохождения нейтрофила через стенку капилляра.
Наряду с адгезивностью, активация нейтрофилов в очаге воспаления проявляется хемотаксисом, фагоцитозом, бактерицидностью и апоптозом - запрограмированной гибелью нейтрофила после его выхода в ткани.
183
Хемотаксис обусловлен рядом факторов. Это моноцитарный цитокин
йнтерлейкин-8 (ИЛ-8), метаболиты арахидоновой кислоты - лейкотриены, активированные компоненты системы комплемента, факторы, продуцируемые базофилами, нейтрофилами, Т-лимфоцитами-эффекторами ГЗТ и мн. др.
Фагоцитоз - важнейший механизм защитного действия воспалительной
реакции впервые описанный И.И. Мечниковым.
Бактерицидные механизмы нейтрофила разнообразны. Одним из важнейших является так называемый "респираторный взрыв" - резкое увеличение
потребления О2 (в 10-15 раз) и внемитохондриального окисления. Образующиеся при этом свободные радикалы перекиси водорода, супероксидного аниона,
гидроксильного радикала, синглетного кислорода повреждают микробы. Особо
мощным повреждающим действием обладает система: миелопероксидаза + перекись водорода + галогены.
По современным представлениям, возможно, наиболее важным бактерицидным фактором является радикал NO -.
Хорошо известно бактерицидное действие таких факторов, как лизоцим,
катионные белки, лактоферрин. Недавно были изучены пептиды с "антибиотическим" действием, так называемые дефензины.
Экзоцитоз нейтрофилов способствует проявлению их внеклеточной бактерицидности. В результате экзоцитоза, апоптоза выходят также гидролитические лизосомальные ферменты. Это приводит к активации систем ограниченного протеслиза, а также простагландин-тромбоксановой системы и накоплению
БАВ . Если ферментов очень много, может происходить расплавление тканевых
структур и формирование таких форм гнойного воспаления, как абсцессы и
флегмоны.
Преимущественная эмиграция нейтрофилов характерна для начального
периода воспаления, они выполняют исключительно важную работу по очистке
тканей от патогенных возбудителей и необратимо поврежденных фрагментов
самих тканевых структур.
Постепенно начинает преобладать эмиграция моноцитов.Моноцит, покидая сосуд, превращается в экссудативный моноцит-макрофаг, затем в незрелый
и зрелый макрофаг. Зрелые макрофаги по структурно-функциональным характеристикам разделяются на фагоцитарные и секреторные макрофаги.
В отличие от нейтрофилов, которые располагаются преимущественно в
центре очага воспаления, макрофаги размещаются по периферии, образуя
своеобразный вал (предшественник грануляционного вала), отграничивающий
очаг воспаления от неповрежденной ткани. Сначала формируется нейтрофильно-макрофагальный вал, затем макрофагальный и наконец, макрофагальнофибробластный вал, который непосредственно трансформируется в грануляционную ткань.
Фагоцитарная активность макрофагов значительно ниже активности
нейтрофилов, хотя они, так же как нейтрофилы, осуществляют "респираторный взрыв" и неокислительную бактерицидную активность.
Главной их функцией является отграничение очага воспаления, предотвращение дальнейшей агрессии и создание условий для антигенной стимуляции
184
для Т- и В-лимфоцитов, тем самым макрофаги участвуют в формировании специфического иммунного ответа на системном уровне.
Выделяя цитокины (ИЛ-1, ФНО, ИЛ-6, ИЛ-8), колониестимулирующие
факторы - КСФ и многие другие, макрофаги осуществляют аутокринную (в отношении других макрофагов), паракринную (в отношении других клеток, находящихся в непосредственной близости: эндотелиоцитов, нейтрофилов, лимфоцитов, тромбоцитов и др.) и эндокринную (системную на уровне всего организма) регуляторные функции. Например, ИЛ-1 и ФНО (в несколько меньшей
степени) стимулируют тепловой центр - вызывая лихорадку, ИЛ-1 - костный
мозг, мобилизуя из костного мозга уже созревшие нейтрофилы в периферическую кровь - эффект в течение 24 часов, ИЛ-1, ИЛ-3, колониестимулирущие
факторы усиливают миелопоэз и появление незрелых форм нейтрофилов в крови, ИЛ-1 стимулируют поступление в кровь стрессовых гормонов: кортикотропина и глюкокортикоидов
ПРОЛИФЕРАЦИЯ
Пролиферация - завершающая стадия воспаления - характеризуется увеличением числа стромальных и, как правило, паренхиматозных клеток ,
также образованием межклеточного вещества в очаге воспаления. Эти процессы направлены на регенерацию альтерированных и замещение разрушенных
тканевых элементов. Существенное значение на этом этапе воспаления имеют
различные БАВ, стимулирующие пролиферацию клеток (митогены).
Пролиферативные процессы при остром воспалении начинаются вскоре
после воздействия флогогенного агента на ткань и более выражены по периферии зоны воспаления. Одним из условий оптимального течения пролиферации
является затухание процессов альтерации и экссудации.
Размножение соединительнотканных элементов сопровождается активным биосинтезом коллагена. Молодая грануляционная ткань постепенно заменяется зрелой фиброзной, а затем рубцовой тканью.
Таким образом, воспалительная пролиферация постепенно переходит в
процесс регенерации, репарации, восстановления ткани, поврежденной или даже разрушенной воспалительной альтерацией.
Однако процесс рубцевания носит обратный, управляемый характер. Дело в том, что при контактном взаимодействии коллагеновые волокна тормозят
деление фибробластов и коллагеногенез в них, и даже способствуют деградации и гибели фибробластов.
Коллагенокластические фибробласты, напротив, при контакте с коллагеном стимулируются.
Таким образом, осуществляется обратная связь, приостанавливающая
разрастание рубцовой ткани в очаге воспаления и приводящая к обратному развитию грубого фиброзного рубца. Нарушения равновесия в этой системе могут
проявиться в недостаточно активном размножении клеток и сниженном биосинтезе коллагена, что клинически проявляется в форме вялотекущих нагноительных процессов и незаживающих трофических язв. Избыточная неконтролируемая активность приводит к грубым деформирующим и нарушающим
185
функции рубцам и склерозированию паренхиматозных органов, например, при
хронических инфекционных поражениях (пневмония и склероз легких) или интоксикациях (хронический гепатит и цирроз печени при алкоголизме).
Размножение эндотелиоцитов играет решающую роль в формировании
сети грануляционной ткани.
Пролиферация макрофагов наблюдается при ГЗТ и хроническом специфическом инфекционном воспалении, протекающем в форме инфекционных
гранулем. Происходит не только деление макрофагов, но и их дифференцировка: они превращаются в эпителиоидные и гигантские многоядерные клетки
Лангганса.
Пролиферация и дифференцировка фибробластов и эндотелиоцитов стимулируется многочисленными факторами роста, которые продуцируются многими клеточными элементами: макрофагами (ИЛ-1, фактор некроза опухолей,
макрофагальный фактор роста и пр.), тромбоцитами (тромбоцитарный фактор
роста и др.), эндотелиоцитами, Т-лимфоцитами-эффекторами, а также ростовыми факторами плазмы.
Общие проявления воспаления
Воспаление, являясь местным процессом, всегда приводит к перестройке
системной нейроэндокринной и иммунной регуляции. На уровне целостного
организма реакция на воспаление проявляется так называемым «ответом острой
фазы», включающим болевой синдром, лихорадку, лейкоцитоз, диспротеинемию, повышение скорости оседания эритроцитов, интоксикацию. Все эти изменения опосредованы системными эффектами медиаторов воспаления.
Болевой синдром.Патогенез:
а) Действие на нервные окончания таких медиаторов, как гистамин, калликреин, каллидин.
б) Механическое сдавление болевых рецепторов, особенно если воспаление
развивается в замкнутых полостях: пульпит, гайморит, панариций.
в) Повышение осмотического давления.
г) Действие Н+ на болевые рецепторы.
д) Прямое действие патогенного агента на болевые рецепторы.
Лихорадка развивается в результате действия ИЛ-1, ИЛ-6 и ФНО, называемых в данном случае «эндогенными пирогенами» (см. главу «Лихорадка»).
Лейкоцитоз - это увеличения количества лейкоцитов в определенном объеме
крови. Его развитие обусловлено продукцией клетками-участниками воспаление колониестимулирующих факторов, усиливающих лейкопоэз. Например,
макрофаги синтезируют ГМ-КСФ, Г-КСФ и М-КСФ, которые стимулируют
пролиферацию и дифференцировку клеток-предшественниц гранулоцитов и
моноцитов. Это приводит к увеличению соответствующих популяций лейкоцитов в периферической крови и обеспечивает их достаточный уровень в очаге
воспаления.
186
Диспротеинемия характеризуется повышением в крови концентрации белков глобулиновой фракции при снижении концентрации альбуминов. Общее
название этих белков «белки острой фазы воспаления», синтез их происходит в
гепатоцитах, стимулированных ИЛ-1, ИЛ-6 и ФНО. В соответствии с выполняемыми функциями белки острой фазы можно разделить на следующие группы:
1.
Антиоксидантные: церулоплазмин, амилоид А, гаптоглобин, Среактивный белок, транскобаламин,α2-макроглобулин.
2.
Антимикробные: С-реактивный белок, лактоферрин, компоненты комплемента.
3.
Регуляторы гемостаза и антигемостаза: VII, VIII и IX факторы свертывания, плазминоген, антитромбин III.
Ускорение СОЭ является следствием диспротеинемии, так как снижение
концентрации низкомолекулярных альбуминов и увеличение концентрации высокомолекулярных глобулинов приводит к дестабилизации взвеси эритроцитов
и ускорению их агглютинации и осаждения.
Возможность возникновения и характер развития воспаления в значительной мере определяется состоянием реактивности организма. В зависимости
от выраженности ответа организма на флогогенный агент воспаление может
быть:
1.Нормэргическое - выраженность ответа адекватна силе и характеру действия агента, вызвавшего его.
2. Гиперегическое - выраженность воспалительного ответа значительно превосходит необходимый и достаточный для элиминации флогогенного фактора
объем. Воспаление протекает бурно, с повреждением собственных тканей. Такое воспаление характерно для аллергических реакций.
3.Гипоэргическое - воспалительный ответ выражен недостаточно, приобретает затяжной характер с преобладанием процессов альтерации. Такое воспаление характерно для лиц с иммунологической недостаточностью.
Влияние различных систем организма на характер воспалительной
реакции
1. Гормональная регуляция:
а) усиливают воспаление: СТГ, ТТГ, дезоксикортикостерон, инсулин, тироксин, они стимулируют рост фибробластов, повышают проницаемость капилляров.
б) ослабляют воспаление: АКТГ, глюкокортикоиды, женские половые
гормоны. Они задерживают высвобождение лизосомальных гидролаз, уменьшают проницаемость сосудов, тормозят эмиграцию.
2. Нервная система.
Применение местных анестетиков (новокаин, дикаин и др.) дает при воспалении терапевтический эффект.
3. Роль возраста.
а) У новорожденных, ввиду слабого включения сосудистого русла не выражена экссудация, воспаление протекает по альтеративному типу.
187
б) В раннем детском возрасте недоразвитие иммунной системы создает
тенденцию к генерализации воспалительного процесса, септическим осложнениям (тому способствует и недостаточность в этом возрасте пролиферативной
реакции).
в) В пожилом возрасте имеется тенденция к хроническому течению воспалительного процесса.
Принципы противовоспалительной терапии
Схема лечения воспаления базируется на этиотропном, патогенетическом,
саногенетическом и симптоматическом принципах.
Этиотропная терапия - подразумевает устранение, прекращение, уменьшение силы и/или длительности действия на ткани и органы флогоген-ных факторов. Например, извлечение травмирующих инородных тел,
уничтожение инфекционных агентов (путем применения антимикробных,
противопаразитарных и антигрибковых препаратов различных групп).
Патогенетическая терапия - имеет целью блокирование механизма развития
воспаления, при этом воздействия направлены на разрыв звеньев патогенеза
воспаления. К данному виду терапии относятся физиотерапевтические процедуры, стимулирующие артериальную гиперемию, процессы резорбции жидкости; применение антигистаминных препаратов, иммуномодуляторов и
др.
Саногенетическая терапия - направлена на активацию общих и местных механизмов компенсации, регенерации, защиты, восстановления и устранения повреждений и изменений в тканях, вызванных флогогенным агентом, а также
последствий его влияния. Например, стимуляция иммунных и пролиферативных реакций, развитие артериальной гиперемии, фагоцитоза и
др.
Симптоматическая терапия - лечение, направленное на предупреждение или
устранение симптомов, сопровождающих воспаление (с этой целью применяют, например, болеутоляющие, анестезирующие лекарственные средства, вещества, нормализующие функцию органов и систем).
ЛИХОРАДКА
Температурный гомеостаз состоит из равновесия процессов теплопродукции и теплоотдачи. Теплопродукция зависит от интенсивности процессов
метаболизма и мышечной работы. Теплоотдача складывается, как сумма потерь тепла следующими путями:
- Теплоизлучение. Излучение организмом инфракрасных лучей зависит от температуры окружающей среды и температуры поверхности тела. При температуре окружающей среды выше температуры тела теплоотдача данным путем затруднена.
- Теплопроводность – отдача тепла от тела с более высокой температурой к телу с более низкой температурой при прямом контакте. Теплопроводность зависит от площади соприкосновения.
188
- Испарение – отдача тепла путем потоотделения.
- Тепловые потери с мочой и калом обеспечивают незначительную часть теплопотерь.
Лихорадка - типовая защитно-приспособительная реакция высших теплокровных (гомойотермных) животных и человека на воздействие пирогенных
раздражителей, проявляющаяся повышением температуры тела независимо
от температуры окружающей среды, изменениями обмена веществ и физиологических функций организма.
В филогенетическом отношении лихорадка является более поздним проявлением реакции организма на действие патогенного агента и появляется
только при высокой степени развития центральной нервной системы и со
сформировашейся функцией терморегуляции.
Непосредственной причиной лихорадки являются пирогены.
Пирогенами называют те вещества, которые, попадают в организм извне
или образуются внутри него, вызывают лихорадку.
Пирогенны бывают:
1. Первичные
-экзогенные – вирусы, бактерии
-эндогенные – продукты распада тканей
2. Вторичные – образуются в организме в ответ на действие первичного
пирогена. К ним относятся: цитокины организма: интерлейкин-1 (ИЛ-1), интерлейкин-6 (ИЛ-6), интерлейкин-8 (ИЛ-8), фактор некроза опухолей (ФНО), интерферон (ИФН), а также симпатическая нервная система и гормоны, участвующие в стрессе.
Патогенез лихорадки
В основе развития лихорадочной реакции лежит функциональная перестройка центра терморегуляции гипоталамуса. После контакта ретикулоэндотелиальных клеток с экзо- и эндопирогенами начинается выработка цитокинов. Под действием цитокинов происходит повышение «установочной точки» (Set-point) центра терморегуляции гипоталамуса на более высокий температурный уровень (выше 37оС). Цитокины в начале действуют на терморецепторы нервных клеток ядер преоптической зоны переднего гипоталамуса. Возбуждение нейронов этой зоны передается на промежуточные интернейроны, а затем на нейроны центров вегетативной иннервации органов теплопродукции и
теплоотдачи. Интернейроны также выполняют функцию тормозных нейронов,
их активность возрастает в последнюю стадию лихорадки. Возбуждение центров парасимпатической иннервации вызывает увеличение теплоотдачи, а возбуждение центров заднего гипоталамуса способствует активации процессов
теплопродукции. В процессе взаимодействия цитокинов и их рецепторами в
преоптической области гипоталамуса активируется фосфолипаза А2. Под действием этого фермента из клеточных мембран высвобождается арахидоновая
кислота, метаболитом которой является простагландин Е2. Простагландин Е2
вызывает накопление 3-5 цАМФ в термочувствительный клетках Этот процесс
189
сопровождается повышением возбудимости клеток к ацетилхолину и увеличением их электрической активности. Это возбуждение передается нервным
клеткам заднего гипоталамуса организующего торможение теплоотдачи и соответственно развитие 1 стадии лихорадки.
Стадии лихорадки
Лихорадочный процесс всегда протекает в три стадии, в соответствии, с
чем температурная кривая состоит из трех частей. В 1 стадии (stadium incrementi) температура тела повышается, во 2 (stadium fastigi) – удерживается некоторое время на повышенном уровне, в 3 (stadium decrementi) - снижается до
исходного уровня.
Стадия повышения температуры (stadium incrementi). Происходит перестройка терморегуляции так, что теплопродукция превышает теплоотдачу.
Эта стадия может протекать по следующим вариантам:
Ознобный вариант лихорадки. Резко увеличена теплопродукция, за счет спазма
периферических сосудов - субъективное чувство холода. Импульсы идут в гипоталамус, а оттуда через латеральную часть ретикулярной формации реализуются в ритмичных сокращениях мышц - дрожании. При этом идет распад АТФ
с образованием тепла. Спазм периферических сосудов резко сокращает теплоотдачу.
Безознобный вариант лихорадки. Увеличение теплопродукции сопровождается
повышением теплоотдачи (кожные покровы гиперемированы, больной "пышет
жаром"), однако теплопродукция превышает теплоотдачу.
В первом периоде лихорадки, особенно при ознобном варианте, больной
бледен (сужение сосудов кожи), что свидетельствует об уменьшении теплоотдачи, наряду с этим мышечная дрожь, озноб резко увеличивают теплопродукции в скелетных мышцах, что приводит к быстрому повышению температуры
тела. В этом периоде отмечается общее недомогание, боли в мышцах, головная
боль.
Стадия стояния повышенной температуры (stadium fastigi).
Теплопродукция равна теплоотдаче. Высокий уровень температуры тела,
поднявшийся в первой стадии, остается таким некоторое время. Теплоотдача
увеличивается в результате расширения периферических сосудов, что сопровождается чувством жара. Кожные покровы гиперемированы, дыхание учащено, озноб и дрожь отсутствуют, диурез ограничен.
По степени повышения температуры во II стадии лхорадки различают
следующие её виды: субфебрильная – до38 ºС; фебрильная – 38-39ºС; пиретическая – 39-41ºС; гиперпиретическая – выше 41ºС.
Стадия снижения температуры (stadium incrementi).
Характеризуется относительным преобладанием теплоотдачи над теплопродукцией. Установочная точка центра терморегуляции опускается на более
низкий уровень, и температура тела воспринимаются гипоталамусом как повышенная. Усиливается потоотделение, диурез, расширяются периферические
сосуды.
Снижение температуры тела может быть:
190
- литическим (в течение нескольких суток);
- критическим (за час-два).
При критическом снижении температуры наблюдается резкое расширение кровеносных сосудов, что это может привести к острой сосудистой недостаточности, коллапсу. Особенно эти явления опасны для детей.
Типы температурных кривых
При заболеваниях, которые сопровождаются лихорадкой, показатели
температуры тела имеют суточные колебания. Так, максимум подъема температуры в 5-7 часов вечера, минимум в 4-6 часов yтpa. По характеру этих колебаний различают нижеперечисленные температурных кривых:
Преходящего типа (febris ephemera) — однократное кратковременное
повышение температуры продолжительностью несколько часов. Этот тип характерен при нетяжёлом течении псевдотуберкулёза, при задержке молока у некормящих родильниц (молочная лихорадка).
Постоянного типа (febris continua)- температура тела повышается до высоких цифр и остается на них некоторое время, колебания составляют не более
1 градуса. Данный тип может наблюдаться при крупозной пневмонии, брюшном и сыпном тифе.
Послабляющего типа (febris remittens) - температура тела повышается до
высоких цифр и остается на них некоторое время, отмечаются выраженные
размахи суточных колебаний от 1 до 3 градусов, Данный тип характере до нормы температура не опускается. Данный тип характерен для брюшного тифа,
бронхопневмонии, туберкулеза, экссудативного плеврита, вирусных инфекций
и асептических лихорадок.
Перемежающегося типа (febris intermittens) — температурная кривая
имеет большие размахи со снижением утренней температуры до нормы и ниже.
Наблюдается при малярии, остром гепатите, туберкулезе и сепсисе.
Возвратного типа (febris recurrens) - на кривой данного типа наблюдаются периоды высокой температуры и периода снижения темпетаруры тела до
нормы и ниже, длящиеся несколько суток. Например — возвратный тиф.
Истощающая, изнуряющая, гектическая (febris hectica) — для данной
кривой характерно длительное течение и большие суточные колебания температуры от 3 до 5 градусов. Характерна для сепсиса, тяжелого туберкулезе, злокачественных опухолей.
Неправильная, атипическая (febris atypica): температурная кривая с
неравномерным беспорядочным чередованием периодов высокой и низкой
температуры. Встречается при сепсисе.
Нарушение обмена веществ при лихорадке
При развитии лихорадки происходит нарушение всех видов обменов веществ.
Углеводный обмен – гипергликемия, уменьшение содержания гликогена в печени.
191
Жировой обмен – нарушается всасывание и усвоение жиров. Липемия, кетонемия.
Белковый обмен – увеличение распада белков в тканях, увеличение содержания
небелкового азота в крови.
Нарушения физиологических функций при лихорадке
Центральная нервная система
У больного отмечаются головная боль, сонливость, апатия, разбитость.
Зависимости между степенью подъема температуры и выраженностью нарушений работы головного мозга нет (часто при низкой температуре отмечаются
бред, галлюцинации, а при высоких - нет). Глубокие нарушения ЦНС являются,
видимо следствием интоксикации при инфекционном процессе.
Эндокринная система
Лихорадка - это стрессовая реакция, она сопровождается активацией системы гипоталамус - гипофиз - надпочечники. Усиленная выработка глюкокортикоидов, адреналина, тироксина способствует гликогенолизу и увеличению
содержания глюкозы в крови, усиливается мобилизация жира из депо для покрытия энергетических затрат. Однако путь утилизации жира может привести к
развитию метаболического ацидоза.
Активация функции надпочечников может привести к усиленной выработке альдостерона.
Изменения эндокринной регуляции обеспечивают все изменения обмена
веществ, которые поддерживают относительно высокий уровень терморегуляции в организме.
Сердечно-сосудистая система
Развивается тахикардия как результат повышения тонуса симпатической
нервной системы. По правилу Либермейстера при повышении температуры на
1°С наблюдается учащение сердечного ритма на 8 – 10 ударов в минуту. Тахикардия обеспечивает необходимый минутный объем крови для поддержания
артериального давления, особенно в стадии стояния температуры, когда имеет
место расширение сосудов. Постоянная тахикардия приводит к нарушению питания сердечной мышцы, так как с учащением сердечных сокращений возрастает суммарное время систолы и сокращается время диастолы, а кровоснабжение сердечной мышцы происходит в период диастолы.
Артериальное давление несколько увеличено в стадии повышения температуры за счет сокращения периферических сосудов. В стадии стояния температуры оно выравнивается. В результате расширения периферических сосудов
в стадии снижения температуры может наблюдаться резкое падение артериального давления. Такое падение артериального давления обусловлено рядом факторов:
1. Миокардиодистрофия в результате тахикардии и интоксикации;
2. Резкое снижение периферического сопротивления из-за расширения
сосудов;
3. Стимуляция нервно-мышечного аппарата сердца теплой кровью снижается.
Система внешнего дыхания
192
Отмечается тахипное при достаточно высокой глубине дыхания. Повышенная вентиляция поддерживает адекватный кислородный режим в организме
и является компенсаторным механизмом в борьбе с метаболическим ацидозом.
Функция почек и водно-солевой обмен
В первой стадии кратковременно отмечается усиление диуреза за счет
повышения артериального давления и расширения сосудов. В результате активации ренин-ангиотензиновой системы повышена продукция альдостерона, которая ведет к задержке натрия и вторичной задержке воды. В этой стадии больной одутловатый, черты лица сглажены. Периоду падения температуры предшествует нормализация функции надпочечников: уменьшается выработка альдостерона, усиливается диурез. Одновременно отмечаются потеря натрия и
нарушение минерального обмена.
Система пищеварения
Повышение тонуса симпатической нервной системы, гиперкатехоламинемия угнетают секреторную функцию пищеварительных желез. Гипосаливация вызывает сухость во рту, эпителиальный покров губ высыхает и трескается,
создаются условия для размножения патогенной микрофлоры в полости рта. В
пазухах полости рта задерживаются остатки пищи, слущенный эпителий, бурно
размножается микрофлора, что способствует развитию тяжелого язвенного
стоматита. У больных развивается жажда, пропадает аппетит в результате снижения секреции пищеварительных желез и психогенного фактора (вид, вкус,
запах не вызывают желания есть). Все эти процессы вызывают нарушение пищеварения. Усилить секрецию можно за счет дачи бульонов, отваров, соков.
Голодание приводит к развитию метаболического ацидоза и резко ухудшает
состояние лихорадящего больного.
Рациональное питание - обязательное, условие в лечении лихорадящих
больных.
Усиление процессов всасывания в толстом кишечнике ведет к запорам,
каловому завалу, аутоинтоксикации.
Значение лихорадки
Лихорадка для высших теплокровных животных и человека это защитноприспособительная реакция, но при этом лихорадка в определенных условиях
может иметь и отрицательное значение для организма.
При развитии ряда заболеваний повышение температуры тела способствует борьбе организма с патогенным агентом. Высокая температура тела препятствует размножению большинства патогенных микроорганизмов (кокков,
спирохет, вирусов). Она также может снижать резистентность некоторых бактерий к лекарственным препаратам. При лихорадке стимулируются обменные
процессы в клетках, повышается их функциональная эффективность. При лихорадке в клетках печени отмечено повышение процессов фосфорилирования,
усиление мочевинообразования и увеличение выработки фибриногена. Наблюдается повышение барьерной и антитоксической функции печени. Возрастает
фагоцитарная активность лейкоцитов и фиксированных макрофагов, в связи, с
193
чем патогенный агент быстрее исчезает из крови. Возрастает также интенсивность продукции антител. Повышение активности иммунокомпетентных клеток
к антигенному раздражению под влиянием высокой температуры особенно ярко обнаруживается у пойкилотермных животных, у которых при низкой температуре образование антител не происходит.
Отрицательное значение лихорадки может быть связано с дополнительной нагрузкой на ряд органов и систем, особенно на сердечно-сосудистую. В
некоторых случаях обнаруживается индивидуальная повышенная чувствительность организма к высокой температуре. Критическое снижение температуры
может привести к развитию коллапса.
Принципы жаропонижающей терапии
Жаропонижающая терапия – терапия, направленная на снижение температуры тела при лихорадке. Понижение температуры достигается с помощью
лекарственных веществ, воздействующих на центр терморегуляции, а также на
процессы теплопродукции и теплоотдачи. Широко используются ненаркотические анальгетики: ацетилсалициловая кислота, амидопирин. Они воздействуя
на центр терморегуляции, увеличивают процессы теплоотдачи за счет расширения сосудов кожи и повышения потоотделения. Ингибирование ими синтеза
простагландинов Е также вызывает температуры. Алкалоиды некоторых растений (например, хины), оказывающее угнетающее действие на окислительные
процессы, в тканях уменьшая, таким образом, интенсивность процессов теплопродукции и снижая температуру тела.
Пиротерапия
Пиротерапия — введение в организм с лечебной целью агентов, резко
повышающих температуру тела: чужеродных белков, вакцин, пирогенала и
т.п.
Данный вид терапии применяется при лечении возвратного тифа, малярии, сифилиса, костно-суставного и легочного туберкулеза Пиротерапия применялась в онкологии, в связи с противоопухолевым действием некоторых цитокинов-участников острофазного ответа, в частности, ФНО.
Лихорадку следует отличать от другого случая повышения температуры
тела — перегревания. Перегревание или гипертермия — a результат декомпенсации или поломки механизмов поддержания температурного гомеостаза,
при стойкой недостаточности теплоотдачи, по отношению к теплопродукции.
При перегревании наблюдается экстремальное напряжение механизмов терморегуляции, при котором способность организма к теплоотдаче недостаточна,
что приводит к патологическому повышению температуры. Гипертермия может
быть экзогенной и возникает при действии на организм физических и химических факторов, затрудняющих теплоотдачу и активирующих теплопродукцию.
Например, действие высокой температуры окружающей среды в комбинации с
высокой влажностью и отсутствием конвекции делает условия теплоотдачи
крайне невыгодными, приводя к ее недостаточности. Перегревание не может
длиться долго, так как приводит к необратимым нарушениям водно-солевого
194
гомеостаза и денатурации белков. Денатурация факторов свёртывания и белков
эритроцитов ведёт к геморрагическому синдрому и гемолизу. Уже при температуре более 42,2°С наступают нарушения в работе нейронов, у пострадавшего
нарушается ориентация, возникает бред, повышается внутриклеточное содержание кальция, развиваются судороги. Нейроны впадают в некробиоз. Крайняя
степень декомпенсированного перегревания носит название теплового удара.
Это понятие следует отличать от солнечного удара. Солнечный удар сопряжён
с действием на непокрытую голову и кожу ультрафиолетовых лучей, а тепловой происходит при действии лучей инфракрасных и может наступить даже в
полной темноте. При солнечном ударе ультрафиолетовые свободнорадикальное
повреждение клеток и наблюдается массивное освобождение цитокинов и простагландинов, особенно, в крови мозговых оболочек и тканях головы. Эти сигналы нарушают нормальную работу терморегуляторной области гипоталамуса.
Поражение мозга, при солнечном и тепловом ударе приводит к нарушению
центральных механизмов терморегуляции, в результате чего прекращается потоотделение, что обостряет состояние. Температура тела 43,3°С при перегревании считается абсолютно смертельной.
Ни одно из этих расстройств не наблюдается при протекании лихорадки.
Лихорадка, в отличие от гипертермии, может длиться достаточно долго и, при
этом, наносит обратимый ущерб гомеостазу, так как её механизмы экономны и
основаны на временном смещении равновесия теплопродукции и теплоотдачи,
с последующим его восстановлением на новом уровне. (Подробнее смотри тему
«Патогенное действие факторов внешней среды»).
ПАТОФИЗИОЛОГИЯ ТКАНЕВОГО РОСТА.ОПУХОЛИ
Онкологические заболевания составляют вторую по значению причину
смертности населения Земли и повинны в каждом четвертом смертельном случае в странах с высоким жизненным уровнем населения. В странах со среднем
и низким жизненным уровнем онкологическая патология обусловливает 10%
общей смертности. По данным ВОЗ в мире более 57,2 млн больных неопластическими заболеваниями.
Учение о новообразованиях представляет собой важнейшую часть более
широкой патофизиологической доктрины, описывающей причины и механизмы
недостаточного или избыточного роста и размножения клеток – то есть гипербиотических и гипобиотических процессов.
Гипобиотические процессы в организме представлены атрофией – то
есть уменьшением объема клеток и их числа, приводящем к уменьшению веса и
объема органов ( тканей). При атрофии развивается и гипофункция соответствующего органа. Атрофия может охватывать весь организм. Она может касаться отдельных органов ( например, атрофия коры надпочечников при гипофизэктомии), а также отдельных тканей. Врожденный гипобиотический процесс, аналогичный атрофии органа, именуется гипоплазией. Гипоплазия чаще
касается отдельных органов, но может относиться и ко всему телу ( гипофизарный нанизм ) к какой – либо системе органов (например, костной системе – при
хонродисплазии). Крайне выраженная гипоплазия, когда имеется лишь орган-
195
ный зачаток, известно под названием аплазии ( например, гипоплазия тимуса
при синдроме Незелова и аплозия тимуса – при синдроме Ди Джорджа). Если
отсутствует и органный зачаток – такой врожденный гипобиотический процесс
характеризуют как агенезию (пример – агенезия почки). Атрофия – важный
приспособительный механизм, участвующий в онтогенетических изменениях
органов и тканей. Физиологическая атрофия, основанная на запрограммированной гибели клеток и аутофагоцитозе, лежит в основе рассасывания провизорных органов плода и онтогенетического устранения клеток. Естественная
атрофия в порядке регрессии гиперплазии сопровождает изменения размера органов и числа клеток в них, связанные с беременностью, лактацией, натуральными биоритмами. Эта форма адаптации основывается на усилении интенсивности апоптотических процессов в соответствующих органах. Такую физиологическую атрофию эволюционной, в противоположность старческой, инволюционной физиологической атрофии. При патологической атрофии ускоренный апоптоз сохраняет свое значение. Однако, по сравнению с атрофией
физиологической, большое значение приобретает ускоренная гибель клеток путем некробиоза и недостаточные темпы размножения клеток, вследствие нехватки ресурсов и / или блокады ростостимулирующих сигналов. В основе разнообразных вариантов патологической атрофии лежат следующие механизмы,
ускоряющие гибель и тормозящие размножение клеток: хроническая гипоксия, дефицит или блокада ростостимулирующих сигналов (например, при
нехватке тропных гармонов, при денервации, при бездействии, при аутоиммунном поражении соответствующих ростовых рецепторов). Ярким примером
последней формы служит атрофия коры надпочечников при наличии аутоантител к рецепторам АКТГ, блокирующих его ростовое действие. Избыток апоптогенных сигналов ( например, атрофия при раневой и инфекционной кахексии, связанных с гиперпродукцией ФНОα, ИЛ – 1. Различные формы атрофии
характеризуются некоторыми общими признаками. В атрофичных органах чаще обнаруживаются апоптотические тельца. Уменьшаются размер клеток и
степень их гидротации, возрастает скорость аутоокисления липидов и лизосомальной аутофагии, а, как следствие этого – растет концентрация пигмента липофусцина. В органах может происходить замещение атрофичной паренхимы
переживающими в условиях патологии элементами стромы или адипоцитами (
атрофический фиброз и заместительное ожирение). Атрофия может провоцироваться неоплазиями – за счет механического сдавления тканей, ишемии и
системной раковой кахексии.
Гипербиотические процессы в организме представлены гипертрофией, гиперплазией, регенерацией и опухолевым ростом.
Гипертрофия – компенсаторно - приспособительный процесс увеличения
размеров клеток и числа функционирующих внутриклеточных структур. При
гипертрофии увеличиваются размеры и функциональные возможности соответствующих органов. Основной механизм гипертрофии – гиперфункция и соответствующий избыток ростостимулирующих и /или дефицит ростоограничивающих сигналов. Например, в молочной железе гипертрофия имеет место при
лактации и соответствующем избытке пролактина ( «рабочая» гипертрофия),
196
в оставшемся надпочечнике она происходит при унилатеральной адренолэктомии и избытке АКТГ («заместительная» гипертрофия), в аденогипофизе гипертрофируются тиреотрофы после удаления щитовидной железы – из-за дефицита тормозного влияния тиреоидных гармонов на их рост (« корреляционная» гипертрофия).
Гипертрофия возможна и без гиперфункфии, вследствие патологического избытка ростовых сигналов. Например, ростостимулирующие аутоантитела к органу могут вызвать его гипертрафию, как это происходит при «идеопатической» гипертрафии миокарда, переносимой от донора к реципиенту сывороткой ее иммуноглобулинами.
При чистой гипертрофии, наблюдаемой в органах, где клетки не вступают
в митоз, например – в зрелом миокарде, не меняется количество клеток. В этом
случае гипертрофия проходит три типичные стадии:
- стадию увеличения интенсивности функционирования внутриклеточных
структур;
-стадию увеличения числа структур и нормализации интенсивности их
функционирования (« сформировавшаяся гипертрофия»);
-стадия ускорения клеточной гибели и снижения эффективности работы органа, в целом («чрезмерная гипертрофия и декомпенсация» - например, в случае
гипертрофии миокарда – с исходом в кардиосклероз).
В реальных ситуациях гипертрофия, в меру митотических возможностей органов и тканей, комбинируется с гиперплазией.Это происходит,например, при
воссоздании поврежденныз органов («регенерационная» гипертрофия).Такие
органы, как миокард и почка активно гипертрофируются, но слабо регенерируют.
Гиперплазия – увеличение размеров органа или ткани вследствие увеличения числа его клеток. Дифференцировка клеток и тканевая структура органа
при этом остаются нормальными.
Гипертрофия и гиперплазия составляют основу для такого гипербиотического процесса как регенерация. Регенерацией называется возрождение (восстановление) утраченных тканей, органов и отдельных частей организма. Регенерация существует физиологическая ( в процессе сомообновления тканей) и
патологическая (при убыли клеток из-за повреждения). Процессы регенерации
присущи всем живым существам, но полная регенерация (восстановлением
исходной ткани ее микроархитектуры) наиболее выражена у простых животных. Чем сложнее животные – тем способность к полной регенерации меньше.
У млекопитающих и птиц регенерируют лишь части органов и тканей. В связи с
этим, у нах большую роль играет неполная регенерация:
- в виде фиброплазии, то есть возникновения регенерата, представленного, в
основном, соединительной тканью;
- в виде атипической регенерации – то есть с восстановлением ткани того же
вида, но без воспроизведения ее оригинальной микроархитектуры (например,
при узловатой);
- в виде метаплазии – то есть с отклонением направлении дифференцировки
(см. ниже).
197
Наиболее хорошо регенерирует эпителиальная ткань и рыхлая соединительная
ткань. Велики регенераторные потенции других видов соединительной ткани
(костной, плотной соединительной, а также костного мозга и крови). Слабее регенерируют хрящ.
Регенераторные возможности мышечной ткани ограниченны, а в нервной ткани нейроны, хотя и обладают регенераторными возможностями, регенерируют
очень слабо.
Метаплазия-аномалия дифференцировки клеток того или иного органа(ткани).При метаплазии камбиальные клетки дифференцируются не в характерные для данной локализации, а в иные, нетипичные для органа, но вполне
зрелые клетки. Примером может служить образование кости и костного мозга в
рубцовой капсуле вокруг туберкулёзного очага творожистого некроза. Метаплазия обратима, а клетки, подвергшиеся ей, не проявляют нарушения нормальных межклеточных взаимоотношений, т.е. отсутствуют признаки тканевого атипизма.
Дисплазия- представляет собой аномалию как дифференцировки, так и
созревания. При дисплазии существует частичная потеря контроля за дифференцировкой клеток и некоторые нарушения их тканевой организации. В клетках при дисплазии имеются цитологические аномалии. Дисплазия частично обратима. Для самой по себе дисплазии нехарактерен избыточный рост ткани.
Однако в катамнезе при дисплазии повышена частота возникновения неоплазии
соответствующей локализации. В связи с этими особенностями, дисплазию характеризуют как преднеопластический (предопухолевый) рост.
Неоплазия- синоним понятия «опухолевый рост». Опухолевый рост —
типовая форма нарушения тканевого роста. Возникает под действием канцерогена.Проявляется патологическим разрастанием ткани. Характеризуется атипизмом роста, обмена веществ, структуры и функции.
Опухолевая клетка- есть, прежде всего, клетка генетически измененная и передающая свои особенности клону своих потомков. Опухоль образуется в организме в результате превращения нормальных клеток в опухолевые, в которых нарушается регуляция деления. В таких клетках отсутствует
или недостаточно эффективно подавляется клеточное деление, что обусловливает неудержимое размножение опухолевых клеток, или в них начинается
самоподдерживающаяся стимуляция деления (аутокринный механизм — деление клетки стимулирует фактор, производимый ею самой).
Опухолевая ткань отличается беспредельным ростом. Этот процесс заканчивается только со смертью организма. В культуре ткани рост поддерживается бесконечно долго в отличие от нормальной ткани в связи с тем, что
отсутствует «лимит Хейфлика» . Способность опухолевых клеток беспредельно размножаться передается по наследству как доминантный признак
соматической наследственности и проявляется не только в организме, но и
в культуре опухолевой ткани, а также при трансплантации опухоли.
Опухоль растет «сама из себя», т.е. увеличивается в результате размножения даже одной-единственной малигнизированной клетки.
198
Опухолевая ткань отличается от исходной ткани, из которой она произошла, по структуре, биохимическим, физико-химическим и другим признакам. Эти изменения выражают анаплазию — возврат к эмбриональному состоянию, а также метаплазию — приобретение свойств другой ткани.
Рост опухоли может быть экспансивным и инфильтративным. При
экспансивном росте окружающая здоровая ткань по мере роста опухоли раздвигается, при инфильтративном - опухолевые клетки прорастают между
нормальными клетками и через сосудистую стенку. Попадая в лимфу или
кровь, они переносятся в другие органы и могут образовывать новые очаги
опухолевого роста (метастазы). Экспансивный рост характерен для доброкачественных опухолей, а инфильтрирующий с образованием метастазов — для
злокачественных опухолей.
В отличии от других гипербиотических явлений, неопластическому процессу
свойственна необратимость. По сути, при неоплазии клетки, в той или иной
степени, утрачивают признаки, свойственные им как элементам многоклеточного организма. Реализуются программы, определяющие автономное «одноклеточное или колониальное», некооперирующееся соседями повреждения.
Итак, под новообразованиями (неоплазиями) можно понимать беспредельный, нерегулируемый, автономный рост клеток, характеризующийся необратимостью, а на молекулярном уровне- расстройством функции генов, регулирующих пролиферативные процессы- протоонкогенов и опухолевых супрессоров.
Неоплазия развивается многошаговым путем, на основе ступенчатых генетических изменений моноклона клеток.
Классификация неоплазм и онкологическая терминология
Существуют условное и относительное деление опухолей на злокачественные
и доброкачественные.
Доброкачественные. Морфологически они соответствуют зрелым, гомотипическим опухолям с незначительной степенью атипии, четко отграничены
от окружающих тканей, имеют обычно форму узла или полипа (на слизистой
оболочке). Эти опухоли растут опухоли экспансивно с большей или меньшей
скоростью, раздвигая окружающие ткани, иногда сдавливая их, но обычно не
повреждая; в некоторых случаях они инкапсулируются. Доброкачественные
опухоли, как правило, не оказывают выраженного неблагоприятного воздействия на организм. Их клиническое значение невелико. Исключение составляют
опухоли, локализация которых становится фактором, угрожающим жизнедеятельности организма (например, опухоль головного мозга, сдавливающая нервные центры).
Злокачественные. Морфологически они соответствуют незрелым, гетеротидическим опухолям, характеризуются выраженной катаплазией клеток, как
правило, растут инвазивно (т.е. с прорастанием и замещением окружающих
тканей, по путям наименьшего механического сопротивления) и метастазируют ( т.е. дают аутотрансплантанты на удалении от места первичной локализации), имеют форму узлов, полипов, инфильтратов, образуют перифокальные
199
очаги воспаления, оказывают генерализованное действие на весь организм,
нарушая его гомеостаз, растут быстро.
Атипизм у злокачественных неоплазм присутствует как тканевой, так и
клеточный, у добракачественных же может выявляться только тканевой. У злокачественных новообразований больше выражена биохимическая анаплазия,
они вызывают в организме носителя больше паранепластических явлений.
Клиническое понятие о злокачественности не всегда совпадает с биологическими свойствами опухолевой клетки в смысле степени ее анаплазии (анаплазия — стойкая дифференцировка клеток с изменением их структуры и биологических свойств, как бы возврат к эмбриональному состоянию). Иногда злокачественное течение имеют опухоли с достаточно дифференцированными клеточными элементами.
Анаплазия может быть биологической (утрата клетками всех функций,
кроме функции размножения), биохимической (утрата клетками части ферментных систем, характерных для исходных клеток) и морфологической (изменение
внутриклеточных структур, формы и размеров клеток и ядер).
Относительные условные критерии доброкачественности и злокачественности новообразований.
Критерий
«Доброкачественные» «Злокачественные»
неоплазмы
неоплазмы
Поверхность
Размер
Гладкая, бывает кап- Неровная, без капсулы
сула
Любой
Не бывают очень большими
Скорость роста
Меньше
Прогноз
Редко обусловливают
смерть больного, даже если не лечатся
Характер роста
Экспансивный, чаше
эндофитный
Степень дифферен- Выше, нет катаплацировки
зии, клетки мономорфны
Митотический
ин- Соответствует
нордекс
мальной ткани происхождения
Кровеносные сосуды Нормальные
Дегенеративные из- Минимальные
менения
Больше
Обычно без лечения тяжелый
или фатальный
Инфильтративний,экзофитный
Ниже.Выражена катаплазия,
клетки плеоморфны
Бывает повышен, описывается
патологические митозы
Многочисленные, анормаольные, может отсутствовать эндотелиальная выстилка
Выражены, бывает некроз,
кровоизлияния
200
Метастазирование
Нет
Типично
Генетический аппа- Типичны нормальный Типичны повышение содеррат
кариотип и содержа- жания ДНК и аномалии кариние ДНК
отипа
Название опухоли строится следующим образом: к названию ткани, из
которой состоит паренхима опухоли, прибавляется окончание «ома» или «бластома» (например, глиома, миома, мезетелиома и т.д.). Используются также и
специальные названия для обозначения некоторых видов опухолей (например,
эпителиальная опухоль — рак, соединительнотканная опухоль — саркома).
Классификация новообразований по зародышевым зачаткам и типу клеток, из
которых они исходят:
1.Из прогаметных тотипотентных клеток, то есть клеток, принципиально
способных дифференцироваться по любому пути. Доброкачественные опухоли
тотипотентного генеза- зрелые тератомы. В большинстве своём, зрелые тератомы доброкачественные, примером может служить дермоидная киста яичника. Незрелые тератомы всегда высоко злокачественные
2.Опухоли из эмбриональных полипотентных клеток возникают из элементов органных зачатков. Опухоли данной группы носят название – эмбриомы или бластомы они всегда исключительно злокачественные, именуются по
названию соответствующего органного зачатка (ретинобластома, нефробластома, гепатобластома, нейробластома, медуллобластома, пинеобластома,
эмбриональная рабдомиосаркома).
Значение эмбриом обусловлено тем
фактом, что они возникают в первые годы жизни и , таким образом, встречаются исключительно, у детей.
3.Опухоли из дифференцированных клеток, утративших часть или все признаки дифференцировки и подвергшихся анаплазии, составляет большинство
новообразований у взрослых. Доброкачественные опухоли эпителиального
происхождения называют аденомами – если они происходят из эпителия желез
и папилломами – и если их источник – кроющий эпителий. Злокачественные
опухоли эпителиального происхождения – карциномы (аденокарциномы –
если относятся к железистому эпителию). Рак – синоним понятия «карцинома».
Доброкачественные опухоли мезенхимального генеза обозначают по названию
соответствующей ткани или органа (гемангиома, липома). Злокачественные
опухоли мезенхимального происхождения называют, прибавляя к обозначению
ткани (органа) суффикс – саркома (ангиосаркома, липосаркома).
Эксперипентальное воспроизведение опухолей
Методами экспериментального моделирования опухоли являются ее индукция, эксплантация и трансплантация.
Индукция опухоли химическими веществами. В 1775 г. хирург лондонского
госпиталя Персиваль Потт описал профессиональное злокачественное заболевание рак кожи мошонки у трубочистов. Однако, несмотря на очевидную связь рака у
трубочистов с поражениями кожи сажей и смолой, попытки воспроизвести подобную опухоль в эксперименте долгое время заканчивались неудачей. В 1916 г. япон-
201
ские ученые Ишикава и Ямагива впервые смогли вызвать опухоль у животных. На
протяжении 6 мес. они смазывали кожу кроликов каменноугольной смолой, и
только после этого у животных развился рак кожи. Позже канцерогенные вещества
были получены в чистом виде, установлена канцерогенность веществ, относящихся к различным классам химических соединений.
Индукция опухоли вирусами. В 1908 г. Эллерман и Банг впервые вызвали лейкоз у курс помощью бесклеточного фильтрата из лейкозных лейкоцитов. В 1910 Раус помощью бесклеточного фильтрата, полученного из саркомы курицы, вызвал
развитие саркомы у здоровых кур. Так впервые были получены доказательства вирусной этиологии лейкозов и опухолей.Однако в последующие десятилетии выявить опухолеродные вирусы у млекопитающих не удавалось за исключением папилломы Шоупа и фактора молока Биттнера. Шоуп обнаружил у диких кроликов
бородавчатые разрастания на коже (папилломы), которые удалось перевить здоровым животным посредством бесклеточного фильтрата. Фактор молока был открыт
Биттнером (1936). Имеются линии мышей с высокой степенью заболеваемости раком молочной железы (высокораковые) и низкой степенью (низкораковые). Однако, если у самки высокораковой линии забрать новорожденных мышат до первого кормления и отдать для кормления самке низкораковой линии, частота появления рака у них будет резко снижена. И наоборот, при кормлении высокораковой
самкой мышат низкораковой самки частота опухоли у них значительно повышается. Биттнер доказал, что в молоке высокораковых мышей имеется фактор, вызывающий у потомства рак молочной железы. В 1950 г. Л.Гросс после многих
безуспешных попыток вызвать лейкоз у взрослых мышей ввел бесклеточный фильтрат из лейкозных клеток новорожденным мышам и получил у них лейкоз. Таким
образом было получено подтверждение гипотезы о вирусной этиологии развития
опухоли у млекопитающих и установлено значение при этом индукции опухали онко-генными вирусами, в которых теперь открыты онкогены.
Индукция опухоли физическими факторами. Опухоль удается воспроизвести с
помощью ионизирующей радиации, в том числе рентгеновских лучей, радиоактивных изотопов, а также ультрафиолетовых лучей.
Эксплантация опухоли. Выращивание опухоли в культуре ткани вне организма. В лабораториях имеется большое количество постоянно пассируемых
штаммов опухолевых клеток, свойства которых хорошо изучены, что позволяет проводить опыты на одинаковом материале. Культура тканей дает возможность индуцировать опухоли вне организма химическими канцерогенами
и онкогенными вирусами. Данный метод особенно ценен тем, что позволяет
изучать индукцию опухолей и опухолеродных вирусов на человеческих тканях. Пассируемые или индуцированные в культуре ткани опухолевые клетки
при подсадке здоровому животному растут в его организме и образуют злокачественную опухоль.
Трансплантация опухоли. Впервые российский ученый М.А.Новинский в
1876 г. успешно трансплантировал опухоль взрослой собаки щенкам. Фактически данным опытом было положено начало экспериментальной онкологии.
Метод трансплантации широко используется и в настоящее время.
202
Этиология опухолей
Причинами опухолей являются канцерогенные агенты химической, биологической и физической природы, а главным условием, способствующим реализации их действия (фактором риска) — снижение эффективности антиканцерогенных механизмов противоопухолевой защиты организма.
Химические канцерогенны
По данным ВОЗ, более 75% случаев злокачественных опухолей человека
вызвано воздействием химических факторов внешней среды. К возникновению
опухолей приводят преимущественно факторы сгорания табака (примерно
40%); химические агенты, входящие в состав пищи (25–30%) и соединения, используемые в различных сферах производства (около 10%). Известно более
1500 химических соединений, обладающих канцерогенным эффектом. Из них
не менее 20 определённо являются причиной опухолей у человека. Например, к
ним отнесены 2–нафтиламин, бензидин, 2–аминотиофенил, вызывающие рак
мочевого пузыря у работников анилинокрасочной и резиновой промышленности; компоненты синтеза поливинилхлорида, индуцирующие опухоли печени;
бис–(хлорметил)–эфир, приводящий к возникновению рака бронхов и лёгких.
Большинство канцерогенов относится к нескольким классам химических веществ.
Органические химические канцерогены
• Полициклические ароматические углеводороды.
Наибольшей канцерогенной активностью среди них обладают 3,4-бензпирен,
20-метилхолантрен, диметилбензантрацен. Ежегодно в атмосферу промышленных городов выбрасываются сотни тонн этих и подобных им веществ.
• Гетероциклические ароматические углеводороды.
В эту группу входят дибензакридин, дибензкарбазол и другие соединения.
• Ароматические амины и амиды.
К ним относятся 2–нафтиламин, 2–аминофлюорен, бензидин и др.
• Нитрозосоединения. Наиболее опасные среди них — диэтилнитрозамин,
диметилнитрозамин, нитрозометилмочевина.
• Аминоазосоединения
Высокоэффективными
канцерогенами
среди
них
считаются
4диметиламиноазобензол и ортоаминоазотолуол.
• Афлатоксины — продукты метаболизма (производные кумаринов) плесневых грибов, в основном аспергилл Aspergillus flavus (отсюда название производимых ими веществ).
• Прочие органические вещества с канцерогенной активностью: эпоксиды,
пластмассы, уретан, четырёххлористый углерод, хлорэтиламины и другие.
Неорганические канцерогены
1.Экзогенные: хроматы, мышьяк и его соединения, кобальт, окись бериллия,
асбест и ряд других.
2.Эндогенные.Эти соединения образуются в организме в результате физико-химической модификации продуктов нормального обмена веществ. Полагают, что такими потенциально канцерогенными веществами являются жёлчные
203
кислоты, эстрогены, некоторые аминокислоты (тирозин, триптофан), липопероксидные соединения.
Физические канцерогенные факторы
К ним относятся:
• радиоактивное излучение веществ, содержащих 32Р, 131I, 90Sr и др.;
• рентгеновское излучение;
• поток нейтронов;
• ультрафиолетовые лучи.
У лиц, хронически, периодически или однократно подвергавшихся воздействию указанных агентов, часто возникают различные злокачественные новообразования. Так, у врачей-рентгенологов нередки лейкозы (в 8–9 раз чаще,
чем у других врачей). У пациентов, лечившихся препаратами, содержащими
радиоактивные вещества, с более высокой частотой, чем в общей популяции,
возникают новообразования (например, опухоли печени у пациентов, которым
неоднократно вводили рентгеноконтрастное вещество торотраст). У людей,
подвергшихся воздействию радиации при нарушении технической безопасности или во время аварий на атомных реакторах, во время ядерных испытаний, а
также при бомбардировке Хиросимы и Нагасаки, онкологическая заболеваемость намного выше, чем в общей популяции.
Канцерогены биологической природы
К канцерогенам биологической природы относят онкогенные (опухолеродные) вирусы.
По типу вирусной нуклеиновой кислоты онкогенные вирусы подразделяют на ДНК-содержащие и РНК-содержащие.
• ДНК– содержащие вирусы
Гены ДНК-онковирусов способны непосредственно внедряться в геном
клетки-мишени. Участок ДНК онковируса (собственно онкоген), интегрированный с клеточным геномом, может осуществить опухолевую трансформацию
клетки. Не исключают также, что один из генов онковируса может играть роль
промотора клеточного протоонкогена.К ДНК– содержащим онковирусам относят некоторые аденовирусы, паповавирусы и герпесвирусы.
• РНК–содержащие вирусы
РНК–содержащие вирусы (ретровирусы). Интеграция вирусных РНК–
генов в клеточный геном происходит не непосредственно, а после образования
ДНК–копий. Такая ДНК–копия может интегрироваться в геном клетки, экспрессироваться и обусловить её трансформацию в опухолевую.
Условия, способствующие возникновению опухолей
• Наследственные факторы.
204
Существует не менее 300 так называемых семейных форм злокачественных заболеваний. В ряде случаев генетическая природа предрасположенности к
возникновению опухолей определена. К числу наиболее значимых относятся
следующие:
- аномалии генов, контролирующих процесс репарации ДНК.Это определяет
повышенную чувствительность к канцерогенным воздействиям;
- аномалии генов-супрессоров опухолевого роста (выявлены при: новообразованиях толстой кишки и поджелудочной железы, множественном канцероматозе
- аномалии генов, ответственных за межклеточное взаимодействие, например
кадгерина.Уменьшение экспрессии E-кадгерина — один из главных молекулярных механизмов, способствующих инвазии и метастазированию раковой
опухоли.
- другие генные и хромосомные дефекты: мутации гена рецептора андрогенов
вызывают рак молочной железы у мужчин; различные хромосомные дефекты
зарегистрированы при лейкозах; аномалии хромосом 8 и 9 выявляются при
наследственных формах меланом кожи.
• Низкая активность механизмов противоопухолевой защиты организма
(см. ниже).
Патогенез опухолей ( механизмы канцерогенеза)
Канцерогенез - длительный процесс накопления генетических повреждений. Латентный период (время от начальный изменений в клетке до первых
клинических проявлений) может длиться до 10—20 лет. Возникновение опухоли - это многостадийный процесс, включающий 3 этапа (стадии):
I этап - инициация (трансформация) — приобретение исходной нормальной клеткой способности беспредельно размножаться. Все теории, исторически
подготовившие базу для открытия молекулярных механизмов канцерогенеза,
исходили из общей посылки, что превращение нормальной клетки в опухолевую (трансформация, или инициация) является результатом стойких изменений в геноме клетки - мутации одного из генов, регулирующих клеточное
размножение. Вследствие этого клетка становится инициированной (потенциально способной к неограниченному размножению), но требующей для проявления этой способности ряда дополнительных условий. Инициирующими
факторами служат различные канцерогены, вызывающие повреждения ДНК.
Каковы же современные представления о молекулярных механизмах канцерогенеза? На сегодня установлено, что в нормальных клетках в ДНК имеется участок гомологичный по иуклеотидному составу онкогену вирусов, а точнее —
для каждого из 20 известных ретровирусных онкогенов в геноме нормальных и
опухолевых клеток различных видов животных имеется свой клеточный аналог. В нормальных клетках клеточный аналог вирусного онкогена неактивен и
назван протоонкогеном. В опухолевых клетках он активен и называется клеточным онкогеном.
Переход неактивного клеточного онкогена (протоонкогена) в активный
клеточный онкоген происходит под влиянием химических, физических и био-
205
логических канцерогенов. Выделяют 4 основных механизма активации протоонкогенов:
1. Включение (вставка) промотора. Промотор — это участок ДНК, с
которым связывается РНК-полимераза, инициируя транскрипцию онкогена.
Проявлению активирующего действия промотора способствует его расположение рядом с про-тоонкогеном («в непосредственной близости»). В роли промоторов для протоонкогенов могут выступать ДНК-копии определенных участков
онкорнавирусов, а также «прыгающие гены», которые представляют собой мобильные сегменты ДНК, способные перемещаться и встраиваться в разные
участки генома клеток.
2. Амплификация, т.е. увеличение числа (копий) протоонкогенов, которые в норме обладают небольшой активностью. В итоге общая активность
протоонкогенов значительно возрастает, что в конце концов может привести к
опухолевой трансформации клетки.
3. Транслокация протоонкогенов. Установлено, что перемещение протоонкогена в локус с функционирующим промотором превращает его в клеточный онкоген.
4. Мутации протоонкогенов. Введение в геном клетки хотя бы одной
копии клеточного онкогена (мутация) сопровождается активацией протоонкогенов.
Вслед за превращением протоонкогенов в активные клеточные онкогены
начинается экспрессия активных клеточных онкогенов. Она проявляется в увеличении синтеза онкобелков или в синтезе структурно измененных онкобелков.
Затем начинается превращение (трансформация) нормальной клетки в опухолевую благодаря следующим механизмам:
а) онкобелки соединяются с рецепторами для факторов роста и образуют комплексы, постоянно генерирующие сигналы к делению клеток;
б) онкобелки повышают чувствительность рецепторов к факторам роста или
понижают чувствительность к ингибиторам роста;
в) онкобелки сами могут действовать как факторы роста.
Говоря о трансформации неопухолевых клеток в опухолевые, следует
остановиться на гипотезе Хьюгса, которая в известной степени отвечает на вопрос, каким образом опухолевая клетка становится «бессметрной», т.е. утрачивает лимит Хейфлика и приобретает способность к постоянному делению. Согласно этой гипотезы, регуляция деления в каждой клетке осуществляется системой, состоящей из трех регуляторных генов:
1. Ген-инициатор клеточного деления, кодирующий синтез белка — инициатора клеточного деления.
2. Ген-репрессор I, который кодирует синтез белка — репрессора I. Репрессор I
выключает функционирование гена-инициатора клеточного деления.
3. Ген-репрессор II, кодирующий синтез белка — репрессора II. Репрессор II
выключает функционирование гена-ре-прессора I.
При активации гена-репрессора I синтезируется репрессор I, который выключает ген-инициатор клеточного деления, в результате этого прекращается
синтез белка-инициатора клеточного деления, и деление клеток прекращается.
206
В свою очередь, ген-репрессор I находится под контролем гена-репрессора II, который кодирует синтез репрессора II, а он ингибирует ген-репрессор I.
И далее, компоненты белка инициатора клеточного деления способны выключать (репрессировать) ген-репрессор II.
Таким образом, система регуляции клеточного деления работает по
принципу обратной связи, что обеспечивает ей автономность и определенную
интенсивность клеточного деления. «Обратная связь» в работе системы генов,
регулирующих клеточное деление, заключается в репрессии гена-репрессора II
компонентами инициатора клеточного деления.
При повреждении гена-репрессора I (воздействие радиации или химических канцерогенов) белок репрессор I не синтезируется, а значит ген-инициатор
клеточного деления все время продуцирует инициатор клеточного деления — в
итоге отмечается постоянное бесконечное деление опухолевых клеток. Это так
называемый мутационный канцерогенез.
Некоторые канцерогенные факторы, например, вирусы, могут создавать
устойчивое нарушение нормальной регуляции генома соматической клетки хозяина путем интеграции с геном-репрессором II этой клетки. В результате этого
инициатор клеточного деления может выключить только ген-репрессор II хозяина, а на вирусном гене, интегрированном рядом с геном-репрессором II в
клетку хозяина, будет продолжаться синтез репрессора II — в итоге будет происходить безудержное деление клеток (опухолевых). Такой канцерогенез называется эпигеномным (геном клетки хозяина не подвергается мутации!).
II этап — промоция, или активизация опухолевых клеток. Трансформированные клетки длительное время могут оставаться в ткани в неактивной
форме, а дополнительное воздействие ко канцерогенных факторов запускает
амплификацию онкогенов, активирует новые протоонкогены, вызывает дополнительные генные и хромосомные аберрации, обусловливает включение промотора. Промоторы - множество химических веществ, которые сами не вызывают повреждения ДНК и не являются канцерогенами, но их постоянное воздействие на инициированные клетки приводит к возникновению опухоли.
Вследствие этого опухолевые клетки, до этого находившиеся в латентном состоянии, начинают интенсивно размножаться, образуя первичный опухолевый
узел. Главное в промоции -стимуляция клеточного деления, вследствие чего создается критическая масса инициированных клеток, что обусловливает высвобождение инициированных клеток из-под тканевого контроля и способствует мутационному процессу.
III этап - опухолевая прогрессия, или стойкие качественные изменения
свойств опухоли в сторону малигнизации, возникающие по мере ее роста. Опухолевая прогрессия — это не просто увеличение опухоли в размерах, это качественное изменение ее части с появлением по существу новой опухоли, обладающей ранее отсутствовавшими свойствами, что может быть связано с отбором клеточных клонов, а также с мутацией опухолевых клеток. Прогрессия
опухоли осуществляется посредством отбора клеточных популяций с их непрерывным развитием в направлении все большей автономии, деструктивного ро-
207
ста, инвазивности, способности к образованию метастазов и поразительную
приспособляемость к меняющимся условиям существования.
Опухолевая прогрессия в отличие от дифференцировки нормальных тканей происходит независимо и несопряженно (В.С. Шабад, 1980), а поэтому развитие опухоли никогда нельзя считать завершенным. Прогрессия касается и
первичных, и вторичных признаков. Первичным или «неотъемлемым» признаком опухоли является нерегулируемый рост, а остальные свойства: скорость
роста, инвазивность опухоли, метастазирование и т.д., это «вторичные» свойства или признаки, которые как раз и изменяются в ходе прогрессии.
Трансформации нормальных клеток в опухолевые, промоции и опухолевой прогрессии способствуют ряд факторов: снижение антибластомной резистентности и противоопухолевого иммунитета (иммунодепрессия, иммунодефицит), ослабление «кейлонного надзора» за опухолью, эндокринный дисбаланс, гормонально-метаболические нарушения и др.
Опухолевый атипизм
Для опухолей характерен атипизм - отличия опухолевых клеток от нормальных. Он проявляется в относительной автономности роста, особенностях
размножения, дифференцировки, метаболизма, структуры, функции и антигенного набора опухолевых клеток.
1. Одной из причин относительной автономности роста опухоли, увеличения ее массы является усиленная экспрессия канцерогенами ряда протоонкогенов (гомологов онкогенов ретровирусов), кодирующих синтез опухолевой
клеткой онкопродуктов, которые нередко гомологичны факторам роста, их рецепторам и белкам, участвующим в пострецепторной передаче митогенного
сигнала. Опухолевые клетки обладают способностью продуцировать собственные факторы роста путем так называемой аутокринной секреции. Это α- и βтрансформирующие факторы, эпидермальный фактор роста, инсулиноподобные факторы роста I и II.
Эти факторы или регуляторные пептиды, продуцируемые самой опухолевой клеткой, обеспечивают утилизацию энергетических и пластических субстратов из окружающей среды и включают механизмы деления опухолевой
клетки. Продуцируемые опухолью ростовые факторы стимулируют последующий рост массы опухоли и снижают потребность новообразования в экзогенных факторах роста. Полагают, что именно аутокрииная секреция факторов роста лежит в основе относительной автономности опухоли, ее независимости от
регуляторных внешних факторов.
2. Метаболический и энергетический атипизм. До настоящего времени
не удалось выявить качественных изменений метаболизма опухолевых клеток,
которые отличали бы их от нормальных. Все обнаруженные изменения в опухолевых клетках носят количественный характер и касаются изменений концентрации соединений, активности ферментов, размера транспорта метаболитов и других величин. Эти изменения метаболизма опухолевых клеток являются следствием нарушения регуляторных процессов в них, причем величина изменений метаболизма прямо связана со скоростью роста опухоли.
208
Особенности метаболизма углеводов. Типичным для опухолевых клеток
является анаэробный гликолиз — расщепление глюкозы до лактата в присутствии кислорода. Причиной активации анаэробного гликолиза считается недостаток коферментов, особенно НАД, КоА-SН и тиаминпирофосфата, что препятствует аэробному распаду глюкозы в опухолевой клетке. Весьма характерно, что распад углеводов до пирувата и его превращение в лактат происходит в
присутствии кислорода (этот феномен получил название отрицательного эффекта Пастера). Если имеется недостаток глюкозы (главного энергетического
субстрата опухолевых клеток), о чем свидетельствует гипогликемия, встречающаяся при разнообразных опухолях, то они способны окислять и другие субстраты.
Наиболее часто гипогликемия является следствием продукции инсулиноподобных факторов роста (ИФР-1 и ИФР-II) самой опухолью. Гены инсулина
кодируют образование проинсулина (неактивный предшественник инсулина),
структура которого сходна с двумя инсулиноподобными факторами роста, которые образуются в печени. Наибольшая концентрация ИФР-1 выявлена в печени, нервной системе, глазу, легких, сердце, скелетных мышцах, яичках, тимусе, лимфоузлах, жировой ткани, поджелудочной железе.
Кроме того, причинами паранеопластической гипогликемии могут быть:
повышенная продукция соматостатина и ингибиторов инсулиназы, торможение
гликогенолиза в печени, блокирование глюконеогенеза и повышенное потребление глюкозы опухолью.
Для опухолевых клеток характерно низкое содержание митохондрий, что
уменьшает интенсивность тканевого дыхания и изменяет способ ресинтеза
АТФ, а именно: увеличивается доля АТФ, образуемой в ходе гликолиза и
уменьшается доля АТФ, синтезируемая в процессе тканевого дыхания. Общая
продукция АТФ в опухолевой клетке снижена по сравнению с нормальной.
Усиление гликолиза в опухолевых клетках обусловливает их высокую выживаемость в условиях гипоксии.
С увеличением размеров опухоли прогрессивно ухудшается ее васкуляризация, что также усиливает анаэробный гликолиз. В опухолевых клетках активируется обмен глюкозы по пентозофосфатному шунту через аэробную (при
участии глюкозо-6-фосфатдегилрогеназы) и анаэробную (при участии
трансальдолазы и транскетолазы) ветви этого процесса, что обеспечивает повышенную продукцию рибозо-5-фосфата как основного продукта для синтеза
нуклеотидов и нуклеиновых кислот.
В опухолевых клетках в несколько раз увеличивается активность гексокиназы, фосфофруктокиназы и пируваткиназы — гликолитических ферментов
(в итоге накапливаются недоокисленные продукты), а активность ферментов
глюконеогенеза (глюкозо-6-фосфатаза, фруктозо-1,6-дифосфатаза, фосфоенолнируваткарбоксилаза и пируваткарбоксилаза) несколько снижена. И тем не менее, глюконеогенез в опухолевых клетках протекает с большей скоростью, чем
в нормальных. Субстратом для этого процесса являются аминокислоты. Следует отметить, что ферменты глюконеогенеза обладают большим сродством к
субстратам и хуже поддаются гормональной регуляции.
209
Содержание гликогена в опухолевых клетках снижено, но это не типично
для них, так как уменьшение содержания гликогена отмечается и в нормальных
клетках при быстром росте и регенерации тканей. Скорость гликогенолиза
снижена.
Для злокачественного роста типичным является сниженный ответ гликемии на инсулин и сниженная в соответствии с этим толерантность к глюкозе.
Учитывая, что синтез и высвобождение инсулина из клеток поджелудочной
железы при опухолевом росте не меняется, нарушение следует искать на
уровне рецепторов клеточных мембран.
Особенности белкового метаболизма. Обмен белков нарушается не только в опухолевых клетках, но и в организме, пораженном злокачественным ростом. На уровне опухолевых клеток интенсифицируется синтез онкобелков
(«опухолеродных» или «опухолевых» белков), которые обусловливают появление у опухолевых клеток характерных биологических свойств: бесконтрольность деления, утрата лимита Хейфлика, иммортализация (бессмертие) др.
Синтез онкобелков программируется активными клеточными онкогенами
и в очень малых количествах — их неактивными предшественниками, именуемыми протоонкогенами. Активные онкогены выявляются только в опухолевых
клетках, а протоонкогены — во всех нормальных клетках. В опухолевых клетках отмечается уменьшение синтеза и содержания гистонов — белковсупрессоров синтеза ДНК.
На увеличение скорости белкового синтеза в опухолевых клетках влияет
повышенная проницаемость цитоплазматических мембран для некоторых ключевых субстратов этого процесса. Опухолевые клетки представляются «пастью, открытой для белков». Они изымают необходимые, незаменимые аминокислоты из крови без какой-либо регулировки этого процесса, влияя тем самым на состояние здоровых клеток. Результатом этого становится не только
быстрый рост опухолевых клеток, но и отрицательный азотистый баланс организма, что, как правило, сопровождается быстрым снижением массы тела и
развитием кахексии. Кроме того, угнетаются процессы дезаминирования и переаминирования.
Изменения белкового состава крови у лиц с опухолевым процессом можно разделить на 2 группы:
1. Изменение количественного соотношения естественных белков плазмы крови.
2. Появление белков новых типов, связанных с возникновением или течением
опухолевого роста.
Снижается синтез и концентрация сывороточного альбумина и повышается
синтез α1,α2 и β- глобулинов. Прежде всего, это относится к α1- гликопротеиду,
α1-антитрипсину, церулоплазмину и трансферрину, в увеличении содержания
которых в сыворотке крови существенную роль играют внутриклеточные гидролазы, освобождающиеся, при распаде опухолевых и неопухолевых клеток.
Развитие злокачественного роста в некоторых органах сопровождается
появлением белков, синтез которых имел место только в эмбриональном пери-
210
оде: альфа-фетопротеин, канцероэмбриональный антиген и хорионгонадотропин. Альфа-фетопротеин синтезируется эмбриональными гепатоцитами и находится в сыворотке эмбриона. В сыворотке крови взрослого человека этот белок обнаруживается при гепатоцеллюлярном раке печени, тератобластоме яичка и яичника. Он способен специфически связывать стероидные гормоны и
один из изоферментов щелочной фосфатазы.
Повышенное содержание хорионгонадотропина отмечается во время беременности, но если его содержание возрастает без беременности, то следует
искать трофобластические опухоли.
Наиболее изученным при канцерогенезе и развитии опухоли оказался
метаболизм нуклеотидов и нуклеиновых кислот. Установлено, что одним из
первых проявлений злокачественной трансформации является экспрессия генов, ответственных за кодирование ключевых ферментов анаболических и катаболических процессов. При этом вначале значительно повышается активность ферментов, участвующих в анаболических процессах, поэтому в опухолевых клетках повышается синтез нуклеиновых кислот, отмечается их избыточное накопление, что характерно для злокачественного роста. Активность
ферментов, участвующих в катаболических процессах, вначале опухолевого
роста снижается (а в организме повышаются, и усиливаются катаболические
процессы), а затем повышается.
Особенности метаболизма липидов. В организме, пораженном злокачественным ростом, липиды выполняют роль источника энергии и субстратов для
образования сложных липидов, участвующих в построении и в обмене фосфолипидов цитоплазматических мембран. В первом случае в метаболизме не
наблюдается никаких отклонений: липолиз происходит обычными путями и регулируется гормонами, но постепенно запасы нейтрального жира иссякают.
Поскольку при этом не отмечается как правило повышения в крови кетоновых
тел, можно полагать, что процесс их распада является аэробным.
Структурные липиды, фосфолипиды, образующие цитоплазматические
мембраны в опухолевых клетках, по своему качественному составу принципиально не отличаются от таковых и в нормальных клетках. Отмечается лишь некоторое упрощение их полисахарид кого компонента. Обнаруживаются также
количественное различие в представительстве отдельных видов фосфолипидов,
входящих в мембраны различных опухолевых клеток.
Общее содержание фосфолипидов в опухолевых клетках повышено,
ускорен их метаболический оборот. Это связано с быстрым синтезом и делением клеток, для которого необходимым условием является быстрый синтез липидных компонентов мембран. Отсюда и ускоренный метаболизм липидов в
микросомальной фракции, где их молекулы и образуются. Аналогично изменяется синтез холестерина.
Весьма характерен для опухолей феномен «субстратных ловушек». Он
заключается в усиленном захвате и использовании субстратов для энергообразования (глюкозы), для построения цитоплазмы (аминокислот — отсюда «ловушка азота») клеточных мембран (холестерина), для защиты от свободных радикалов и стабилизации мембран (антиоксидант α-токоферол). Эта особенность
211
повышает выживаемость опухолевых клеток при контакте их с нормальными
клетками в условиях инвазивного роста и метастазирования.
3. Физико-химический атипизм проявляется увеличением содержания в
опухолевых клетках воды и некоторых электролитов. Увеличение содержания
воды облегчает диффузию субстратов метаболизма внутрь клетки и его продуктов наружу. Далее, в опухолях в пересчете на сухую массу или на белковый
азот повышается содержание ионов натрия и кальция (в опухолевой клетке), в
меньшей степени — калия и значительно снижается концентрация магния.
Увеличение содержания калия в опухолевой клетке препятствует в определенной мере развитию внутриклеточного ацидоза в связи с усилением гликолиза и накоплением молочной кислоты. Концентрация ионов водорода увеличивается в периферической, растущей зоне опухоли благодаря интенсивному
гликолизу и уменьшается в некротизирующейся зоне, обычно расположенной
центрально, благодаря выходу из распадающихся структур опухолевых клеток
больших количеств калия и белка.
В организме-носителе опухоли отмечается тенденция к развитию алкалоза. Полагают, что механизм его развития связан с компенсаторным перераспределением (в ответ на резорбцию из опухоли в кровь лактата) щелочных катионов из тканей в кровь.
В некротически измененной опухоли высвобождаются жирные кислоты,
которые связываются с ионами кальция, образуя соли (мыла) и тем самым способствуют увеличению ионов кальция в опухолевой ткани. Снижение ионов калия характерно для опухолей, отличающихся высокой продукцией муцинов
(например, аденокарцинома яичников), которые связывают ионы калия. При
быстрой потере массы тела и при развитии кахексии вследствие разрушения
большого количества клеточных структур калия много выделяется с мочой.
Изменения концентрации кальция обычно вторичны и сопровождают
опухоли эндокринных желез или метастазы в кости. Часто отмечается недостаточность железа, что играет важную роль в возникновении железодефицитной
анемии.
Повышается величина отрицательного заряда поверхности опухолевых
клеток вследствие накопления на ней анионов нейраминовой кислоты, что способствует увеличению их взаимного отталкивания и проникновению по межклеточным щелям в нормальные ткани. Повышается также электропроводность
и снижается вязкость клеточных коллоидов.
В последние годы установлено, что опухолевые клетки излучают митогенетические лучи — ультрафиолетовые лучи с длиной волны 190—325 нм.
Они генерируются всеми клетками, но наиболее интенсивно — делящимися.
Эти лучи способны стимулировать деление соседних клеток. Они были открыты А.Г. Гурвичем и получили название митогенетических лучей Гурвича. В
крови животных, страдающих опухолями, обнаруживаются вещества, ингибирующие митогенетическое излучение опухолевых клеток. Их назвали тушителями митогенетических лучей.
4. Морфологический атипизм делят на тканевой и клеточный. Тканевой
атипизм сам по себе, без клеточного атипизма, характерен только для добро-
212
качественных опухолей и заключается в нарушении нормального соотношения
тканевых структур, в неравномерности волокнистых или мышечных пучков, в
образовании неправильных и неравномерных железистых ходов, в отсутствии
выводных протоков у опухолей железистого характера.
Клеточный атипизм. Опухолевая клетка сама по себе не несет черт специфичности, но по совокупности структурно-функциональных качеств она отличается от нормальной клетки организма, т.е. она атипична. Морфологическая атипия опухоли может выражаться в нарушении органотипической, гистотипической и цитотипической дифференцировки.
Для доброкачественных опухолей характерны два первых признака; для
злокачественных опухолей характерным является в первую очередь нарушение
цитотипической дифференцировки, отражающее появление опухолевого роста
на уровне клетки и ее органоидов. На светооптическом уровне морфологические признаки атипии клеток выражаются в их полиморфизме или мономорфизме. Полиморфизм касается ядер, ядрышек. Выявляется гиперхроматоз
ядер,
«комковатый»
хроматин,
полиплоидия,
нарушение
ядерноцитоплазматического индекса (из-за укрупнения ядра), обилие митозов с преобладанием среди них патологических.
Наряду с атипией, проявляющейся дедифференцировкой, анаплазией, катаплазией, отмечаются признаки дифференцировки опухолевых клеток с образованием в них специфических структур. Дифференцировка опухолевых клеток
всегда неполная, атипичная и афункциональная, но продукты дифференцировки позволяют установить тканевую принадлежность опухоли, а нередко — и ее
гистогенез.
Дифференцировка выражается не только в появлении структур, характерных для нормальных клеток данной ткани и органа. Она сопровождается
изменениями функции клеток и проявляется в форме выработки специфических
структурных белков (коллагена, миозина), секретов (слизи), гормонов (паратгормон, глюкагон), изменений активности ферментов (фосфорилазы) и др.
Ультраструктура опухолевой клетки. Специфических электронномикроскопических изменений, характерных для опухолевых клеток, не обнаружено. Описываемая обычно дезорганизация цитоплазмы, преобладание в ней
свободных рибосом, увеличение ядра, инвагинация ядерной оболочки и изменения митохондрий встречаются далеко не во всех опухолях, а если и выявляются, то далеко не во всех клетках данной опухоли. Все это свидетельствует,
по мнению академика Д.С. Саркисова, о том, что опухолевая клетка совершает
не «шаг назад», а «шаг в сторону», что Р.Вепеке назвал «катаплазией».
Катаплазия (приставка «ката» означает движение вниз) - появление слабодифференцированных или недифференцированных клеток, похожих на эмбриональные. Опухоль может утрачивать частично или полностью тканеспецифпчес-кие признаки.
Было бы принципиальной ошибкой пытаться описать ультраструктурную организацию опухолевой клетки вообще, т.е. какой-то средней, единой для всех
опухолей клетки. И тем не менее выделяют 2 особенности опухолевых клеток:
ультраструктурную органоспецифичность и ультраструктурный полиморфизм.
213
Крайне редко опухоли имеют мономорфную ультраструктуру. Они весьма разнообразны — в одной и той же опухоли встречаются клетки, находящиеся на
разных уровнях дифференцировки и функционального созревания. Вот поэтому-то в опухолях можно выявить 2 группы клеток:
5. Антигенный атипизм опухоли состоит в разнонаправленных изменениях антигенного состава ее клеток: антигеном упрощении и появлении новых
антигенов. Под антигенным упрощением понимают утрату опухолевыми клетками антигенов, имеющихся в исходно нормальных клетках. В опухолевых
клетках появляются новые, отсутствовавшие в нормальных клетках, антигены.
Существует две гипотезы, объясняющие возникновение новых антигенов в
опухолевых клетках:
а) новые антигены (неоантигены) возникают вследствие соматической мутации
генома клетки;
б) новые антигены являются результатом реактивации гех участков генома, которые в ходе развития (дифференцировки) были ингибированы.
Как известно, большинство клеточных антигенов локализуется в цитоплазматической мембране и имеет природу интегральных белков. Обычно, это
гликопротеиды, проникающие через всю толщу мембраны, а на поверхности
оканчивающиеся цепью или цепями олигосахаридов. Именно эти олигосахариды принимают участие в обеспечении таких жизненно важный функций, как
адгезия, контактное инициирование и отличие своих белков от чужих.
При злокачественной трансформации может происходить отщепление
выступающих над поверхностью опухолевой клетки антигенных структур под
влиянием протеаз, и тогда на поверхность выходят детерминантные группы,
локализующиеся глубже — криптоантигены. Кроме того, выявляется обеднение поверхностных углеводных структур трансформированных клеток. Такая
упрощенная поверхностная структура менее всего способна различать другие
подобные обедненные структуры. Это приводит к утрате контактного торможения (ингибирования), суть которого заключается в том. что клетки, входя в
контакт с клетками того же вида, перестают делиться.
В зоне злокачественного перерождения па поверхности клеток не только
возникают новые антигены; но одновременно с этим идет процесс исчезновения некоторых, ранее присутствовавших поверхностных антигенов. Они могут
попадать в кровь, и это будет иметь большое значение для диагностики опухолей. Из типично опухолевых антигенов, освобождающихся с поверхности клетки и выходящих в кровь, с диагностической целью можно использовать такие
антигены, как:
— α1-фетопротеин. Это гликопротеин (м.м. около 70 кД), образующийся в печени эмбриона. Его синтез прекращается после рождения и содержание его в
крови находится на столь низких величинах, что можно обнаружить только радиоиммунным методом. Повышение его содержания характерно для рака печени, а также для тератом различной природы и локализации;
— канцероэмбриональный антиген. Это также гликопротеид (м.м. 180—200
кД); выделено 3 различных вида данного антигена. В физиологических условиях он имеется в клетках слизистой пищеварительного тракта и с их поверхности
214
постоянно выделяется в просвет кишечника. В крови его очень мало (следы) и
он выявляется иммунохимически. Концентрация этого антигена в крови возрастает при раке прямой кишки, толстого кишечника, печени, бронхов, доброкачественных полипах кишечника, язвенном колите. Содержание этого антигена
может быть также повышено и при всех состояниях, которые сопровождаются
повышенной секрецией слизи: хронический бронхит, при курении.
Утрата опухолевыми клетками одних антигенов (органоспецифических) и
появление в них эмбриональных антигенов (к которым не образуются антитела,
поскольку они воспринимаются иммунной системой как свои) способствует
«антигенной маскировке» опухолевых клеток и «неузнаваемости» их иммунной
системой.
Кроме того, опухолевые клетки несут на своей поверхности туморассоциированные трансплантационные антигены — ТАТА. Именно эти антигены
вызывают каскад реакций иммунной системы, результатом которых является
торможение роста опухоли или цитолиз трансформированных клеток.
Биологичесчкие особенности злокачественных опухолей
1. Инфильтративный рост (от лат. infiltratio — проникновение; син.:
инвазивный). Он заключается в проникновении опухолевых клеток в окружающие ткани с деструкцией их. В отличие от этого доброкачественные опухоли
растут экспансивно, т.е. сдавливая и раздвигая (отодвигая) окружающие такни.
При этом сдавленные опухолью паренхиматозные элементы ткани атрофируются, наступает коллапс стромы, часто ее количество нарастает и вокруг опухоли образуется как бы капсула (псевдокапсула). Некоторые злокачественные
опухоли (например, канальцевый рак почек, рак околощитовидной железы,
фибросаркома) также растут экспансивно.
Инфильтративному росту способствует ряд факторов:
а) приобретение опухолевыми клетками способности отшнуровываться от опухолевого узла и активно перемещаться;
б) способность опухолевых клеток продуцировать «канцероагрессины» — вещества белковой природы, проникающие в окружающие нормальные ткани и
стимулирующие хемотаксис и благодаря этому инвазию в них опухолевых клеток;
в) уменьшение сил клеточной адгезии, что значительно облетает отделение
(отшнуровывание) опухолевых клеток и их последующее движение. Доказано,
что уменьшение клеточной адгезии обусловлено снижением содержания кальция, деполимеризацией белковых и полисахаридных компонентов межклеточного вещества, накапливающимися в опухолях ферментами: гиалуронидазой, протеазами, а также повышение поверхностного заряда опухолевых клеток;
г) уменьшение контактного торможения.
2. Метастазирование (от греч. metastasis — перемена места, перемещение, перенос) — перенос опухолевых клеток за пределы первичной опухоли в
различные органы и ткани с образованием вторичных опухолевых узлов той же
гистологической структуры. При изучении метастазирования следует выявлять
215
возможные пути распространения опухолевых клеток из первичного очага и
механизм формирования метастаза. Возможны следующие пути распространения опухолевых клеток:
1)лимфогенное- по лимфатическим сосудам (например, метастазы рака з регионарные лимфатические узлы, а оттуда с током лимфы з грудной лимфатический
проток и в другие органы и ткани);
2)гематогенное -по кровеносным сосудам;
Механизм образования метастазов во многом еще не изучен. Не выяснены причины избирательной локализации метастазов мри опухолях различной
локализации и структуры. Например, для рака легкого характерны метастазы в
головной мозг, кости, надпочечники; для канальцевого рака почки — в кости,
прорастание вдоль почечных вен и нижней полой вены с образованием внутри
этих сосудов массивных опухолевых конгломератов; для рака печени - обширные внутриорганныс метастазы с прорастанием почечных вен и внутрисосудистым ростом опухоли. В 80—90% случаев опухолевые клетки можно обнаружить в крови (даже в ранние сроки опухолевого процесса). Однако, большинство опухолевых клеток погибает в крови, а оставшиеся способствуют метастазированию.
В развитии лимфогенных, гематогенных и гемато-лимфо-генных метастазов различают три стадии;
1). Стадия инвазии — проникновение опухолевых клеток через стенку сосудов
в их просвет. Она обеспечивается теми же факторами, что и инфильтративный
рост. Важную роль играет также «неполноценность» структуры сосудов опухолей, которые устроены по типу капилляров. Их стенка состоит из одного слоя
эндотелия и базальной мембраны.
2). Стадия клеточной эмболии — перенос с током крови пли лимфы опухолевых клеток, проникших в сосуды, остановка («заклинивание») их в просвете
микрососуда с последующим образованием на их поверхности нитей фибрина.
Это превращает клеточный эмбол в тромбоэмбол, прикрепляющийся к эндотелию. Развитию клеточной эмболии благоприятствует сличение количества
и активности Т-киллеров, призванных уничтожать опухолевые клетки, а также
уменьшение канцеролитической активности крови и лимфы, т.е. снижении их
способности лизировать чужеродные клетки, включая и опухолевые.
3). Стадия проникновения опухолевых клеток из клеточного тромбоэмбола через стенку сосудов в окружающие ткани, безудержное деление этих опухолевых клеток с образованием новых опухолевых узлов. В эту стадию происходит
направленное движение опухолевых клеток из сосуда в нормальную ткань. В
механизме развития этой стадии определяющую роль играют те же факторы,
что и в развитии первой стадии (инвазии). Кроме того, важное значение в развитии третей стадии принадлежит снижению активности механизмов аллогенного ингибирования роста опухолевых клеток и феномен антигенной маскировки опухолевых клеток.
3. Рецидивирование (от лат. recidivas — возврат, повторное развитие болезни). Рецидив - возникновение опухоли на том же самом месте после ее хирургического удаления или излечения с помощью лучевой терапии или химио-
216
терапии. В одних случаях рецидивы возникают в связи с тем, что в операционном поле остаются неудаленные комплексы опухолевых клеток из-за технических трудностей и нечетких границ опухоли (прямой рецидив или процидив), в других — рецидив возникает в связи с прорастанием в рубец или культю
удаленного органа (например, желудка) метастаза из соседнего лимфатического
узла, незамеченного во время операции.
Рецидив возможен и в связи с оставлением части органа, в котором сохранились условия для возникновения новой опухоли на почве прогрессирующих пред опухолевых процессов, что согласуется с теорией опухолевого ноля.
Это так называемый ложный рецидив — речь идет фактически о новой опухоли, возникшей в зоне бывшей операции.
Кроме того, имеются факты, свидетельствующие о том, что причиной рецидива может быть проникновение нуклеиновых кислот (ДНК онкогенов) в
клетки окружающих нормальных тканей. Рецидивированию способствует
также и иммунодепрессия, возникающая в части случаев после операции.
Рецидивы (как и метастазы) по своей структуре оказываются катаплазированными по сравнению с первичной опухолью, то есть в них происходят те или
иные явления прогрессии.
4. Кахексия (от греч. kakos — плохой, дурной + hexis - состояние) —
синдром истощения и слабости организма, возникающий при раках желудочнокишечного тракта Кахексия, возникающая при злокачественных опухолях, получила название «раковой» кахексии. Она служит одним из проявлений
парнеопластического синдрома, возникает в периоде, близком к терминальному, характеризуется потерей массы тела в основном из-за усиленного распада
белков скелетных мышц (частично миокарда), а также истощения жировых депо. Кахексия сопровождается отвращением к пище и изменением вкусовых
ощущений. Важную роль в развитии раковой кахексии играют следующие факторы:
— повышенный расход энергии (иногда до 50%), обусловленный гормональным дисбалансом;
— нарушение нервно-эндокринной регуляции обмена веществ и усиленное образование АТФ за счет гликолиза (что существенно повышает расход энергетических субстратов);
— ингибирование липопротеинлипазы, катализирующей накопление липидов в
организме;
— угнетение процессов синтеза РНК, обеспечивающих синтез белков и дифференцировку адипоцитов;
— образование особого полипептидного гормона «кахектина», известного также как TNF (tumor necrosis factor - фактор некроза опухолей); TNF секретируется макрофагами и опосредует воспалительные реакции, а поскольку практически все клетки организма имеют к нему рецепторы, то его эффекты могут быть
многообразны - шоковое состояние, снижение АД, расстройства липидного и
углеводного обмена, метаболический ацидоз, анорексия и истощение организма
вплоть до гибели;
217
— снижение синтеза каталазы, что способствует накоплению продуктов свободно-радикального перекисного окисления;
— сопутствующие опухоли осложнения: боль, кровотечение, нарушение функций желудочно-кишечного тракта;
— повышенное улавливание опухолью из крови субстратов (глюкозы, аминокислот и др.).
Кахексия может наблюдаться и при некоторых доброкачественных опухолях определенной локализации. В частности, при опухолях желудочнокишечного тракта вследствие развития непроходимости или резкого нарушения
секреторной, моторной и всасывательной функции; в головном мозге в области
трофических центров — из-за нарушения нервно-гормональной регуляции обмена веществ и энергии.
Взаимоотношения опухоли и организма
Возникновение и развитие опухоли в организме не является абсолютно
автономным процессом. Сам факт трансформации нормальной клетки в опухолевую и дальнейший её рост - реакция организма на действие различных факторов внешней и внутренней cреды: канцерогенов химической, биологической
и физической природы.
Взаимодействие опухоли и организма осуществляется при участии всех
физиологических систем - нервной, эндокринной, ИБН, кровообращения и других. Воздействие организма на новообразование может осуществляться через
изменение кровоснабжения и иннервации новообразования и окружающих его
тканей, действия на него БАВ (гормонов, медиаторов, цитокинов и других),
факторов системы ИБН (Ig, лимфоцитов, мононуклеарных фагоцитов, гуморальных факторов системы неспецифической защиты организма) и других.
Результат взаимодействия опухоли и организма может быть различным.
Гибель бластомных клеток. Это наблюдается наиболее часто. В организме среди большого числа постоянно образующихся различных клеток–
мутантов имеются и опухолевые. Однако они, как правило, сразу же обнаруживаются и уничтожаются при участии факторов системы ИБН.
Латентное, «дремлющее» состояние опухолевых клеток. Они делятся,
образуя сравнительно небольшой клон бластомных клеток, не имеющих стромы. Трофика их обеспечивается за счёт диффузии веществ, содержащихся в
межклеточной жидкости. Как правило, при этом не наблюдается признаков инвазии клеток бластомы в окружающую нормальную ткань. Такую форму опухолевого роста обозначают как неинвазивную, или «рак на месте» — cancer in
situ. Подобное состояние может наблюдаться в течение ряда лет. Оно может завершиться либо гибелью клеток бластомы (при активации факторов системы
ИБН), либо интенсификацией её роста — приобретением способности к инвазии в окружающие ткани, метастазированию и другим (при снижении эффективности факторов системы ИБН).
Прогрессирующее формирование новообразования с нарастанием степени
его атипизма. При этом условно можно выделить две разновидности воздействия опухоли на организм: местное и общее.
218
Местные эффекты новообразования характеризуется следующими феноменами:
- инвазивным ростом, сочетающимся со сдавлением и деструкцией окружающей нормальной ткани, нарушением микрогемо- и лимфоциркуляции, развитием недостаточности ткани или органа;
- образованием и выделением в межклеточную жидкость метаболитов, в том
числе обладающих свойствами БАВ (гормонов, факторов роста, ферментов,
иммунодепрессантов и др.), способных вызвать дисфункцию органов;
- подавлением активности местных факторов системы ИБН — фагоцитирующих клеток, лимфоцитов, лизоцима, ИФН и других, что способствует прогрессии опухолевого роста, а также развитию воспаления.
Системное влияние новообразований проявляется развитием ряда общих неспецифических синдромов. Их называют паранеопластическими. К
наиболее клинически значимым паранеопластическим синдромам относятся
кахексия, иммунопатологические состояния,психоневрологические синдромы
(психозы,
слабоумие,
невропатии,
нейро-трофические
расстройства),эндокринопатии(являются результатом нарушения продукции, инкреции
и эффектов гормонов, выделяемых как гормональноактивными опухолями так
и непоражёнными бластомой эндокринными железами),тромбогеморрагические
синдромы,анемии и др.
Антибластомная резистентность организма
Воздействие канцерогенных этиологических факторов само по себе недостаточно, чтобы вызвать опухоль. Далеко не всякий возникший в организме
клон опухолевых клеток превращается в злокачественную опухоль. Организм
располагает определенными средствами противодействия. На первых этапах
действует система естественной неспецифической резистентности, способная
элиминировать небольшое количество опухолевых клеток. К ней относятся
естественные киллеры (это крупные гранулярные лимфоциты, составляющие
до 2,5% от всей популяции периферических лимфоцитов и не зависимые от тимуса, которые после их активации интерфероном могут лизировать опухолевые
клетки) и макрофаги, способные образовывать кислородные радикалы и перекись водорода, которые разрушают опухолевые клетки.
Возможность преодолеть опухолевыми клетками барьера естественной
резистентности определяется их способностью продуцировать факторы, подавляющие эту систему (например, простагландины Е), и их устойчивостью к перекиси водорода.
Возникновению опухоли способствует снижение антибластомной резистентности. Под антибластомной резистентностью понимают устойчивость организма к возникновению и развитию опухолей. При нарушении соотношения
между действием канцерогенных факторов и антибластомной резистентностью
в сторону преобладания влияния канцерогенных факторов и снижения антибластомной резистентности возникает опухоль.
219
Все механизмы антибластомной резистентности в зависимости от характера воздействия или точки приложения применительно к основным звеньям
опухолевого процесса подразделяют на:
1. Антиканцерогенные — действующие на канцерогены, препятствуя
взаимодействию этих факторов с клетками, органеллами, молекулами. Среди
них различают:
а). Антиканцерогенные механизмы, действующие против химических канцерогенов. Они включают:
— инактивацию канцерогенов с помощью реакций окисления (например, окисление полициклических ароматических углеводородов оксидазами печеночных
микросом), реакций восстановления с помощью микросомальных редуктаз
(аминоазо-красители), реакций конъюгации с глюкуроновой кислотой;
— элиминацию экзо- и эндогенных канцерогенов из организма в составе желчи,
мочи и кала;
— ингибирование свободных радикалов антиоксидантами: альфа-токоферолом,
глутатионом, унитиолом и др.;
— пино- и фагоцитоз канцерогенов с последующим их обезвреживанием;
— образование антител против канцерогенов (канцерогены являются гаптенами).
б). Антиканцерогенные механизмы, действующие против физических канцерогенов. Они включают:
— реакции торможения образования свободных радикалов;
— реакции инактивации свободных радикалов и перекисей липидов и водорода. Через эти продукты, как полагают, реализуют свое опухолеродное влияние
физические канцерогены. В этих реакциях участвуют витамин Е, селен, глутатион и ряд ферментов: супероксиддисмутаза, глутатионпероксидаза и каталаза.
в). Антиканцерогенные механизмы, действующие против онкогенных вирусов.
Они включают:
— ингибирование онкогенных вирусов интерферонами. Интерферон усиливает
литическую активность естественных киллеров , а также активирует цитотоксические Т-лимфоциты (они разрушают опухолевые клетки и тем самым ограничивают вирусную репликацию);
— нейтрализацию онковирусов специфическими антителами.
2. Антитрансформационные механизмы. Они включают механизмы,
препятствующие трансформации нормальной клетки в опухолевую:
а). Антимутационные механизмы. Они реализуются благодаря наличию в клетке ферментных систем репарации ДНК, которые устраняют повреждения,
«ошибки» ДНК (генов) и поддерживают таким образом генный гомеостаз.
б) Антионкогенные механизмы, которые осуществляются антионкогенами —
специальными клеточными генами, являющимися антагонистами онкогенов.
Они подавляют деление клеток и стимулируют их дифференцировку
3. Антицеллюларняе механизмы. Они препятствуют превращению образовавшихся отдельных клеток в клеточную колонию — опухоль. Эти механизмы включаются с момента образования опухолевых клеток. Они направлены на
уничтожение отдельных опухолевых клеток и опухолей в целом. Эти механиз-
220
мы осуществляются благодаря антигенной и клеточной чужеродностк опухолей. Выделяют две группы антицеллюлярноых механизмов: иммуногенные и
неиммуногенные.
а). Иммуногенные антицеллюлярные механизмы осуществляются благодаря
специфическим и неспецифическим иммуногенным механизмам:
— специфические иммуногенные механизмы включают цитотоксическое повреждение, ингибирование роста и уничтожение опухолевых клеток, вопервых, иммунными Т-лимфоцитами -киллерами; во-вторых, иммунными макрофагами с помощью целой группы факторов, секретируемых этими макрофагами (макрофаглизина, лизосомальных ферментов, рос-тингибирующего
компонента интерферона, фактора некроза опухоли) с участием комплемента; в
третьих, К-клетками, имеющими рецепторы к иммуноглобулинам, благодаря
чему они проявляют сродство и цитотоксичность к опухолевым клеткам, покрытым IgG. К К-клеткам относятся «нулевые» лимфоциты (ни Т-, ни Влимфоциты), лишенные характерных маркеров Т- и В-лимфоцитов.
— иеспецифические иммуногенные механизмы. К ним относят неспецифическое цитотоксическое повреждение, ингибирование роста и лизис опухолевых
клеток, во-первых, натуральными (природными) киллерами (NK-клетки), которые подобно К-лимфоцитам являются разновидностью нулевых лимфоцитов,
во-вторых, неспецифически активированными Т-лимфоцитами с помощью
лимфокинов и, в-третьих, с помощью неспецифически активированных (бактериями или их эндотоксинами) макрофагов;
б). Неиммуногенные антицеллюлярные механизмы включают:
— туморнекротический фактор. Он продуцируется моноцитами, тканевыми
макрофагами, Т- и В-лимфоцитами, гранулоцитами, тучными клетками;
— интерлейкин I (ИЛ-1) —стимулирует К-клетки, Т-киллеры, активирует макрофаги, стимулирует синтез ИЛ-2, который индуцирует размножение и рост Тлимфоцитов (включая Т-киллеры), стимулирует синтез -интерферона;
— аллогенное торможение — подавление и уничтожение опухолевых клеток
окружающими нормальными клетками, обусловленное цитотоксическим действием гистонесов местимых антигенов, метаболитов и различием конфигурации поверхности мембран;
— кейлонное ингибирование. Кейлоны (хейлоны) — это тканеспецифические
ингибиторы размножения клеток, в том числе и опухолевых;
— контактное торможение; в его реализации участвуют цАМФ (активирует
контактное торможение) и цГМФ (ингибирует контактное торможение и стимулирует деление клеток);
— лаброцитоз. Канцерогенез сопровождается локальным увеличением тучных
клеток — лаброцитов. Они продуцируют гепарин, который ингибирует образование фибрина на поверхности фиксированных и циркулирующих в крови опухолевых клеток, а это препятствует развитию метастазов из-за торможения превращения ракового клеточного эмбола в клеточный тромбоэмбол;
— регулирующее влияние гормонов. Они оказывают активное регулирующее
влияние на антибластомную резистентность: это влияние зависит от дозы гормона и вида опухоли.
221
ПАТОФИЗИОЛОГИЯ ОБМЕНА ВЕЩЕСТВ
Современное понимание медико-биологических проблем основано на
взаимосвязи атрибутивных свойств всех живых организмов – структуры, функции и обмена веществ.Живой организм –макромолекулярная система, осуществляющая обмен веществ, энергии и самовоспроизведение.В основе процессов жизнедеятельности лежат химические превращения, в которые вовлекаются в организме природные химические соединения, химические реакции
высвобождения, запасания и использования энергии, химические реакции ,
обеспечивающие регуляторные функции.Изменение химических основ жизнедеятельности свойственно патологическим процессам, заболеваниям отдельных
органов и систем.Эти знания необходимы врачу для определения путей исправления или устранения этих нарушений.Изучение отклонений базируется на
представлениях о естественном течении процессов.
Обмен веществ – сложная система химических реакций, связанных между
собой через пластические компоненты, энергетическое обеспечение и общие
регуляторы.Биохимическая схема обмена веществ включает цепи, каскады, и
циклы, связанные метаболическими путями.
•
•
•
•
Посредством обмена веществ осуществляется:
Извлечение энергии.
Получение структурных блоков (моносахара, аминокислоты, жирные кислоты) и синтез полимеров, соответственно индивидуальной генетической программе организма.
Синтез и инактивация регуляторных молекул и молекул, необходимых для
выполнения специфических функций данной клетки.
Разрушение генетически чужеродных генетических соединений.
Этапы обмена веществ:
• Питание.
• Пищеварение.
• Транспорт.
• Межуточный обмен (внутриклеточные превращения химических веществ)
включает два типа реакций:
- Катаболизм — процесс расщепления органических молекул до конечных
продуктов. Конечные продукты превращений органических веществ — СО2,
Н2О и мочевина. В процессы катаболизма включаются метаболиты, образующиеся как при пищеварении, так и при распаде структурно-функциональных
компонентов клеток. Реакции катаболизма сопровождаются выделением энергии (экзергонические реакции).
222
- Анаболизм объединяет биосинтетические процессы, когда строительные блоки соединяются в сложные макромолекулы. В анаболических реакциях используется энергия, освобождающаяся при катаболизме (эндергонические реакции).
• Выделение конечных продуктов метаболизма.
Болезни как ферментопатии.
Скорость метаболических реакций зависит от концентрации и активности
определенных ферментов. В каждом метаболическом пути имеется такая реакция от которой зависит скорость всего пути – лимитирующая реакция и соответственно имеется фермент, который катализирует лимитирующую реакцию –
ключевой фермент. Активность ферментов контролируются гормонами, доступностью кофакторов (витамины, ионы), ограничивается ростом концентрации одного из продуктов реакции.
Ферментопатия (энзимопатия) – отсутствие какого-либо фермента, снижение или нерегулируемое усиление его активности, приводящее к метаболическим расстройствам.
• Наследственные (врожденные) ферментопатии – возникают в результате мутации генов, кодирующих синтез фермента.
• Приобретенные – возникают при нарушении биосинтеза белковой части
фермента (белковое голодание, дефицит аминокислот, энергии), недостатке
коферментов (гипо- и авитаминозы, нарушение обмена ионов), накоплении
ингибиторов ферментов (соединения ртути, свинца, цианиды).
Ферментопатии приводят к метаболическим блокадам.
Виды метаболических блокад:
I. Дефицит конечного продукта метаболизма.
Патология возникает, если жизнедеятельность организма нарушается без
конечного продукта заблокированной метаболической цепи.
а) Выпадение какого-либо из факторов системы гемокоагуляции (после
активации они, как известно, приобретают протеолитическую активность)
влечет за собой задержку в образовании фибрина и кровоточивость.
б) Низкая активность ферментов, катализирующих присоединение йода к
тирозину в клетках щитовидной железы (в этом процессе принимают участие
окислительно-восстановительные ферменты, цитохромы, пероксидазы и др.),
приводят к недостаточной выработке трийодтиронина и тироксина. Развивается синдром гормональной недостаточности - гипотиреоз (микседема).
в) В коре надпочечников при недостатке соответствующих ферментов из
холестерина не образуются в достаточном количестве кортикостероиды (чаще
страдает выработка глюкокортикоидов). Развиваются проявления кортикальной
недостаточности.
г) Если недостаточно активен фермент тирозиназа, в организме снижается
выработка пигмента - меланина.. Развивается местный (витилиго) или системный альбинизм: обесцвечивание кожи, волос, радужной оболочки (иногда светобоязнь, глухота, немота, эпилепсия, олигофрения).
223
II. Избыток исходного или промежуточного продуктов метаболизма
В качестве примера можно привести болезни накопления.
а) Гликогенозы, например болезнь Гирке - дефицит глюкозо-6фосфатазы. Блокируется образование глюкозы из гликогена и последний
накапливается в гепатоцитах, эпителии почечных канальцев. Возникает функциональная недостаточность печени и почек. Кроме того, имеет место длительная гипогликемия, отрицательно влияющая на функции и формирование ЦНС.
Характерны: постоянная слабость и отставание в развитии ребенка.
б) Болезнь Гоше - накопление цереброзидов в печени и селезенке вследствие падения активности фермента, ответственного за их распад.
в) Акаталазия - отсутствие каталазы, фермента расщепляющего перекись водорода (один из продуктов, постоянно образующихся в процессе перекисного
окисления липидов). Для этого заболевания характерны воспалительные процессы и изъязвления различных слизистых оболочек.
г) Врожденная недостаточность метгемоглобинредуктазы способствует
накоплению в крови метгемоглобина и развитию гемической гипоксии.
д) В качестве приобретенной ферментопатии с тяжелейшими последствиями
можно рассматривать инакгивацию холинэстеразы различными токсическими
препаратами (фосфорганическими соединениями - табун, зарин, заман, некоторые инсектициды, судилищный боб и др.). В организме в избытке накапливается ацетилхолин и (иногда в течение нескольких минут или даже секунд) наступает смерть от грубейших расстройств функции нервной системы.
III. Усиление альтернативных путей метаболизма
В организме, как правило, те или иные вещества могут метаболизироватъся по нескольким различным путям. Путь, по которому метаболизируется
большая часть данного продукта, называют главным путем метаболизма. Пути,
по которым превращаются незначительные количества, обозначаются, как альтернативные.
Если заблокирован главный путь, то включение вещества в обмен по альтернативному пути может резко усиливаться. Усиление альтернативного пути
может повлечь за собой различные нарушения жизнедеятельности. Например,
при сахарном диабете (см. ниже):
1. Альтернативные пути гликолиза:
а) сорбитоловый шунт;
б) синтез глюкуроновой кислоты;
в) синтез гликопротеидов (полигликанов).
2. Альтернативные пути превращения ацетил-КоА:
а) синтез кетоновых тел;
б) синтез холестерина с вытекающими из этого тяжелейшими последствиями для организма;
3. При гипоксии тормозится аэробный путь энергообразования (ЦТК) и
резко нарастает анаэробный гликолиз. Это приводит к накоплению пировиноградной и молочной кислот, развивается метаболический лакгоацидоз;
224
4. Если в коре надпочечников тормозится синтез глюкокортикоидов, происходит стимуляция синтеза половых гормонов, чаще андрогенов, и развивается адреногенитальный синдром.
IV. Усиление побочных метаболических путей
Накопление исходного продукта может привести к тому, что это вещество включается в химические преобразования, которые в нормальных условиях не происходят или происходят в ничтожных объемах. При этом могут образовываться токсические для организма метаболиты.
а) Классическим примером является фенилкетонурия.
Из-за отсутствия соответствующей гидроксилазы не происходит превращение фенилаланина в тирозин.
Из фенилаланина образуются фениллакгат и фенилпируват, продукты
очень токсичные для ЦНС. У таких детей развивается очень тяжелая форма
слабоумия.
Своевременное выявление заболевания и применение диеты, не содержащей фенилаланина, предотвращает развитие олигофрении.
б) При алактазии в кишечнике не вырабатывается лактаза. Не происходит
расщепление молочного сахара. Этот сахар подвергается брожению, образуются СО2 и органические кислоты, развивается бродильная диспепсия, метеоризм,
диаррея, обезвоживание, истощение больного.
V. Развитие вторичных блоков.
Накапливающийся исходный продукт может блокировать какой-то фермент в какой-нибудь другой метаболической цепи и в этой заблокированной
цепи могут наблюдаться все те последствия, которые рассматривались выше.
а) При галактоземии из-за отсутствия фермента галактозофосфат - уридилтрансферазы тормозится превращение галактозо-1-фосфата в УДФ-галактозу.
Накапливающийся галактозо-1-фосфат, ввиду своего структурного сходства с
глюкозо-1-фосфатом, блокирует фермент фосфоглюкомутазу и тем самым
препятствует взаимопревращению глюкозы и гликогена. У детей с галакгоземией развивается кахексия, цирроз печени, катаракта и олигофрения.
б) Избыток мочевой кислоты в организме приводит к образованию аллоксана - уреида мезоксалевой кислоты, который блокирует выработку инсулина в поджелудочной железе и способствует развитию сахарного диабета.
При любой форме патологии, связанной с ферментопатиями и метаболическими блоками, клинические проявления обуславливаются не каким-либо
одним из пяти перечисленных механизмов, а их сочетанием, с преобладанием
одного из них.
Особо можно выделить недостаточность ферментов, определяющих активный энергозависимый транспорт веществ через различные клеточные мембраны - так называемых пермеаз.
Такие нарушения могут возникать при дефиците соответствующих ферментов, недостатке энергии - АТФ, нарушениях гормональной регуляции (глюкоза-инсулин, натрий-альдостерон) и по другим причинам.
225
Последствия могут проявляться на всех трех условно выделяемых уровнях обмена веществ.
1. На уровне пищеварения и всасывания (ЖКТ) нарушения транспорта из
просвета кишечника во внутреннюю среду приводит к:
а) накоплению невсосавшихся продуктов в кишечнике и к таким, например, последствиям, как бродильная или гнилостная диспепсия;
б) дефициту невсосавшихся веществ в крови (гипогликемия при снижении транспорта глюкозы, дефицит ароматических аминокислот при болезни
Хартнупа).
2. На уровне межуточного обмена (кровь - клетки органов и тканей) - будет наблюдаться:
а) избыток каких-либо продуктов в крови;
б) энергетический и пластический дефицит клеточных элементов, ионный
дисбаланс клеток (при сахарном диабете, когда нарушается транспорт глюкозы
- гипергликемия и энергетический дефицит клеток).
3. На уровне экскреции - выделения метаболитов из организма:
а) задержка, накопление каких-то веществ во внутренней среде (гиперазотемия при почечной и печеночной недостаточности, гипербилирубинемия при
печеночной недостаточности);
б) снижение содержания каких-либо метаболитов в крови (гипогликемия
при нарушении реабсорбции глюкозы, дефицит аминокислот при нарушении
реабсорбции аминокислот - аминоацидурия, потеря натрия, воды и калия - гиповолемия и гипокалиемия при нарушении реабсорбции этих факторов в почечных канальцах).
Изменения метаболизма и функционирования физиологических систем
организма могут быть также связаны с нарушением продукции неферментных
белков:
• Белки плазмы крови.
• Белки системы гемостаза и антигемостаза.
• Гемоглобин.
• Иммуноглобулины.
• Белки системы комплемента.
• Белки калликреин-кининовой системы.
Энергетический обмен – совокупность химических процессов организма, направленных на получение свободной (тепловой) и связанной (химической, макроэргической) энергии, необходимой для жизнедеятельности организма.
Главная общая энергетическая характеристика жизнедеятельности организма – основной обмен (ОО).
Основной обмен — количество энергии, необходимое для поддержания
нормальных функций организма при минимальных процессах обмена веществ
226
в условиях абсолютного мышечного покоя, лежа, натощак, при температуре
18°С.
ОО обеспечивает уровень максимально экономичного функционирования
организма при сохранении базовых анаболических процессов.
На уровень основного обмена влияют возраст, пол, сезон, климат, время
суток, специфически динамическое действие пищи, профессия, рефлекторные
влияния.
Причинами нарушения основного обмена могут быть расстройства нервно-эндокринной регуляции, лихорадка, голодание, инфекции, интоксикация,
авитаминозы, изменения деятельности сердца, дыхания, печени и др.
Организм получает энергию в результате катаболизма пищевых веществ,
в нормальных условиях – в основном углеводов и липидов. Массивное использование белка в энергетических целях возможно лишь в крайне вынужденных
ситуациях (например, при голодании).
Катаболизм – трёхэтапный процесс, каждый из этапов приводит к освобождению определённой энергии, которая сразу частично рассеивается в виде
тепла.
Первый этап – гидролитический, проходит в ЖКТ и в лизосомах без участия кислорода, освобождает 1% всей энергии субстратов. Эта энергия полностью переходит в первично рассеянное тепло и не запасается.
Второй этап – бескислородное цитоплазматическое расщепление (гликолиз, липолиз) приводит к получению универсального катаболита – ацетилкоэнзима А. При этом освобождается до 30% энергии субстратов и около 43 %
её запасается в виде АТФ, остальная энергия рассеивается.
Третий этап – аэробный, наиболее эффективный путь катаболизма, осуществляемый в митохондриях. Освобождается до 70% энергии субстратов, которые окисляются до конечных метаболитов – СО2 и Н2О. При этом до 60%
энергии запасается в виде макроэргических соединений.
Запасённая энергия необходима организму для жизнедеятельности и для обеспечения анаболических процессов (биосинтез).
Нарушение энергетического обеспечения клетки может быть вследствие:
• Нарушений количественного поступления субстратов в организм
• Нарушений переваривания и всасывания в ЖКТ
• Нарушений транспорта питательных веществ через клеточные мембраны
• Нарушений процессов катаболизма в клетке
• Нарушения нервно-эндокринной регуляции
Механизм нарушения энергетического обмена связан с разобщением окисления и фосфорилирования, что ведет к гиперпродукции свободной энергии в
организм и перегреванию. Инфекционно-токсические факторы увеличивают
проницаемость мембран митохондрий, способствуют выходу субстратов цикла
Кребса в гиалоплазму, окислению их ферментами
гиалоплазмы, увеличению свободного окисления, гиперпродукции АТФ и тепла.
227
Голодание — это типический патологический процесс, развивающийся
когда организм не получает пищи совсем, получает ее в недостаточном количестве или не усваивает ее вследствие болезни.
При голодании прежде всего включаются приспособительные механизмы, происходит своеобразная ферментативная адаптация
организма к отсутствию питательных веществ и переход на эндогенное питание.
Голодание по своему происхождению может быть физиологическим (периодически повторяется у некоторых видов животных в связи
с особыми условиями их обитания или развития) и патологическим.
Различают голодание полное и неполное. Полное голодание может быть
без ограничения воды, с ограничением или вовсе без воды (абсолютное голодание). Неполное количественное голодание развивается в том случае, когда в организм поступают все питательные вещества, но в недостаточном по калорийности количестве. Неполное количественное голодание наблюдается, если при
достаточной калорийности пищи в ней нет какого-либо компонента или он
есть, но в недостаточном количестве (жиры, белки, углеводы, витамины). Часто
эти два вида голодания комбинируются.
Причины голодания могут быть внешними и внутренними:
• Внешние причины - отсутствие пищи.
• Внутренние причины - пороки развития у детей, заболевания органов
пищеварительной системы, инфекционные процессы, анорексия.
При голодании продолжительность жизни уменьшают те внешние условия, которые увеличивают потерю тепла, повышая энергетические затраты организма на поддержание температуры тела (низкая температура окружающей
среды, высокие влажность и скорость движения воздуха, активные движения).
Из внутренних условий на продолжительность жизни при голодании влияют: пол, возраст, общее состояние организма, количество и качество жировых
и белковых резервов, а также интенсивность обмена веществ.
По клиническим проявлениям полное голодание можно разделить на четыре периода:
• безразличия, когда животное ведет себя относительно спокойно и особых
изменений не обнаруживается;
• возбуждения, нарастающего по мере усиления чувства голода;
• угнетения (самый длительный) - животное становится вялым, безразличным, большую часть времени лежит, свернувшись в клубок;
• параличей и гибели животного.
На основании состояния обмена веществ и энергии в голодании можно
выделить три периода:
• Неэкономного расходования энергии. В общих чертах обмен веществ в
первом периоде голодания характеризуется усиленным расходованием
углеводов, в связи с чем дыхательный коэффициент повышается. Содержание гликогена в печени быстро снижается, но он полностью не исчезает, образуясь за счет усиления процессов гликогенеза. В связи с угнете-
228
нием секреции инсулина в печени ослабляется эффективность цикла
Кребса, снижается уровень окислительного фосфорилирования, что отражается на энергетическом обмене клеток. Конкурентное торможение
глюкокортикоидами скорости гексокиназной реакции снижает усвоение
глюкозы клетками печени. В начале первого периода голодания основной
обмен повышен. К концу первого периода по мере перехода на экономное
расходование энергии снижается на 10-20% и остается на этом уровне и
во втором периоде, несколько увеличиваясь лишь в третьем. Снижение
основного обмена при голодании отражает глубокую перестройку обменных процессов, направленную на экономное расходование энергетических ресурсов. Определенную роль при этом играет угнетение функции
щитовидной железы и островков Лангерганса. Показательно, что параллельно снижению массы тела и печени снижается содержание митохондриального белка. Выделение азота с мочой уменьшается. Ослабевает интенсивность дезаминирования переаминирования аминокислот в печени,
снижается биосинтез аминокислот из кетокислот и аммиака. Уменьшается образование цитруллина и аргинина из их предшественников и снижается синтез мочевины. Однако все эти процессы не могут сбалансировать
распад белка - развивается отрицательный азотистый баланс.
• Максимального приспособления Во втором, самом длительном периоде
голодания, дыхательный коэффициент снижается до 0,7, что отражает
преимущественное окисление жира. Около 80% энергии организм получает за счет окисления жира, 3% — путем окисления глюкозы и 13% —
белка. Мобилизация жира из депо приводит к липемии. Печень подвергается жировой инфильтрации. В результате недоокисления жира усиливается образование кетоновых тел, которые вместе с продуктами недоокисления белков вызывают развитие метаболического ацидоза. Вследствие
этого увеличивается выделение аммонийных солей с мочой. Основной
обмен в этот период снижен, азотистый баланс отрицательный. Вместе с
тем сохраняется возможность синтеза жизненно необходимых белковых
структур за счет распада ряда других белков.
• Тканевого распада, интоксикации и гибели. Третий, терминальный,
период характеризуется резким усилением распада белков жизненно важных органов. Белки расходуются в качестве энергетического материала.
Дыхательный коэффициент равен 0,8. Увеличивается выведение с мочой
азота, калия, серы, фосфора. Азот, калий и фосфор содержатся в моче в
таких же соотношениях, как и в протоплазме мышечных волокон. Это
свидетельствует о распаде не только легко мобилизуемых, но и стабильных белков мышц. Возникают деструктивные изменения в митохондриях.
В связи с задержкой хлоридов и повышением тканевой осмотической
концентрации происходит задержка воды. Нарушение трофики тканей и
снижение общей резистентности проявляется иногда в виде пролежней и
участков некроза на коже и слизистых, возникновением кератита. Теплопродукция в течение всего голодания сохраняется на минимальном
уровне и снижается лишь к концу третьего периода. Теплоотдача сокра-
229
щается. Температура тела мало меняется, оставаясь на нижней границе
нормы, и лишь в конце снижается до 30-28°С.
Полное голодание без воды протекает так же, как и голодание с водой,
но более тяжело и менее длительно. Если вода не вводится извне, она черпается
из тканей — это оксидационная вода. Наибольшее количество воды освобождается от жировых отложений — 100 г жира при окислении отдают 112 г воды, а
белки и углеводы примерно вдвое меньше. При этом образуется много продуктов обмена, требующих для выделения еще больше воды, и создается порочный
круг, ускоряющий гибель животного.
Лечебное голодание – ограничение питания с лечебной целью.
Применяется при алиментарно-конституциональном первичном ожирении, аллергических атопических заболеваниях, иммунопатологических кожных
болезнях, эссенциальной гипертензии, подагре, ревматоидном полиартрите, болезнях системы пищеварения.
Лечебное голодание используют для снижения массы тела и повышения
адаптационных возможностей организма.
Состоит из начального периода, периода разгрузки и периода восстановления общей длительностью не более 15 дней.
ПАТОФИЗИОЛОГИЯ
УГЛЕВОДНОГО
ОБМЕНА.
САХАРНЫЙ
ДИАБЕТ.
В пищеварительном тракте конечными продуктами переваривания углеводов являются глюкоза, фруктоза и галактоза. Основной углевод, циркулирующий в крови — глюкоза (нормальный уровень глюкозы в плазме крови – 3,35,5 ммоль/л).
Транспорт глюкозы через клеточную мембрану. Глюкоза присоединяется
к белкам-переносчикам, которые транспортируют глюкозу через клеточную
мембрану внутрь клетки посредством облегченной диффузии. Главный активатор трансмембранного переноса глюкозы – инсулин. Под влиянием инсулина
скорость и количество глюкозы, транспортируемой через клеточные мембраны,
значительно возрастают.
Фосфорилирование глюкозы. Поступившая в клетки глюкоза фосфорилируется ферментом глюкокиназой.
Накопление гликогена и гликогенолиз. После поступления в клетки глюкоза сразу же используется для образования энергии или накапливается в виде
гликогена (большой полимер из молекул глюкозы). Все клетки тела способны
запасать некоторое количество гликогена, но только гепатоциты, скелетные
мышечные волокна и кардиомиоциты могут депонировать большие запасы гликогена. Большие гликогеновые молекулы преципитируются в форме плотных
гранул. Процесс образования гликогена — гликогенез. Гликогенолиз — процесс расщепления гликогена с образованием глюкозы, происходит под влияни-
230
ем фосфорилазы. В состоянии покоя этот фермент находится в неактивированном состоянии. Активация фосфорилазы происходит под влиянием адреналина
и глюкагона.
Выделение энергии из глюкозы. При полном окислении одной молекулы
глюкозы может образоваться 38 молекул АТФ, из них 2 в ходе гликолиза, 2 в
цикле лимонной кислоты и 34 при окислительном фосфорилировании.
Анаэробное выделение энергии. Возникают случаи, когда кислород недоступен, или его слишком мало для клеточных процессов окисления глюкозы. В
этих условиях небольшие количества энергии могут быть выделены в клетках
путём гликолиза, поскольку химические реакции расщепления глюкозы до пировиноградной кислоты не нуждаются в кислороде. Образуется 2 молекулы
АТФ и молочная кислота.
Регуляция расщепления глюкозы. Гликолиз и окислительное фосфорилирование — процессы регулируемые. Оба процесса постоянно контролируются в
соответствии с потребностями клеток в АТФ. Этот контроль имеет отношение к
механизмам обратной связи между концентрациями АТФ и АДФ. Одним из
элементов контроля энергии является ингибирующее влияние АТФ на ферментативные процессы, протекающие на начальных этапах гликолиза. Излишки
АТФ останавливают гликолиз с последующим торможением углеводного метаболизма. АДФ же, напротив, повышает активность гликолитических процессов.
Как только АТФ используется тканями, ингибирующее влияние АТФ на ферменты гликолиза уменьшается. Одновременно с этим возрастает активность
ферментов вследствие формирования АДФ. Когда клеточные запасы АТФ
вновь переполняются, ферментативные процессы замедляются.
Глюконеогенез. Когда запасы углеводов в организме становятся ниже
нормального уровня, то умеренное количество глюкозы может образовываться
из аминокислот и из глицериновой части жиров в процессе глюконеогенеза.
Приблизительно 60% аминокислот в белках организма могут легко превращаться в углеводы. Низкий уровень углеводов в клетках и уменьшение содержания глюкозы в крови – главные стимулы увеличения интенсивности глюконеогенеза (регулируется глюкокортикоидами).
Нарушение углеводного обмена.
Нарушение обмена углеводов происходит при расстройстве:
• Расщепления и всасывания углеводов в пищеварительном тракте. Основными причинами являются тяжелые повреждения кишечника, дефицит амилолитических ферментов, нарушение фосфорилирования глюкозы в клетках
кишечной стенки (недостаточность гексокиназы). При уменьшении всасывания углеводов возникает гипогликемия и снижение массы тела, осмотическая диарея.
• Синтеза, депонирования и распада гликогена. Снижение синтеза гликогена
происходит при тяжелом поражении печеночных клеток, когда нарушается
их гликогенобразовательная функция (гепатиты), при гипоксии. Распад гликогена усиливается при стрессе (активация симпатической нервной системы), тяжелой мышечной работе, голодании, увеличении гормонов, стимули-
231
рующих гликогенолиз. При уменьшении в организме гликогена развивается
гипогликемия, накопление кетоновых тел, интоксикация, потеря пластического материала клетками. Усиление синтеза гликогена приводит к его избыточному накоплению в печени и других органов и тканях и их повреждению. Это характерно для гликогенозов – ферментопатии (наследственный
дефицит ферментов, катализирующих распад или синтез гликогена), наследуются по аутосомно-рецессивному типу.
• Обмена углеводов органах и тканях. При гипоксии (происходит анаэробный
путь окисления углеводов, накопление молочной и пировиноградной кислоты, ацидоз), при гиповитаминозе В1 (дефицит кокарбоксилазы, являющейся
простетической группой ферментов углеводного обмена).
Нарушение нейрогуморальной регуляции.
Нарушение гормонального звена регуляции приводит к развитию гипогликемии или гипергликемии. Инсулин обладает гипогликемическим эффектом. Контринсулярные гормоны (глюкагон, адреналин, глюкокортикоиды, соматотропин, тиреоидные гормоны) – гипергликемическим эффектом.
Влияние нервной системы на обмен углеводов опосредуется гормонами:
активация симпатической нервной системы приводит к увеличению синтеза адреналина, парасимпатической – инсулина и глюкагона, гипоталамогипофизарного пути – глюкокортикоидов.
Гипогликемия – синдром, развивающийся при понижении содержания
глюкозы в крови ниже 3,8 ммоль/л. Причиной данного синдрома может быть
понижение поступления глюкозы в кровь из печени и/или кишечника, повышение её утилизации тканями и выведения из крови, а также комбинация этих
механизмов.
Виды гипогликемии:
• Инсулиновая – при передозировке инсулина у больных сахарным диабетом,
при наличии инсулиномы (доброкачественной инсулинпродуцирующей
опухоли).
• От недостаточности контринсулярных гормонов – гипопитуитаризм, гипокортицизм, гипотиреоз, острая недостаточность функций мозгового вещества надпочечников.
• От недостаточности расщепления гликогена – гликогенозы, при печеночной
недостаточности (хронические гепатиты, цирроз печени).
• Алиментарная – общее и углеводное голодание, кишечная и энзимопатическая мальабсорбция углеводов, транзиторная гипогликемия новорожденных.
• При снижении реабсорбции глюкозы в проксимальном отделе канальцев
возникает глюкозурия - при отравлении монойодацетатом и флоридзином.
• Аутоиммунные формы – инсулиномиметическое действие аутоантител к инсулиновым рецепторам.
• Длительная физическая нагрузка.
Последствия гипогликемии:
232
• Гипогликемическая реакция – острое временное снижение глюкозы крови
до нижней границы нормы. Возникает в результате избыточной секреции
инсулина через 2-3 суток после начала голодания, либо через несколько
часов после нагрузки глюкозой и проявляется легким чувством голода,
мышечной дрожью, тахикардией.
• Гипогликемический синдром – стойкое понижение глюкозы крови, сопровождающееся расстройствами жизнедеятельности организма. Проявления связаны с избыточной секрецией катехоламинов (чувство голода,
мышечная дрожь, потливость, тахикардия) и с расстройствами функций
ЦНС (головная боль, головокружение, спутанность сознания, заторможенность, нарушения зрения).
• Гипогликемическая кома – развивается при резком снижении глюкозы
крови, потеря сознания, значительные нарушения жизнедеятельности организма. От момента развития до смерти (при отсутствии адекватной помощи) проходят минуты.
Причины: передозировка инсулина, прием алкоголя, чрезмерное
физическое и психическое напряжение.
Патогенез. Возникает нарушение энергообеспечения нейронов и клеток
других органов из-за недостатка глюкозы, повреждаются мембраны и ферменты, возникает ионный дисбаланс, нарушается создание потенциалов покоя и
действия. При снижении уровня глюкозы крови стимулируется выброс гормонов обратной регуляции (адреналин, глюкагон, кортизол, соматотропин), но
при гиперинсулинемии образовавшаяся глюкоза быстро утилизируется тканями
и уровень глюкозы продолжает падать. Появляются симптомы гипогликемии,
обусловленные ответной реакцией на снижение глюкозы и компенсаторными
реакциями на гипогликемию.
Лечение направлено на ликвидацию гипогликемии (введение в организм
глюкозы), лечение основного заболевания, блокирование патогенетических
звеньев гипогликемической комы, устранение симптомов (головной боли, тахикардии).
Гипергликемия – синдром, характеризующийся увеличением глюкозы
крови выше нормы.
Причины: эндокринопатии, переедание, неврологические и психогенные
нарушения, патология печени.
Эндокринопатии приводят к гипергликемии в результате дефицита инсулина (его эффектов) или избытка контринсулярных гормонов (их эффектов).
Избыток глюкагона может возникать в результате гиперплазии α-клеток
островков поджелудочной железы, что приводит к стимуляции глюконеогенеза
и гликогенолиза.
Избыток глюкокортикоидов возникает при гипертрофии или опухолях коры
надпочечников, гиперсекреции кортикотропина, что приводит к активации
глюконеогенеза и ингибированию активности гексокиназы.
Избыток катехоламинов (феохромоцитома) активирует гликонеогенез.
233
Избыток тиреоидных гормонов возникает при диффузном или узловом
гормонально-активном зобе и приводит к усилению гликогенолиза и глюконеогенеза, торможению гликогенеза, активации всасывания глюкозы в кишечнике.
Избыток соматотропина (аденома аденогипофиза) активирует гликогенолиз и тормозит утилизацию глюкозы.
Недостаток инсулина см. в разделе сахарный диабет.
К неврологическим и психогенным расстройствам относятся психическое
возбуждение, стрессы, каузалгии, при которых активируется симпатическая и
гипоталамо-гипофизарная системы – гормоны этих систем приводят к гипергликемии.
Переедание (длительное избыточное потребление легкоусваемаемых углеводов с пищей) приводит к увеличенному всасыванию глюкозы, избыток углеводов в кишечнике стимулирует гликогенолиз в гепатоцитах.
Печеночная патология – вследствие печеночной недостаточности гепатоциты не способны синтезировать гликоген из глюкозы.
Последствия гипергликемии:
• Гипергликемический синдром – значительное повышение уровня глюкозы
(свыше 10,5 – 11,5 ммоль/л), сопровождающееся расстройствами жизнедеятельности. Проявляется глюкозурией, полиурией, полидипсией, гипогидратацией и артериальной гипотензией.
• Гипергликемическая кома.
Гликогенозы – типическая форма нарушения углеводного обмена наследственного или врожденного происхождения, характеризующаяся избыточным
накоплением гликогена в клетках, приводящее к нарушению жизнедеятельности организма.
Развиваются вследствие мутации генов, кодирующих синтез ферментов
расщепления или образования гликогена. Это приводит к отсутствию или низкой активности ферментов гликогенолиза или синтеза гликогена. В основном
гликогенозы наследуются по аутосомно-рецессивному типу.
Дефект фермента
Глюкозо-6-фосфатаза
Альфа-1,4-глюкозидаза
Амило-1,6-глюкозидаза
D-1,4-глюкано-α-глюкозилтрасфераза
Гликогенфосфорилаза миоцитов
Гликогенфосфорилаза гепатоцитов
Фосфогюкомутаза
Тип гликогеноза
1-й тип (болезнь Гирке)
2-й тип (болезнь Помпе)
3-й тип (болезнь Кори)
4-й тип (болезнь Андерсен)
5-й тип (болезнь МакАрдля)
6-й тип (болезнь Гирса)
7-й тип (болезнь Томпсона)
234
Фосфофруктомутаза
Киназы фосфорилазы в гепатоцитах
8-й тип (болезнь Таруи)
9-й тип (болезнь Хага)
Сахарный диабет.
Сахарный диабет – заболевание, которое развивается в результате гипоинсулинизма (относительной или абсолютной инсулиновой недостаточности) и
характеризуется нарушением всех видов обмена веществ и расстройством жизнедеятельности организма.
Сахарный диабет – это хроническое мультигормональное расстройство
всех видов метаболизма, характеризующееся нарастающей гипергликемией,
глюкозурией, развитием осложнений, в основе которых лежат повреждения сосудов, а также нейропатия.
Распространение СД колеблется в разных странах от 1 до 3%, у людей
страдающих ожирением различной степени достигает 15-25%.
Классификация СД.
Первичный сахарный диабет – идиопатическое расстройство механизмов инсулиновой регуляции метаболизма.
• Первичный сахарный диабет Ι типа (инсулинозависимый, гипоинсулинемический, юношеский, ювенильный, ИЗСД):
Ιа – обусловленный комбинацией генетического и средового воздействия
Ιb – первичный, генетически обусловленный, со спонтанным аутоиммунным процессом, без явных признаков экзогенной провокации
Ιс – с первичным поражением В-клеток экзогенными диабетогенами
(химическими, вирусными).
• Первичный сахарный диабет ΙΙ типа (инсулиннезависимый, гиперинсулинемический, взрослых, пожилого возраста, тучных, ИНСД):
ΙΙа – ИНСД у нетучных больных
ΙΙb - ИНСД у тучных больных
ΙΙс – ИНСД в юношеском возрасте
Вторичный сахарный диабет – диабетические (гипергликемические) синдромы, возникающие как следствие других болезней, поражающих поджелудочную железу или систему регуляции метаболизма углеводов:
• Вторичный диабет, вызванный неаутоиммунной деструкцией панкреатических В-клеток при поражении поджелудочной железы (хронический
панкреатит, рак, гемохроматоз, кистозный фиброз, панкреатэктомия,
травма поджелудочной железы)
• Вторичный диабет, вызванный эндокринными расстройствами с гиперпродукцией контринсулярных гормонов (синдром Иценко-Кушинга, акромегалия, феохромоцитома, глюкагонома, гипертиреоидизм, гиперплазия эпифиза)
235
• Вторичный ятрогенный диабет при применении медикаментов (кортикостероиды, АКТГ, оральные контрацептивы, атидепрессанты, фенотиазины, некоторые мочегонные)
• Вторичный диабет при генетически обусловленных синдромах (липодистрофия, гликогеноз Ι типа, атаксия-телеангиэктазия, хромосомные аномалии Дауна, Шерешевского-Тернера и Кляйнфельтера).
Этиология СД.
• Дефицит инсулина (абсолютная недостаточность инсулина).
• Недостаточность эффектов инсулина (относительная недостаточность
инсулина).
Дефицит инсулина возникает под действием биологических, химических,
физических, развитие воспаления поджелудочной железы.
• Генетические дефекты β-клеток (иммунные аутоагрессивные механизмы
вследствие появления чужеродных антигенов – гликопротеины, кодируемые аллелями HLA-DR3, HLA-DR4, HLA-DQ).
• Иммунные факторы (Ig, цитотоксические Т-лимфоциты повреждают βклетки).
• Вирусы, тропные к β-клеткам – коксаки В4, гепатита, кори, ветряной
оспы, эпидемического паротита, краснухи (цитолитическое действие, активация иммунных процессов, развитие воспаления – инсулит).
• Химические факторы (аллоксан, этанол, цитостатики).
• Физические факторы (ионизирующее излучение, механическая травма,
сдавление опухолью).
Недостаточность эффектов инсулина возникает под влиянием нейрогенных
и психогенных факторов, контринсулярных факторов, дефектов инсулиновых
рецепторов и пострецепторных реакций.
• Нейро- и психогенные факторы – стресс-реакции, активация симпатоадреналовой системы (приводит к увеличению в крови катехоламинов,
глюкокортикоидов, тиреоидных гормонов).
• Контринсулярные факторы – активация инсулиназы гепатоцитов (усиливается гидролиз инсулина), антитела к инсулину, увеличение контринсулярных гормонов (катехоламины, глюкагон, СТГ, глюкокортикоиды, тиреоидные гормоны), увеличение белков крови, свызывающих инсулин.
• Дефекты инсулиновых рецепторов – антитела блокирующие или повреждающие инсулинорецепторы, длительный избыток инсулина (приводит
к снижению чувствительности к гормону), гидролазы, свободные радикалы, продукты ПОЛ.
• Нарушение пострецепторных реакций – повреждение мембраны и ферментов клеток, нарушение образования цАМФ.
Факторы риска СД.
236
• Повышение массы тела (возникает инсулинорезистентность).
• Гиперлипидемия (стимулирует продукцию контринсулярных гормонов).
• Артериальная гипертензия (нарушение микроциркуляции в поджелудочной железе).
• Наследственная предрасположенность.
• Стрессы.
•
•
•
•
•
•
•
•
Экспериментальные модели СД.
Панкреатический СД – удаление у собак 9/10 поджелудочной железы.
Аллоксановый СД – введение аллоксана – вещества, избирательно повреждающего β-клетки поджелудочной железы.
Стрептозотоциновый СД – введение стрептозотоцина – антибиотик, избирательно повреждающий β-клетки поджелудочной железы у крыс.
Дитизоновый СД – введение дитизона – связывает цинк, нарушая депонирование и секрецию инсулина.
Иммунный СД – введение антител против инсулина.
Метагипофизарный СД – длительное введение гормонов аденогипофиза
(СТГ, АКТГ).
Метастероидный СД – длительное введение глюкокортикоидов.
Генетические модели СД – выведение чистых линий животных с наследственно обусловленной формой болезни.
Метаболизм углеводов при СД.
Изменения в метаболизме углеводов обусловлены относительным дефицитом инсулина и избытком контринсулярных гормонов и приводят к развитию
гипергликемии (увеличение содержания глюкозы в плазме крови выше 6,66
ммоль/л).
Механизмы гипергликемии:
• В гепатоцитах, миоцитах и липоцитах низкие уровни инсулина приводят к
угнетению гексокиназной реакции, что ведет к понижению продукции глюкозо-6-фосфата, что обуславливает угнетение гликолиза и пентозного цикла.
• Вследствие активации фосфорилазы и ингибирования гликогенсинтетазы в
гепатоцитах усиливается гликогенолиз и глюкоза поступает в кровь.
• Глюкагон активирует ферменты глюконеогенеза и обеспечивает глюконеогенез субстратами (усиливает липолиз, тормозит синтез белка на рибосомах).
• Вследствие дефицита инсулина снижается поглощение глюкозы мышцами
(при обоих типах СД) и жировой тканью (при Ι типе СД) после еды.
• Нарушается цикл Кребса (многие ферменты цикла контролируются инсулином, субстраты цикла используются в глюконеогенезе, снижена концентрация пирувата), в результате снижается окислительное фосфорилирование и
образование АТФ.
237
•
•
•
•
•
•
•
Метаболизм липидов при СД.
Гиперлипацидемия – увеличение содержания в крови свободных жирных
кислот. Связана с активацией липолиза и подавлением липогенеза в жировой
ткани.
Гиперлипопротеинемия – увеличение содержания в крови липопротеидов
очень низкой плотности (ЛПОНП).
Кетонемия – увеличение концентрации кетоновых тел в крови и кетонурия –
нарастание содержания кетоновых тел в моче. В гепатоцитах активируется
β-окисление из-за избыточного поступления жирных кислот и образуется
избыток ацетил-КоА. При отсутствии инсулина ацетил-КоА не включается в
ЦТК, а метаболизируется по альтернативным путям в ацетоуксусную кислоту с последующим образованием β-оксимасляной кислоты.
Гиперхолестеринемия. Еще один путь метаболизма ацетил-КоА – повышение синтеза холестерина.
Атеросклероз. Связан с повышением уровней ЛПОНП и холестерина.
Жировая инфильтрация печени. Является следствием повышения в печени
свободных жирных кислот. При истощении функции гепатоцитов избыток
триглицеридов откладывается в печени.
Похудение – развивается из-за неспособности жировой ткани превращать
свободные жирные кислоты плазмы крови в триглицериды.
Метаболизм белков при СД.
Угнетение биосинтеза белка и усиление его катаболизма. Связано с выпадением анаболического действия инсулина (стимулирует сборку рибосом и
процесс трансляции, усиливает активный транспорт аминокислот в клетки),
общим энергетическим дефицитом (в отсутствии инсулина ЦТК заблокирован), активацией глюконеогенеза (аминокислоты используются для превращения в глюкозу).
Белковый дефицит приводит к задержке физического и умственного развития детей, снижению регенерации тканей (замедление заживления ран,
срастания костей), развитию вторичного иммунодефицита (увеличение чувствительности к инфекциям).
•
•
•
•
Нарушения водно-электролитного обмена при СД.
Гиперосмотическая гипогидратация – развивается в результате полиурии,
которая обусловлена гипергликемией и кетонемией.
Гипонатриемия – возникает на ранней стадии, из-за выхода воды из клеток
по градиенту концентрации.
Гиперкалиемия и гиперфосфатемия– возникают на ранней стадии, из-за выхода калия и фосфора из клеток в связи с распадом тканей, ацидозом и дефицитом инсулина.
Гипернатриемия – возникает вследствие обезвоживания (потери воды превышают потери электролитов).
238
• Гипокалиемия, гипофосфатемия, гипомагниемия – происходят из-за потери
соответствующих ионов при осмотическом диурезе, затем при лечении инсулином.
Нарушения кислотно-щелочного равновесия при СД.
Характерно развитие негазового ацидоза:
• Кетонемический метаболический ацидоз (связан с накоплением кетоновых
тел).
• Лактацидемический метаболический ацидоз (связан с накоплением молочной кислоты вследствие гипоксии).
Клинические проявления СД.
• Гипергликемия – увеличение содержания глюкозы в крови, возникает вследствие нарушения транспорта глюкозы в клетки, снижения синтеза гликогена,
нарушение окисления глюкозы в ЦТК и пентозофосфатном цикле, усиления
глюконеогенеза и гликогенолиза, нарушение функции почек при развитии
диабетической нефропатии.
• Глюкозурия – появление глюкозы в моче. Возникает при концентрации глюкозы в крови, превышающей почечный порог (около 10 ммоль/л), при нарушении реабсорбции глюкозы.
• Полиурия – избыточное образование и выделение мочи. При СД может достигать 4 – 10 литров. Возникает в результате повышения осмотического
давления первичной мочи (накопление в ней глюкозы, азотистых соединений, ионов), что стимулирует клубочковую фильтрацию и тормозит канальцевую реабсорбцию.
• Полидипсия – повышенное употребление жидкости в результате патологической жажды. Возникает из-за гиперосмотической гипогидратации.
• Полифагия – чрезмерное употребление пищи в результате сильного чувства
голода.
Диагностируют СД по уровню глюкозы плазмы крови. Тест толерантности
глюкозы определяется в спорных случаях. Уровень глюкозы определяется спустя 2 часа после стандартного завтрака или приема 75 г глюкозы. Концентрация
глюкозы 7,7 ммоль/л и выше свидетельствует о нарушении толерантности к
глюкозе, выше 11 ммоль/л указывает на сахарный диабет.
Осложнения СД – патологические процессы и состояния, не обязательные для
СД, но обусловленные либо причинами, либо расстройствами развивающимися
при СД.
• Острые – диабетический кетоацидоз с развитием ацидотической комы,
гиперосмолярная и гипогликемическая кома.
• Хронические (поздние) – микроангиопатии, макроангиопатии, невропатии. Признаки часто проявляются через 15-20 лет после выявления гипергликемии.
239
Диабетический кетоацидоз. Характерен для ИЗСД, при котором происходит стимуляция кетогенеза за счет:
- активации липолиза в жировой ткани (нарастает уровень ВЖК в крови,
увеличивается их транспорт в гепатоциты),
- уменьшения активности малонил-КоА в гепатоцитах и активация карнитинацилтрансферазы (увеличивается транспорт ВЖК в гепатоциты, где они
подвергаются β-окислению с образованием кетоновых тел).
Это приводит к нарастанию ацидоза за счет избытка кетоновых тел и развитию кетоацидотической комы.
Гиперосмолярная кома. Характерна для пожилых пациентов. Патогенез
связан с высокой гипергликемией в результате которой повышается осмотическое давление плазмы крови, что приводит к повышению осмотического давления межклеточной жидкости и перемещению жидкости по градиенту давления
из клеток. Возникает обезвоживание клеток. Наиболее подвержена этим изменениям нервная ткань – возникает обезвоживание клеток головного мозга,
угнетение его функций.
Гипогликемическая кома (см. гипогликемия).
Микроангиопатии – патологические изменения в сосудах микроциркуляции. Патогенез – активация превращения глюкозы в сорбитол под влиянием
альдозоредуктазы (накопление сорбитола в сосудистой стенке приводит к ее
уплотнению и утолщению) и неферментативное гликозилирование белков базальных мембран капилляров. Это приводит к уменьшению поступления крови
(ишемия), нарушению транскапиллярного обмена, формированию микротромбов, микрокровоизлияниям.
Ретинопатии – поражения сетчатки глаза, приводящие к снижению остроты
зрения и слепоте. Причина – микроангиопатии и гипоксия тканей глаза.
Макроангиопатии – развитие склеротических изменений в артериях среднего и крупного калибра, приводят к раннему и ускоренному развитию атеросклероза. Патогенез – гликозилирование базальных мембран сосудов, накопление сорбитола, повышение уровня ЛПНП и снижение уровня ЛПВП, активация
синтеза тромбоксана А2 (усиливает вазоконстрикцию и адгезию тромбоцитов),
активация пролиферации гладкомышечных клеток сосудов. Это приводит к
развитию атеросклероза, кальцификации атеросклеротических бляшек, тромбообразованию, закупорке артерий, нарушению кровообращения с развитием инфарктов, инсультов, гангрены (мягких тканей стопы).
Невропатии – поражения различных отделов нервной системы.
Патогенез – гликозилирование белков периферических нервов, образование
антител к измененным белкам, активация превращения глюкозы в сорбитол в
нейронах, снижение интраневрального кровообращения, торможение транспорта миоинозитола в нервные клетки избытком глюкозы (нарушается синтез миелина, нарушается активный транспорт ионов, замедляется проведение нервных
импульсов).
Виды невропатий:
240
• Периферические полиневропатии – поражение периферических нервных
стволов, проявляется нарушениями чувствительности, язвами, эрозиями,
некрозом тканей стоп (синдром диабетической стопы).
• Вегетативная невропатия – поражение вегетативной нервной системы,
проявляется расстройствами функций ЖКТ, дистрофией мочевого пузыря, нарушением регуляции тонуса сосудов и половой функции.
• Радикулопатии – поражение корешков спинного мозга, проявляющееся
повышенной чувствительностью и болевым синдромом по ходу спинальных нервов.
• Мононевропатии – поражение черепных, проксимальных двигательных
нейронов, проявляются преходящими параличами.
Энцефалопатии – дистрофические и дегенеративные повреждения нейронов головного мозга. Возникают из-за понижения энергообеспечения нейронов
(возникновение ишемии), из-за микро- и макроангиопатий. Проявляются нарушениями психической деятельности (расстройства памяти и сна, раздражительность, апатия, быстрая утомляемость) и повреждением вещества головного
мозга (сенсомоторные расстройства, нейродистрофии).
Нефропатии – поражение почек, приводящее к развитию почечной недостаточности. Возникает в результате микро- и макроангиопатий, утолщения базальных мембран клубочков и канальцев, развития интерстициального нефрита
и гломерулосклероза.
Патогенетические принципы лечения сахарного диабета.
• Инсулинотерапия при СД Ι типа.
• Терапия сахароснижающими препаратами при СД ΙΙ типа.
• Диета с ограничением легкоусвояемых углеводов и жиров.
• Учет калорийности диеты.
• Снижение веса при СД ΙΙ типа.
• Регулярность приема пищи.
• Исключение факторов, способствующих декомпенсации диабета (стресс,
инфекции, травмы, операции, гипоксию).
• Ограничение физической нагрузки у пациентов с наклонностью к гипогликемии.
Метаболический синдром («смертельный квартет») – сочетание ожирения,
сахарного диабета, артериальной гипертензии, повышения уровня холестерина в крови,
приводящее к высокому риску развития сердечно-сосудистых заболеваний и смертности
от инфаркта и инсульта.
Метаболический
синдром
Атеросклероз
Инфаркт Инсульт
241
Согласно международным рекомендациям врач имеет право поставить диагноз
«метаболический синдром», если у пациента имеется не менее 3-х из следующих
симптомов:
1. Объём талии >88 см у женщин или >102 см у мужчин
2. Артериальное давление >= 130\85 мм.рт.ст.
3. Сахар крови натощак >= 6,1 ммоль\л
4. Повышение уровня триглицеридов крови >=1,7 ммоль/л
5. Снижение уровня липопротеидов высокой плотности <1 ммоль/л у мужчин, <1,3 ммоль/л у женщин
ПАТОФИЗИОЛОГИЯ БЕЛКОВОГО ОБМЕНА,ОБМЕНА
НУКЛЕОТИДОВ, ЖИРОВОГО ОБМЕНА.
Жиры содержатся во всех тканях человека и относятся к основным и обязательным компонентам пищи человека. Потребности в жире зависят от возраста,
образа жизни, климата и других факторов (80-100 г жира в сутки, из них 25-30 г
должны составлять растительные жиры).
Биологическая полноценность жира обусловлена наличием в нем
витаминов, полиненасыщенных жирных кислот, фосфатидов и других компонентов.
К липидам относятся триглицериды, глицерофосфаты, холестерин и его
эфиры, желчные кислоты, жирные кислоты (ненасыщенные и насыщенные) и
другие соединения.
Липиды входят в состав клеточных мембран, являются главными источниками энергии, растворителями витаминов А, Д, Е, Р, принимают участие в синтезе стероидных гормонов, участвуют в создании защитных термоизоляционных и водоотталкивающих покровов, играют механическую роль (фиксация
почек).
Липиды (простагландины) являются регуляторами функций различных органов, участвуют в передаче нервных импульсов, в создании межклеточных
контактов.
Часть липидов поступает в организм с пищей, часть – синтезируются в организме. С растительными жирами в организм вводятся некоторые полиненасыщенные кислоты (линолевая, линоленовая и другие), которые относятся к незаменимым жирным кислотам, т. к. они в организме человека не синтезируются.
При недостаточном поступлении в организм жиров резко возрастает вероятность онкологических заболеваний.
Регуляция обмена липидов:
• Симпатические влияния, адреналин, норадреналин, соматотропин, тироксин
усиливают липолиз.
• Парасимпатические влияния, глюкокортикоиды и инсулин активируют липогенез.
242
Белки выполняют ряд важных функций в организме: информационная (гормоны), рецепторная (компонент рецепторов), католитическая (ферменты),
структурная, транспортная, источник энергии.
Белки в организме не депонируются, при дефиците используются белки
мышц, кожи, костей, паренхиматозных органов, мозга. При отсутствии поступления белка организм расходует на свои нужды около 4 г белка в сутки.
Потребности организма в белке зависят от возраста, пола, профессии и составляют около 90 г в сутки (или не менее 1 г/кг массы тела), 55 % должно быть
белков растительного происхождения.
Регуляция обмена белка осуществляется нейроэндокринной системой:
• Инсулин, соматотропин и тироксин стимулируют биосинтез белка.
• Глюкокортикоиды усиливают катаболизм белка.
Нарушения белкового обмена.
Показателем уровня белкового обмена является азотистый баланс – суточная разница между поступающим и выделяемым из организма азотом.
Виды азотистого баланса:
• Нулевой – количество поступающего и выделяемого азота одинаковое.
• Положительный – азота больше поступает в организм, чем выделяется
(регенерация тканей, беременность, гиперпродукция соматотропного
гормона).
• Отрицательный – азота меньше поступает в организм, чем выделяется
(голодание, стресс, тяжелое течение СД, гиперкортицизм).
Типические нарушения белкового обмена:
• Несоответствие поступления белка в организм потребностям.
• Нарушение расщепления белка в ЖКТ.
• Нарушение содержания белков в плазме крови.
• Нарушение трансмембранного переноса аминокислот.
• Расстройства метаболизма аминокислот.
• Расстройства конечных этапов катаболизма белков.
Несоответствие поступления белка в организм потребностям.
• Недостаток поступления белка в организм (несбалансированная алиментарная недостаточность белка – квашиоркор, сбалансированная алиментарная недостаточность белка – алиментарная дистрофия).
• Избыток поступления белка в организм.
• Нарушение аминокислотного состава потребляемого белка.
Квашиоркор.
• Снижение массы тела (дефицит белка).
• Гипопротеинемия.
• Гиполипопортеинемия.
• Отрицательный азотистый баланс.
243
•
•
•
•
•
•
Тотальные отеки, асцит.
Иммунодефициты (комбинированные).
Гиперальдостеронизм (в результате гиповолемии).
Гипернатриемия, гипокалиемия, гипофосфатемия, гипомагниемия.
Апатия, гиподинамия.
Задержка физического и умственного развития.
Алиментарная дистрофия (алиментарный маразм).
Полное или частичное белковое голодание приводит к мобилизации белка из
костей, мышц, кожи, из внутренних органов.
• Снижение массы тела.
• Гипопротеинемия, гиполипопротеинемия.
• Отрицательный азотистый баланс.
• Гипогликемия (дефицит углеводов в пище).
• Кетонемия, кетоацидоз (усиление катаболизма липидов).
• Гиперкортицизм (за счет глюкокортикоидов).
• Повышение уровня глюкагона и соматостатина.
• Гиперкалиемия.
• Иммунодефицит (Т-клеточный).
• Задержка физического и умственного развития.
Избыточное поступление белка.
Причины:
• Переедание.
• Несбалансированная диета (пища с высоким содержанием белка).
• Увеличенный синтез белков (СД, гиперпродукция СТГ).
Проявления:
• Положительный азотистый баланс.
• Гиперпротеинемия.
• Диспепсические расстройства (запоры, поносы).
• Дисбактериоз (развитие кишечной аутоинфекции и аутоинтоксикации).
Нарушение аминокислотного состава потребляемого белка.
• Дефицит незаменимых аминокислот – валин, изолейцин, лейцин, метионин, лизин, треонин, триптофан, фенилаланин, которые не могут быть
синтезированы в организме человека, требуется поступление их с пищей.
• Избыток отдельных аминокислот.
Расстройства пищеварения в желудке и кишечнике.
Нарушение расщепления белка в желудке.
Причины:
244
• Гипоацидные состояния (атрофия слизистой оболочки).
• Снижение содержания или активности пепсина.
• Резекция желудка.
Нарушение пищеварения в кишечнике.
Причины:
• Наследственный дефект ферментов, расщепляющих белок.
• Заболевания ЖКТ (язвенный колит, хронический энтерит, дивертикулы
кишечника, панкреатит).
Проявляются нарушением расщеплением белка в кишечнике, нарушение всасывания (синдром мальабсорбции).
Нарушение трансмембранного переноса аминокислот.
Причинами являются мембранопатии различного генеза (наследственные и
приобретенные).
Нарушения транспорта аминокислот могут быть: из кишечника в кровь, из крови в гепатоциты, из первичной мочи в кровь, из крови в клетки органов и тканей.
Нарушение содержания белков в плазме крови.
Нарушения содержания белка в плазме крови называются диспротеинемиями:
• Гиперпротеинемии (гиперсинтетические и гемоконцентрационные).
• Гипопротеинемии (гипосинтетические и гемодилюционные).
• Парапротеинемии.
Гиперпротеинемии – увеличение общего содержания белков в плазме крови.
• Гиперсинтетические (истинные) – гиперпродукция белка (иммуноглобулинов), парапротеинов (В-лимфоцитарные лейкозы, плазмоцитома, миеломная болезнь).
• Гемоконцентрационные (ложные) – гемоконцентрация без усиления синтеза протеинов (ожоговая болезнь, диарея, рвота, усиленное потоотделение).
Гипопротеинемии – уменьшение общей концентрации белка в плазме крови.
• Гипосинтетические (истинные) могут быть первичные или наследственные (болезнь Брутона) и вторичные или приобретенные (печеночная недостаточность, белковое голодание, почечная недостаточность, гипоаминоацидемии, ожоговая болезнь).
• Гемодилюционные (ложные) – обусловлены гиперволемией (гиперальдостеронизм, почечная недостаточность).
Паропротеинемии – накопление аномальных белков. Может наблюдаться
при миеломной болезни (опухолевые плазмоциты продуцируют аномальные
легкие и тяжелые цепи Ig), при лимфомах (синтезируются аномальные IgМ).
245
Расстройства конечных этапов катаболизма белков.
Характеризуются нарушением образования и дальнейшим изменением
мочевины, мочевой кислоты, аммиака, креатинина, а также их выведением из
организма.
Аммиак – обладает цитотоксическими свойствами. В норме инактивируется внутриклеточно, вовлекаясь в реакции аминирования кетокислот.
Мочевина – не обладает токсическими свойствами, образуется в печени
(орнитиновый цикл), выводится почками и потовыми железами. При почечной
недостаточности из мочевины образуется аммиак, что приводит к уремии.
Диспротеинозы – патологические состояния, характеризующиеся изменением физико-химических свойств белков и расстройством их ферментативной, структурной, рецепторной и информационной функции.
Амилоидоз – избыточное накопление избытка аномальных комплексов
белков и полисахаридов в межклеточном пространстве, вокруг сосудов и в их
стенках, приводящее к нарушениям функций органов и тканей.
Гиалиноз – накопление в соединительной ткани органов и тканей неамилоидного белка.
Наследственные нарушения обмена аминокислот.
К наиболее часто встречающимся нарушениям обмена аминокислот относятся: фенилкетонурия (фенилпировиноградная олигофрения), алкаптонурия,
альбинизм.
Фенилкеионурия – наследственная аминоацидопатия, сопровождающаяся
дефицитом фермента фенилаланин-4-гидроксилазы, что приводит к избыточному на5коплению в крови фенилаланина. Недостаток образования тирозина из
фенилаланина приводит к дефициту: меланина (светлые кожа, волосы, глаза),
катехоламинов (гипотензия), серотонина (развитие судорог и тремора). Избыток фенилаланина метаболизируется с образованием токисческих продуктов
(фенилпировиноградная и фенилмолочная кислоты, фенилацетилглутамин),
приводящих к развитию слабоумия.
Алкаптонурия – аутосомно-рецессивная болезнь, дефицит оксидазы гомогентизиновой кислоты. Накопление гомогентизиновой кислоты приводит к
ее превращению полифенилоксидазой в хиноновые полифенолы, которые выводятся почками (на воздухе такая моча темнеет). Отмечается пигментация
склер и ушных раковин, остеохондропатия.
Альбинизм – нарушение образования пигмента меланина из тирозина.
Наиболее часто встречается аутосомно-рецессивная форма альбинизма. Носитель полного альбинизма (альбинос) имеет белую кожу и волосы, розовокрасные глаза. У них возникают фотодерматит (при действии малых доз солнечного света), дневная слепота и фотофобия (при депигментации сетчатки),
первичный гипотиреоз (при дефиците тирозина).
246
Нарушения обмена нуклеиновых кислот.
Нарушения обмена нуклеиновых кислот характеризуются расстройствами
синтеза и разрушения пиримидиновых и пуриновых оснований.
К пиримидинам относятся урацил, тимин, цитозин, метил- и оксиметилцитозин – отвечают за обмен и функционирование ДНК, РНК, нуклеотидтрифосфатов и нуклеотидпирофосфатов.
К пуринам относятся аденин, гуанин, метилгуанин, метиладенин – основные компоненты нуклеиновых кислот, входят в состав макроэргических соединений, поставщики мочевой кислоты (конечный продукт обмена пуринов).
Мочевая кислота образуется в гепатоцитах и энтероцитах с участием ксаниноксидазы, разрушается в кишечнике с образованием глиоксалевой кислоты
и аммиака.
К нарушениям обмена пиримидинов относятся оротацидурия, гемолитическая анемия и аминоизобутиратурия.
К нарушениям обмена пуринов относятся подагра, гиперурикемия, синдром Леша-Найена, гипоурикемия.
Гиперурикемия – состояние, проявляющееся повышенной концентрацией
мочевой кислоты в крови и как следствие в моче.
К наиболее распространенным заьолеваниям относится подагра.
Подагра – нарушение пуринового обмена, характеризующееся хроническим повышением мочевой кислоты в крови, отложением избытка ее солей в
органах, тканях, суставах, уратной нефропатией, нефро- и уролитиазом.
Причины могу быть первичными (генетический дефект ферментов обмена мочевой кислоты) и вторичными (ожирение, сахарный диабет, гиполипопротеинемии, артериальная гипертензия).
Факторы риска:
• Повышенное образование в организме мочевой кислоты (недостаточность гипоксантин гуанин фосфорибозилтрансферазы, поступление пуринов с пищей – мясо, молоко, икра, рыба, кофе, какао, шоколад).
• Усиление распада пуриновых нуклеотидов с образованием уратов (применение цитостатиков, распад АТФ при физической нагрузке).
• Уменьшение выведения молочной кислоты (почечная недостаточность)
Патогенез.
Избыток уратов в плазме крови и межклеточной жидкости способствует активации системы комплемента, кининов, гемостаза, являющиеся хемотаксинами. Это приводит к миграции лейкоцитов в места максимальной концентрации
мочевой кислоты, образующей кристаллы в коже, почках, хрящах, околосуставных тканях. Лейкоциты поглощают кристаллы мочевой кислоты и приводят к выделению и образованию медиаторов воспаления, способствующих хронизации процесса. Повреждение клеток уратами также сопровождается образованием аутоантигенов (активация иммунной системы, аллергические реакции).
В местах отложения уратов клеток (полиморфноядерные нейтрофилы, мононуклеарные фагоциты, лимфоциты, фибробласты), что постепенно приводит к
образованию подагрических гранулем и подагрических тофусов. Тофусы фор-
247
мируются вокруг суставов (ступней, голеностопных, локтевых, лучезапястных),
в почках, коже, хрящах ушных раковин.
Проявления: воспаление различных суставов, сильные боли в местах накопления уратов, лихорадка, повторное возникновение тофусов, почечная недостаточность.
Гипоурикемия – состояние, при котором снижается концентрация мочевой
кислоты в крови ниже нормы.
Связана с недостаточностью ксантиноксидазы и/или сульфитоксидазы и
проявляется образованием кристаллов в тканях почек, вокруг суставов, в мышцах; мышечными судорогами (поражение нервной системы).
Нарушения жирового обмена.
Расстройства в обмене жиров могут возникать при:
• нарушении процессов переваривания и всасывания жиров;
• нарушении транспорта жира и перехода его в ткани;
• нарушении окисления жиров в тканях;
• нарушении промежуточного жирового обмена;
• нарушении обмена жира в жировой ткани (избыточное или недостаточное его образование и отложение);
• нарушении нервной и гуморальной регуляции жирового обмена.
Нарушения переваривания и всасывания жиров.
• Нарушение эмульгирования жиров (недостаточное поступление в кишечник желчи – механическая желтуха, преждевременное разрушение желчных кислот бактериями при нарушении моторной функции кишечника).
• Нарушение гидролитического расщепления жиров (недостаточное поступление панкреатической липазы в двенадцатиперстную кишку, нарушение активации этого фермента при недостатке желчи).
• Нарушение образования липидных мицелл в полости тонкого кишечника
(недостаточное поступление желчи, связывание желчных кислот с лекарственными препаратами, образование кальциевых солей жирных кислот
при избытке поступления кальция).
• Нарушение всасывания мицелл (быстрая эвакуация содержимого тонкого
кишечника – поносы; повреждение слизистой оболочки тонкого кишечника – энтериты, радиационные поражения).
• Нарушение ресинтеза триглицеридов и формирования хиломикронов в
эпителии кишечника (уменьшение количества, подавление активности
ферментов, дефицит АТФ).
Нарушения транспорта жиров в организме.
• Гиперлипопротеинемия – увеличение содержания липопротеидов в плазме крови.
248
• Гиполипопротеинемия – уменьшение содержания липопротеидов в плазме крови.
• Дислипопротеинемия – нарушение соотношения между отдельными липопротеидами плазмы крови.
• Гиперлипацидемия – увеличение содержания свободных жирных кислот
в плазме крови.
Липопротеиды плазмы крови.
• Хиломикроны (ХМ).
• Липопротеиды очень низкой плотности (ЛПОНП).
• Липопротеиды промежуточной плотности (ЛППП).
• Липопротеиды низкой плотности (ЛПНП).
• Липопротеиды высокой плотности (ЛПВП).
Классификация гиперлипопротеинемий.
По происхождению: первичные (наследственные) и вторичные (приобретенные).
По увеличению содержания определенного класса липопротеидов.
По механизмам развития: продукционные (вследствие увеличения образования липопротеидов) и ретенционные (вследствие нарушения утилизации
липопротеидов).
Первичные гиперлипопротеинемии возникают вследствие:
• Генетически обусловленные нарушения структуры белковой части липопротеидов, что приводит к нарушению взаимодействия с рецепторами.
• Наследственные дефекты ферментов (липопротеинлипаза, печеночная
липаза).
• Нарушение структуры клеточных рецепторов к липопротеидам.
Вторичные гиперлипопротеинемии возникают вследствие:
• Эндокринные болезни (сахарный диабет, гипотиреоз).
• Метаболические расстройства (ожирение, подагра).
• Болезни почек (нефротический синдром).
• Болезни печени (обтурационная желтуха).
• Интоксикации (алкоголизм).
Атеросклероз.
Наследственные формы ускоренного атеросклероза обусловлены
дефектами генов алипопротеидов и их рецепторов, ферментов липопротеидного
и холестеринового метаболизма, в печени изменяется скорость выработки и катаболизма циркулирующих в крови
липопротеидов. В разных семьях отмечены разные молекулярные
дефекты, которые ведут к дисбалансу холестерина либо в клетках,
либо циркулирующих в крови ЛП.
249
Атерогенными свойствами обладают холестерин, триглицериды,
насыщенные жирные кислоты. Повышение концентрации атерогенных липопротеидов в крови может вызываться снижением скорости их выведения из
крови в печень; повышением скорости их синтеза; нарушением метаболизма в
плазме, включая образование аномальных модифицированных липопротеидов.
Антиатерогенными свойствами обладают фосфолипиды и полиненасыщенные жирные кислоты. Они ограничивают всасывание пищевого холестерина в тонком кишечнике, стимулируют синтез в печени желчных кислот, тормозят синтез и секрецию различных липопротеидов.
Ожирение.
Нормальное содержание жировой ткани у мужчин 15-20 % массы тела, у
женщин – 20-30 %.
Ожирение – избыточное накопление жира в виде триглицеридов.
Ожирение не является опасным для здоровья, но увеличивает риск возникновения ишемической болезни сердца, атеросклероза, гипертонической болезни, сахарного диабета.
Классификация.
• По степени увеличения массы тела.
Для оценки используют:
Индекс Брока – рост (в см) минус 100.
Индекс физического развития по Пинье.
Индекс массы тела (ИМТ): масса тела (кг) / рост (м2). Норма – 18,5-24,9;
1степень – 25-29,9; 2 степень – 30-39,9; 3 степень – >40.
• По преимущественной локализации жировой ткани – общее и местное
(мужской и женский типы).
Мужской тип (андроидный) – избыточное накопление жира в области живота.
Женский тип (гиноидный) – избыточное накопление жира в области бедер и
ягодиц.
• По преимущественному увеличению количества или размеров адипоцитов.
Гипертрофическое – увеличение массы и размеров адипоцитов. Возникает
после 30-35 лет.
Гиперпластическое – увеличение количества адипоцитов.
Смешанное – гиперпластико-гипертрофическое.
• По происхождению.
Первичное (гипоталамическое) – расстройство системы регуляции обмена
жиров. Возникает при дефиците лептина или снижении его эффектов (повышение аппетита, усиление чувства голода).
250
Вторичное (симптоматическое) – снижение расхода жиров, активация липогенеза. Возникает при увеличении поступления пищи, низкой физической
активности.
Патогенез ожирения.
Нейрогенное ожирение.
Центральный (корковый, психогенный) механизм возникает в результате
расстройств психики, проявляющихся в непреодолимом желании к приему
пищи. Активируется серотонинергическая и опиоидергическая системы,
пища воспринимается как положительный стимул (удовольствие).
Гипоталамический (диэнцефальный, подкорковый) механизм возникает
при повреждении вентромедиального и паравентрикулярного ядер гипоталамуса (сотрясения мозга, энцефалиты, опухоли). Происходит повышение
синтеза и секреции неропептида Y (стимулирует чувство голода и повышает
аппетит).
Эндокринное ожирение.
Лептиновый механизм. Абсолютная или относительная недостаточность
лептина → нарастание чувства голода → повышение аппетита → избыточное потребление пищи.
Гипотиреоидный механизм. Недостаточность эффектов тиреоидных гормонов → снижение интенсивности липолиза → подавление метаболизма в
тканях → снижение энергозатрат организма.
Надпочечниковый механизм. Избыток эффектов глюкокортикоидов (болезнь или синдром Иценко-Кушинга) → активация глюкогенеза → гипергликемия → повышение транспорта и активация гликолиза в адипоцитах →
торможение липолиза.
Инсулиновый механизм. Прямая активация инсулином липогенеза в жировой ткани.
Истощение – патологическое снижение массы жировой ткани ниже нормы
(дефицит жировой ткани составляет 20-25%).
Кахексия – снижение массы жировой ткани с дефицитом более 50%.
Причины разделяют на экзогенные и эндогенные.
К экзогенным относятся:
• Вынужденное или осознанное голодание.
• Низкая калорийность пищи, которая не восполняет энергозатрат организма.
Эндогенные могут быть:
• Первичными (травмы мозга, ишемия гипоталамуса, сильный психогенный стресс).
• Вторичными (мальабсорбция, дефицит глюкокортикоидов, гипоинсулинизм, гиперпродукция соматостатина, гиперпродукция клетками опухоли
ФНОα, гиперпродукция глюкагона).
251
Патогенез экзогенного истощения и кахексии.
Значительный дефицит или отсутствие продуктов питания приводит к
нарушению обмена веществ (недостаточность биологического окисления, снижение анаболизма, усиление катаболизма) и истощению запасов жиров, полиорганной недостаточности, прогрессирующее уменьшение массы тела.
Среди первичных эндогенных форм истощения и кахексии выделяют гипоталамическую, кахектиновую и анорексическую формы.
При гипоталамической форме происходит снижение или прекращение
синтеза нейропептида Y в гипоталамусе, снижение или прекращение транс- и
парагипофизарной активации эндокринных желез, снижение эффективности
метаболических реакций, торможение накопления жира в адипоцитах, прогрессирующее уменьшение массы тела.
При кахектиновой форме наблюдается увеличенная продукция адипоцитами и макрофагами кахектина (ФНОα), подавление синтеза нейропептида Y в
гипоталамусе, угнетение липогенеза, снижение активности липопротеинлипазы
эндотелия, торможение транспорта жирных кислот в адипоциты, активация катаболизма и снижение анаболизма, истощение запасов жира, прогрессирующее
уменьшение массы тела.
При анорксической форме наблюдаются частые, повторные эмоционально негативные стресс-реакции (критическое отношение к своему телу, воспринимаемому как имеющему избыточную массу), приводящие к избыточной продукции подавляющих аппетит веществ (серотонин, холецистокинин), повышению продукции кахектина, снижение синтеза нейропептида Y в гипоталамусе.
Эти все механизмы приводят к прогрессирующему уменьшению массы тела.
Вторичные эндогенные формы истощения и кахексии являются важными
симптомами других болезней.
Кетоз.
Расстройство интеграции липидного и углеводного метаболизма проявляется в виде синдрома кетоза (увеличение содержания в организме кетоновых
тел).
Причины кетоза:
• Инсулинзависимый СД.
• Голодание.
• Жировая диета, обедненная углеводами.
• Алкогольное отравление.
• Отравление ацетоном, аммиаком.
• Подагра и гиперурикемия.
• Стресс.
Накопление кетоновых тел связано с ускорением их образования или
нарушением их утилизации. Ускоренное образование кетоновых тел происходит при усилении липолиза и избыточном поступлении в печень свободных
252
жирных кислот, при ограниченной метаболической доступности углеводов и
кетокислот цикла Кребса.
Стеатоз печени – жировой гепатоз, при котором содержание триглицеридов в печени превышает 10 %, а жировые капли содержатся в более чем 50 %
гепатоцитов.
Основными причинами развития стеатоза являются:
• Токсические вещества, воздействующие на печень (алкоголь, мышьяк,
хлороформ).
• Лекарственные препараты (кортикостероиды, цитостатики, антибиотики тетрациклинового ряда).
• Дисбаланс пищевых факторов (недостаточное поступление с пищей
холина, метионина, избыток поступления холестерина).
• Эндокринно-метаболические нарушения (сахарный диабет, синдром
Иценко-Кушинга).
• Гипоксия (сердечно-сосудистая недостаточность, легочные заболевания).
В основе патогенеза лежат:
• Увеличенное поступление свободных жирных кислот в печень.
• Активация синтеза, ускорение этерификации жирных кислот и замедление окисления жирных кислот.
• Снижение синтеза апопротеинов в печени и нарушение нормальной
сборки ЛПОНП.
• Затруднение выделения ЛПОНП.
ПАТОФИЗИОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
Содержание воды в организме месячного эмбриона - 97,5%, у новорожденного — 70-80%.Содержание воды в организме человека - 60%.
У взрослого внутриклеточная жидкость составляет - 40% веса тела; внеклеточная жидкость - 20% веса тела; в том числе жидкая часть плазмы крови 5% веса тела.
Взрослому человеку необходимо 35г воды на кг массы в сутки, грудному
ребенку – 140г.
Водный баланс в миллилитрах:
Потребление воды: с твердой пищей - 1000,0
с жидкой пищей - 1200,0
Вода образуется при окислении - 300,0
Общее потребление - 2500,0
Выделение воды: с мочой - 1400,0
253
с потом - 600,0
с выдыхаемым воздухом - 300,0
с фекальными массами - 200,0
Общее выделение - 2500,0.
Потеря 60% внеклеточной жидкости несовместима с жизнью. Потеря
жидкости, составляющей 10% веса тела, приводит к снижению ОЦК на 25%.
Вода — основной компонент, обеспечивающий постоянство внутренней
среды организма. В организме вода выполняет множество функций: является
универсальным растворителем, обеспечивает потребности температурной
адаптации за счет теплоемкости, электропроводимости, текучести (способность транспорта веществ, выделение веществ).
У взрослого человека около 2/3 воды находится во внутриклеточном секторе и 1/3 - во внеклеточном .
Внеклеточный сектор включает в себя внутрисосудистую (плазма крови),
интерстициалъную ("организованная" жидкость соединительной ткани, окружающая клетки) и трансцеллюлярную (жидкость, находящаяся в серозных и
других полостях тела - секреты желез пищеварительного тракта, спинномозговая жидкость и др.) жидкости. Интерстициальная и межклеточная жидкости составляют 3/4 всей внеклеточной воды, плазма крови — 1/4.
Клетка и внеклеточное пространство составляют единую гидродинамическую систему, разделены полупроницаемыми мембранами. Во внутриклеточной жидкости протекают все виды обмена веществ. Интерстициальная жидкость и плазма крови являются посредниками между внешней средой и клетками.
Ионный состав разных водных секретов представлен катионами и анионами, обеспечивающими осмотическое давление жидкостей тела. В каждом из
секторов сумма концентраций анионов и катионов делает жидкость водного
пространства электронейтральной (закон электронейтральности).
Осмотическое давление жидкости — способность осмотически активных
веществ связывать воду.
Регуляция водно-электролитного обмена.
У здорового человека содержание жидкости в организме практически
стабильно несмотря на колебания приема солей и воды.
Потеря воды и солей организмом приблизительно равна поступлению воды и солей. Потеря воды составляет: около 1500 мл с мочой, 500 мл с потом,
500 мл с выдыхаемым воздухом. Поступление воды составляет: 1200 мл в составе жидкостей, 1000 мл в составе твердой пищи, 300 мл – эндогенная вода,
образующаяся в результате диссимиляции.
Обмен воды и солей между плазмой крови и внеклеточной средой
осуществляется в капиллярах.
Обмен воды также связан с осмосом – движение воды через полупроницаемую мембрану из области с более низкой концентрацией растворенного вещества в область с более высокой его концентрацией. Осмотическое давление –
давление, которое надо приложить, чтобы прекратить осмотический ток воды.
254
Онкотическое давление – давление, которое создается нефильтруемыми через
капиллярную стенку белками. Вода передвигается туда, где больше концентрация осмотически активных веществ (белки, глюкоза, катион натрия, мочевина).
Фильтрация осуществляется благодаря разнице гидростатического и онкотического давления в артериальном конце капилляра. В венозном конце капилляра, где гидростатическое давление низкое, а давление белков остается
прежним, жидкость будет перемещаться в сосуд и часть покинет интерстициальное пространство через лимфатические сосуды. Существенное влияние на
обмен воды между секторами оказывает проницаемость эндотелия фильтрующих капилляров.
Поддержание объема внеклеточной воды контролируется сложной нейрогуморальной системой, состоящей из рецепторного звена, гипоталамуса, нейрогипофиза, надпочечников и эффекторных органов.
Мочевыделительная система.
Почки являются основным органом, регулирующим количество воды и
электролитов в организме. С мочой выделяются остаточные продукты метаболизма в виде солей и других растворенных веществ. Через сосуды почек проходит до 200 л воды в сутки. Здоровая почка может быстро изменить концентрацию мочи за счет реабсорбции воды.
Регуляцию реабсорбции воды осуществляет антидиуретический гормон,
стимулом для увеличения продукции которого и, следовательно усиления реабсорбции воды, является повышение осмотиского давления крови и межклеточной жидкости. Раздражение осморецепторов, расположенных в стенке правого
предсердия в различных органах и тканях, жидкостью с высоким содержанием
электролитов стимулирует выделение АДГ. Механизм действия АДГ: активизирует гиалуронидазу — фермент межклеточного вещества дистальных почечных канальцев, благодаря чему они становятся более проницаемыми для воды,
что приводит к увеличению реабсорбции жидкости.
Почки регулируют содержание электролитов в организме. В проксимальных отделах канальцев происходит реабсорбция натрия с заменой на водородные ионы. В дистальных отделах канальцев реабсорбция натрия регулируется
гормоном коры надпочечников — альдостероном, под влиянием которого происходит реабсорбция натрия в обмен на калий и аммоний.
Na-уретический фактор синтезируют кардиомиоциты правого предсердия, желудочков сердца, а также некоторые нейроны ЦНС. Действует на клетки
почечных телец, собирательных трубочек почки, клубочковой зоны коры
надпочечников, ГМК сосудов. Контролирует объём внеклеточной жидкости и
гомеостаз электролитов путем угнетения синтеза и секреции альдостерона,
ренина, вазопрессина, оказывает сосудорасширяющее воздействие, усиливает
экскрецию натрия почками. Синтез и содержание его в крови увеличиваются
при сердечной недостаточности.
Система внешнего дыхания.
255
Выделение воды и солей осуществляют также с помощью легких. В
нормальных условиях основного обмена с дыхательной поверхности легких и
дыхательных путей испаряется 400-600 мл воды в сутки. В условиях низкой
влажности атмосферного воздуха при учащении дыхания количество испаряемой воды возрастает. При повышении температуры тела на 10С выделение воды
через легкие возрастает на 500 мл. Теряемая жидкость практически свободна от
электролитов.
Желудочно-кишечный тракт.
В здоровом кишечнике реабсорбируются почти вся вода и электролиты С
калом потеря воды не превышает 200 мл в сутки. Большие потери воды и
электролитов бывают при патологических процессах в желудочно-кишечном
тракте, сопровождающихся поносом, рвотой.
Кожа.
Потеря воды может происходить с поверхности кожи за счет испарения и
потоотделения. В нормальных условиях с потом выделяется 20 мосм/л натрия
и несколько меньше хлора. При повышении температуры тела на 10С потоотделение увеличивается на 500 мл в сутки. При этом возрастает и потеря электролитов.
Классификация нарушений водно-электролитного обмена.
Гипергидратация – нарушение водного баланса, проявляющееся в виде
увеличения содержания жидкости в организме (задержка воды).
Гипогидратация – нарушение водного баланса, проявляющееся в виде
уменьшения содержания жидкости в организме (обезвоживание).
В зависимости от величины осмотического давления во внеклеточной
жидкости гипер- и гипогидратации могут быть:
• Изоосмотическими (осмотическое давление внеклеточной жидкости
остается нормальным).
• Гиперосмотическими (внеклеточная жидкость гиперосмолярна).
• Гипоосмотическими (внеклеточная жидкость гипоосмолярна).
Осмотическое давление внеклеточной жидкости определяет распределение воды между секторами: при снижении осмотического давления внеклеточной жидкости вода перемещается во внутрь клетки и объем клеток увеличивается (развивается вторичная гипергидратация клеток), при повышении осмотического давления внеклеточной жидкости вода перемещается из
клетки во внеклеточное пространство и объем клеток уменьшается (развивается гипогидратация клеток).
Изотоническая гипергидратация.
Причины возникновения: сердечная недостаточность, заболевания почек
(гломерулонефрит, нефротический синдром), печеночная недостаточность, токсикозы беременности, аллергические реакции.
256
Патофизиологические изменения связаны с увеличением объема изотонической жидкости во внеклеточном пространстве, при этом внутриклеточный
сектор остается нормальным.
Проявлением изоосмотической гипергидротации являются отеки. Объем
внеклеточного пространства может увеличиваться на несколько литров.
•
•
•
•
Механизмы развития отеков.
Осмотический. Проявляется накоплением в организме осмотически активных веществ (накопление ионов натрия вследствие его усиленной реабсорбции в почечных канальцах). Этот механизм является компенсаторным при гиповолемии и снижении артериального давления. Отек возникает, если этот синдром развивается на фоне нормального или увеличенного ОЦК (снижение венозного выброса при болезнях сердца, нарушение клубочкового кровотока при болезнях почек, при портальной печеночной гипертензии). При сокращении клубочкового кровотока происходит активация ренин-ангиотензин-альдостероновой системы. В кровь из
юкстагломерулярных клеток поступает ренин. В результате ограниченного протеолиза ангиотензиноген превращается в ангиотензин I и в легких в
ангиотензин II, который активирует выработку альдостерона клубочковой зоной коры надпочечников. Альдостерон усиливает реабсорбцию
натрия в дистальных отделах почечных канальцев, что приводит к гиперосмии внутренней среды. Это приводит к выделению вазопрессина, который увеличивает реабсорбцию воды адекватно натрию. Избыточная
изоосмотическая жидкость накапливается в межклеточных пространствах
и формируются отеки.
Онкотический. Проявляется снижением онкотического давления крови
вследствие гипопротеинемии. Причинами могут быть: нефротический
синдром (потеря белка с мочой), обширные ожоги (плазморея), тяжелые
поносы (избыточное выведение белка), белковое голодание (недостаток
поступления белка). При этом под действием гидростатического давления
жидкость покидает сосуды в артериальном конце капилляра, но в венозном конце не может вернуться в сосуд из-за низкого онкотического давления. При этом развиваются системные отеки (анасарка), накопление
жидкости в серозных полостях (брюшной, плевральной, перикардиальной).
Блокада лимфооттока. Повышение давления в крупных венозных сосудах сопровождается рефлекторным спазмом соответствующих лимфатических сосудов – давление в них повышается и отток лимфы блокируется
(рефлекс Русняка-Петровского). Этот механизм присутствует при наследственной недостаточности лимфатических капилляров – элефантиазис
(слоновость).
Гидростатический (застойный). Проявляется повышением гидростатического давления в капиллярах. Этот механизм играет ведущую роль при
застое малого круга (стеноз левого атриовентрикулярного отверстия, ле-
257
вожелудочковая недостаточность), портальной гипертензии (цирроз печени), правожелудочковой недостаточности (повышение давления в капиллярах печени), переливании жидкостей через крупные вены (подключичная, яремная) под большим давлением. Повышение гидростатического давления способствует фильтрации и препятствует реабсорбции, что
приводит к накоплению жидкости в межклеточном пространстве.
• Эндотелиальный. Проявляется накоплением БАВ, увеличивающих сосудистую проницаемость. Причины: воспаление, ишемия и гипоксия, аллергические реакции, механическая травма, экзотоксины (укусы змей,
насекомых, действие боевых отравляющих веществ). Механизм: БАВ (гистамин, брадикинин, простогландины, тромбоксаны) воздействуют на эндотелиоциты и приводят к их сокращению. Это увеличивает межэндетелиальные щели и белки крови могут проникать в межклеточную жидкость, что приводит к увеличению онкотического давления межклеточной жидкости, которое удерживает воду и препятствует ее возвращению
в сосуд.
Микседематозный отек. Микседема – особый вид отека, характерный для гипотиреоза.
Механизмы возникновения:
• Увеличение гидрофильности соединительной ткани из-за накопление в
ней глюкуроновой и хондроитинсерной кислот, накопление в клетках и
интерстициальной жидкости натрия в связи со снижением выработки
предсердного натрийуретического фактора.
• Задержка жидкости в организме в связи с повышением эффектов АДГ в
условиях гипофункции щитовидной железы
• Связывание жидкости тканевым коллоидом с образованием муцина –
слизеподобного соединения, что приводит к утолщению кожи и подкожной клетчатки (кожа не собирается в складки, сухая, холодная, бледная с
желтоватым оттенком).
Развивается одутловатость (отечность) лица, понижение мимики, отек периорбитальной клетчатки.
Гиперосмотическая гипергидратация.
Возникает при накоплении во внеклеточном пространстве жидкости с повышенным осмотическим давлением.
Причины:
• Введение больших количеств гиперосмотической жидкости
• Питье морской воды
При повышении осмотического давления внеклеточной жидкости нарушается равновесие между клеточным и внеклеточным сектором и вода перемещается во внеклеточный сектор, что приводит к обезвоживанию клеток, повышает
их возбудимость и их гибели.
258
Возникает сильная жажда, симптомы энцефалопатии (возбуждение, головные боли, кома, потеря сознания).
Гипоосмотическая гипергидратация.
Возникает при накоплении во внеклеточном пространстве жидкости с пониженным осмотическим давлением.
Причины:
• Анурическая стадия острой почечной недостаточности
• Введение большого количества гипоосмотических растворов или легкоусвояемых гиперосмотических растворов (5% р-р глюкозы)
• Длительная бессолевая диета
• Усиленное выведении натрия при применении мочегонных препаратов
• Избыток вазопрессина
При понижении осмотического давления внеклеточной жидкости вода
переходит в клетки, где было большее осмотическое давление и развивается
внутриклеточная гипергидратация, набухание клеток, вторичная гипоосмия –
«водное отравление» - нарушение функций клеток и их гибель.
Это проявляется набуханием клеток головного мозга – головные боли,
раздражительность, сонливость, выраженные неврологические расстройства,
сопровождающиеся рвотой, судорогами, нарушением сознания, комой. Могут
проявляться гемолиз и отек легких.
Изоосмотическая гипогидратация.
Патофизиологические изменения связаны с одновременной потерей воды
и электролитов, т.е. изотонической жидкости, по ионному составу близкой к
плазме крови. При этом виде дегидратации в основном уменьшается объем
внеклеточной воды.
Наиболее частыми причинами являются:
• Кровопотеря
• Болезни желудочно-кишечного тракта, длительно протекающие патологические процессы, сопровождающиеся поносом или рвотой, свищи желудка, кишечника, поджелудочной железы - организм теряет большое количество пищеварительных соков
• Депонирование жидкости в кишечнике при непроходимости и перитоните
• Повреждения кожи: при ожогах большой поверхности тела, экссудативном дерматите, отслойке эпидермиса
• Полиурическая стадия острой почечной недостаточности
Для компенсации потери внеклеточной жидкости организм не может использовать внутриклеточную жидкость, т.к. осмотическое давление в клетках и
внеклеточной жидкости одинаковое. Следовательно восполнение ОЦК за счет
клеточной жидкости невозможно и оно осуществляется только за счет внеклеточной жидкости.
259
Потеря больших количеств жидкости, наблюдающееся при острых заболеваниях может привести к выраженным расстройствам кровообращения вследствие быстрого снижения объема циркулирующей крови - снижение венозного
давления, артериального давления, минутного объема сердца. В тяжелых случаях развивается гиповолемический шок.
В зависимости от расстройств гемодинамики выделяют три степени тяжести
изотонического обезвоживания:
- при дефиците жидкости 2л — артериальное давление нормальное, чувство
усталости, тахикардия;
- при дефиците жидкости 5-6 л — низкое артериальное давление, нарушение
сознания, шок;
- при дефиците жидкости свыше 6л — низкое артериальное давление, судороги, смерть.
Гиперосмотическая гипогидратация.
Патофизиологические изменения связаны с недостатком воды и избытком
солей во всех жидкостях организма (осмотическое давление во внеклеточной
жидкости превышает нормальные показатели).
Может возникать при потере воды (через легкие с выдыхаемым воздухом), гипоосмотической жидкости (усиленное потоотделение, гипоосмотическая моча) или недостатке поступления воды в организм:
• При водном голодании – у очень маленьких детей при плохом медицинском уходе; у беспомощных больных, в бессознательном или коматозном
состоянии; в послеоперационном периоде, если больной не может сам себя обслуживать; при нарушениях проходимости пищеварительного тракта и нарушениях акта глотания; в экстремальных условиях внешней среды (в пустынях)
• При полиурии – вследствие осмотического диуреза (при сахарном диабете), вследствие дефицита вазопрессина (при несахарном диабете)
• При гипервентиляции – высокая температура тела (у маленьких детей)
• При необоснованном введении концентрированных растворов электролитов
Повышение осмотического давления в жидкой части крови и внеклеточной жидкости приводит к перемещению воды из внутриклеточного сектора в
межклеточные пространства и в сосудистое русло. В компенсации этого вида
обезвоживания участвует вся жидкость в организме, что позволяет длительно
сохранять ОЦК на нормальном уровне и не допускать развития гиповолемического шока.
Прогрессирующее обезвоживание приводит к обезвоживанию клеток, их
«сморщиванию», функциональным расстройствам и гибели клеток. Наиболее
чувствительны к этому состоянию нервные клетки. Характерны психоневрологические расстройства, больные погибают от нарушений функций головного
мозга – энцефалопатии, комы.
260
Гипоосмотическая гипогидратация.
При этом состоянии в организме уменьшается содержание воды и понижается осмотическое давление во внеклеточной жидкости, возникает когда потери натрия превосходят потери воды. Патофизиологические изменения связаны с дефицитом натрия в плазме крови. Потери натрия могут происходить через почки, пищеварительный тракт кожу.
Причины:
• Недостаточность коры надпочечников – потеря натрия при его недостаточной реабсорбции в почках в связи с гипоальдостеронизмом
• Понижение чувствительности рецепторов почечных канальцев к альдостерону
• Чрезмерное употребление слабительных препаратов
• При обильном потоотделении и интенсивном утолении жажды жидкостью с малым содержанием натрия
• При очень тяжелых поносах, когда токсины блокируют натриевый насос
кишечного эпителия
• При лечении изоосмотической гипогидратации введением гипоосмотической жидкости, например, питьем воды
Характерно быстрое развитие острой сосудистой недостаточности, т.к. при
дефиците натрия снижается тонус резистивных сосудов.
Компенсация ОЦК невозможна, т.к. высокое осмотическое давление внутри
клетки способствует перемещению воды в клетки, вызывая их обводнение,
преимущественно в головном мозге – водное отравление.
Такое перераспределение воды ведет к расстройствам кровообращения —
артериальной гипотензии, тахикардии, снижению тургора тканей, сухости слизистых оболочек. В связи с гипергидратацией клеток появляются неврологические расстройства — головная боль, апатия. В тяжелых случаях наблюдается
анурия, приводящая к вторичной почечной недостаточности. Дефициту натрия
часто сопутствуют гипокалиемия и резкое нарушение кислотно-щелочного
равновесия, усугубляющего психоневрологические расстройства.
ПАТОФИЗИОЛОГИЯ МАКРО- И МИКРОЭЛЕМЕНТОВ.
ПАТОФИЗИОЛОГИЯ ВИТАМИНОВ
Гомеостаз натрия и его нарушения.
Натрий — главный катион внеклеточной среды, где его концентрация
составляет 135-147 ммоль/л (во внутриклеточной жидкости около 10 ммоль/л).
Нарушение обмена натрия тесно связано с нарушением водного
равновесия.
Гипонатриемия может развиваться при избыточном поступлении воды
в организм или потере богатых натрием жидкостей – с мочой, потом, кишечным соком (рвота, понос, при аддисоновой болезни, при длительном применении сульфаниламидных препаратов, салуретиков, усиливающих выведение
натрия почками). При значительной потере натрия наблюдается выход из кле-
261
ток ионов калия, что нарушает деятельность сердца, скелетной и гладкой мускулатуры. Развивается мышечная слабость, потеря аппетита и повышенная
чувствительность к водной нагрузке.
Гипернатриемия может возникать при потери воды (осмотический диурез, применение диуретиков, через ЖКТ – рвота, понос; через кожу – ожоги)
или увеличении поступления (уменьшении выведения) натрия – в случае избыточного потребления соли, введения препаратов натрия с лечебной целью (ацидоз, искусственное питание), при нарушении выведения натрия почками (гломерулонефрит, длительное применение глюкокортикоидов), гиперпродукции
альдостерона, усиливающего реабсорбцию натрия. Избыток солей натрия в организме способствует развитию воспалительных процессов, задержке воды,
развитию артериальной гипертензии.
Гомеостаз калия и его нарушения.
Калий – основной элемент, определяющий нервно-мышечную возбудимость, содержание его во внеклеточной среде составляет 4-6 ммоль/л,
во внутриклеточной жидкости — 160 ммоль/л. Нарушение баланса калия тесно
связано с нарушением обмена натрия. Избыток калия усиливает выведение
натрия и воды из организма, а его недостаток вызывает нарушения, сходные с
эффектом избытка натрия.
Гипокалиемия развивается при недостаточном поступлении калия с
пищей (овощи и молочные продукты), при потере с рвотными массами или поносом, при лечебном применении кортикотропина и глюкокортикоидов, при
гиперальдостеронизме. Гипокалиемия компенсируется за счет перехода калия в
кровь из клеток. Длительная гипокалиемия приводит к снижению содержания
калия в клетках (изменяется потенциал действия возбудимых клеток), что вызывает нарушение нервно-мышечной возбудимости, мышечную слабость, депрессию, утомляемость, понижение моторики желудка и кишечника, снижение
сосудистого тонуса, тахикардию, сердечную недостаточность. Изменение ЭКГ
при гипокалиемии выражается в удлинении и снижении интервала SТ, зубца Т.
Гиперкалиемия может возникать при увеличения поступления в организм
калия (избыточном содержании калия в пище), при снижении выделения калия
с мочой или выхода калия из клеток (при ацидозе, инсулярной недостаточности). Избыток калия удлиняет период деполяризации мембран возбудимых клеток – утрачивается их возбудимость. Задержка калия в организме более опасна,
чем снижение концентрации калия. Проявления – гипотония, парестезии, парезы, тахикардия, аритмии, блокады, спазмы и колики в животе, диарея. Изменения ЭКГ – высокий и узкий зубец Т, увеличение интервала PQ, депрессия ST,
расширение комплекса QRS.
Патофизиология метаболизма железа.
Содержание железа в сыворотке крови у мужчин составляет 14-26 мкМ/л,
у женщин 11-21 мкМ/л. Важная роль железа в организме обусловлена тем, что
оно легко и обратимо окисляется и восстанавливается. Железо выполняет свои
функции в связанной с белками форме.
262
Железосодержащие белки:
• Гемопротеины (гемоглобин, миоглобин, цитохромы, цитохромоксидаза,
гомогентизиноксидаза, пероксидаза, миелопероксидаза, каталаза)
• Железофйлафопротеины – цитохром-с-редуктаза, сукцинатдегидрогеназа,
пролиноксидаза, НАДФ-дегидрогеназа, ацил-КоА-дегидрогеназа, ксантиноксидаза
• Белки, содержащие железо различных молекулярных конфигураций –
трансферрин, ферритин, гемосидерин, мобилферрин, лактоферрин
Железодефицит – состояние, когда содержание железа в организме меньше
нормы.
Причины железодефицита связаны с хроническим превышением расхода
(потерь) железа над его доставкой – отрицательный баланс железа:
• Неадекватное поступление железа с пищей
• Мальабсорбция железа (после резекции желудка, комплексный мальабсорбционный синдром)
• Хроническая кровопотеря – кровотечения из ЖКТ (язвенная болезнь,
опухоли, гастриты, диафрагмальная грыжа), кровохарканье, гельминтозы,
полименорея, меноррагия, метроррагия
• Расход железа на нужды эмбриона или плода при беременности, расход
железа при лактации
• Потеря железа в результате внутрисосудистого гемолиза
• Нарушение включения железа в синтез гема
Патогенез заключается в прогрессирующем использовании и истощении запасов железа в организме, нарушении функций железо-включающих ферментов, рецепторов и других белков.
Крайне выраженная стадия дефицита железа – это железодефицитная анемия.
Для железодефицита также характерно:
• Сидеропенические негематологические симптомы – изменения кожи,
ногтей, волос, слизистой оболочки рта (сухость и трещины кожи, плоские, вогнутые ломкие ногти, ангулярный стоматит), поражение слизистой
верхних дыхательных путей, пищевода, желудка – связаны с дефицитом
железо-зависимых рецепторов и ферментов, участвующих в образовании
поперечных сшивок в коллагеновых белках
• Мышечная слабость – дефицит фермента α-глицерофосфатоксидазы,
нарушение обмена миоглобина
• Поражение эпителиальной ткани – высокая чувствительность к кислороду при дефиците цитохромов
• Поражение нервной системы (парестезии, извращение вкуса) – уменьшение железосодержащих рецепторов
Избыток железа.
Гемохроматоз – патологически повышенная или неподавляемая абсорбция железа из ЖКТ, ускорение выведения железа из энтероцитов в кровь. Железо откладывается во внутренних органах, приводит к гибели паренхиматоз-
263
ных клеток и их замещению соединительной тканью, что приводит к полиорганной недостаточности.
Первичный гемохроматоз – наследственное заболевание с аутосомнорецессивным типом наследования.
Вторичный гемохроматоз – при первичных анемиях с неэффективным
эритропоэзом, при хроническом усиленном гемолизе (талассемии).
Гемосидероз – доброкачественная форма перегрузки железом, при котором оно накапливается в макрофагальных клетках.
Патофизиология обмена важнейших микроэлементов.
Цинк – незаменимый элемент в питании человека. Является компонентом активных центров РНК-полимераз, карбоангидразы, алкогольдегидрогеназы, различных ферментов, необходим для восстановления ретинола в сетчатке,
входит в структуру гормона вилочковой железы тимулина, является минеральным антиоксидантом.
Недостаток цинка (гипоцинкоз) может развиваться при однообразном
растительном питании, ему способствуют энтериты, прием алкоголя, увеличенные потери цинка при хроническом гемолизе. Проявляется задержкой психического развития, низкорослостью, гипогонадотропным гипогонадизмом, половым инфантилизмом, бесплодием, иммунодефицитом, замедляется заживление
ран, возникает раздражительность, депрессия.
Избыток цинка может вызывать острые и хронические отравления. Хроническое воздействие цинка на организм приводит к анемии, нарушению функции цитоскелета клеток крови, неврологические нарушения.
Медь – необходима для эритропоэза и гранулопоэза, участвует в стимуляции созревания ретикулоцитов и захвате железа для включения железа в гем,
регулирует медь-зависимые окислительные процессы в соединительной ткани и
коже, участвует в синтезе нейромедиаторов.
Недостаток меди возникает при парентеральном питании и у младенцев
на молочном вскармливании. Проявления – микроцитарная нормохромная анемия, остеопатия, тормозится рост, нарушаются процессы ороговения в коже.
Дегенерация эластина приводит к расслаивающим аневризмам аорты, вазопатиям.
Кобальт – является ключевым элементом кобамидного кофермента (витамин В12), необходим для превращения метионина и гомоцистеина, стимулирует эритропоэз, оказывает активирующее действие на фосфотазы костей.
При дефиците кобальта возникает В12-дефицитная мегалобластная гиперхромная анемия.
В высоких дозах вызывает отравление, возникает нарушение метаболизма
жирных кислот и пирувата, формируется миокардиодистрофия и сердечная недостаточность, нарушает захват йода в щитовидной железе и способствует зобу.
Марганец – входит в состав ферментов, активирует оксидоредуктазы,
гидролазы и лигазы, участвует в окислительном фосфорилировании, окислении
264
жирных кислот, метаболизме пуринов, холестерина и гликозаминогликанов,
антиоксидантной защите клеток, обладает антиатерогенным эффектом.
Дефицит возникает при нарушении его всасывания и характеризуется задержкой роста, гипогонадизмом, аномалиями скелета, нарушением метаболизма жиров, похудением, дерматитом, кетозом, гипохолестеринемией.
Избыток марганца токсичен – вызывает анемию (конкурирует с медью
при обеспечении кроветворения), вызывает формирование рахитоподобных изменений.
Молибден – компонент активного центра ксантиноксидазы, участвует в
пуриновом метаболизме, активирует некоторые оксидоредуктазы.
При дефиците чаще наблюдается рак пищевода.
Избыток характеризуется высокой встречаемостью подагры и гиперурикемии.
Хром.
При дефиците хрома нарушается рецепция инсулина и снижается толерантность к глюкозе.
Избыток приводит к поражению почек, контактному дерматиту.
Ванадий – участвует в липидном обмене, накапливается в печени и жировой ткани, угнетает синтез холестерина, усиливает катаболизм липидов, оказывает антиатеросклеротическое действие.
При дефиците часто возникает атеросклероз.
Избыток наблюдается при маниакально-депрессивном психозе.
Никель – катализирует многие биохимические процессы, обеспечивает
гемопоэз, участвует в стабилизации структуры РНК.
Дефицит никеля не описан.
Избыток может вызывать контактный дерматит, некроз паренхимы печени, пневмонию в зависимости от пути проникновения в организм.
Стронций – участвует в процессах оссификации.
Избыток вызывает витамин-D-резистентный стронциевый рахит.
Йод – входит в состав тиреоидных гормонов.
Недостаток поступления йода в организм приводит к развитию эндемического зоба.
Избыток йода может вызвать дерматит, способствовать аутоиммунному
тиреоидиту. Радиоактивный йод избирательно поражает щитовидную железу.
Фтор – способствует минерализации костей и зубов.
При снижении содержания фтора увеличивается заболеваемость кариесом зубов.
При избытке фтора и недостатке кальция замедляется минерализация костей. Избыток фтора в постоянно употребляемой воде приводит к развитию эндемического флюороза (дистрофия зубной эмали).
Селен – является сильным антиоксидантом, входит в состав глютатионпероксидазы, фосфолипидглютатион-пероксидазы, некоторых оксидоредуктаз.
Недостаток селена вызывает аритмии, миопатии, азооспермию, некрозы
печени, угри, сердечную недостаточность.
Избыток вызывает острое отравление.
265
Кремний – способствует нормальному раннему росту и минерализации
костей.
Попадание частиц, содержащих двуокись кремния, активирует альвеолярные макрофаги и приводит к формированию гранулем и силикозу легких.
Патофизиология обмена витаминов.
Недостаточное поступление витаминов с пищей приводит к развитию соответствующих заболеваний, называемых авитаминозами и гиповитаминозами.
Глубокий дефицит витамина в организме с выраженной клинической картиной его недостаточности принято называть авитаминозом (рахит, цинга, пеллагра, бери-бери и др.).
Состояние умеренного дефицита витамина, когда наблюдают в
основном общие неспецифические проявления недостаточности витамина
(быстрая умственная и физическая утомляемость, нарушение концентрации
внимания, головокружение, раздражительность, ослабление памяти, головные
боли, апатия, потеря аппетита), называется гиповитаминозом.
Дефицит нескольких витаминов называют полигиповитаминозами, или
полиавитаминозами.
Гипо- и авитаминозы, вызванные недостаточным поступлением
витаминов с пищей, называют экзогенными, или первичными.
Гипо- и авитаминозы, возникающие при нарушении утилизации витаминов или повышенной потребности в них, называются эндогенными, или вторичными.
Третью группу составляют врожденные, или генетически обусловленные
нарушения обмена и функций витаминов.
Важнейшими причинами витаминной недостаточности являются:
• недостаточное поступление витаминов с пищей;
• угнетение кишечной микрофлоры, продуцирующей некоторые витамины;
• нарушение утилизации витаминов (заболевания желудочно-кишечного
тракта, гепатобилиарной системы, поджелудочной железы;
• конкурентная утилизация витаминов кишечными паразитами и
патогенной микрофлорой;
• разрушение ферментами, содержащимися в пищевых продуктах;
• нарушение образования активных форм витаминов при применении лекарственных средств и др.);
• повышенная потребность в витаминах (различные физиологические состояния, например беременность, кормление, климатические и производственные неблагоприятные условия, физическая и нервно-психическая
нагрузка, повышенная экскреция витаминов и др.);
• врожденные нарушения обмена витаминов (нарушения всасывания в кишечнике, транспорта, биосинтеза и превращения в коферментные формы,
нарушение структуры апоферментов, усиление катаболизма витаминов и
нарушения их резорбции в почках и др.).
266
Жирорастворимые витамины.
Нарушения обмена витамина D рассмотрены в патологии фосфорнокальциевого обмена.
Гиповитаминоз А.
Функции витамина А связаны с его участием в фоторецепции и необходимостью для роста, клеточной пролиферации, дифференцировки, для поддержания адекватного иммунологического и гематологического статуса организма.
При гиповитаминозе А нарушается адаптации в сумерках:
• Гемералопия (куриная слепота) - нарушение зрения в сумерках - при дефиците витамина А в палочках и колбочках сетчатки тормозится трансформация светового луча. На свету родопсин, содержащийся в палочках,
поглощает световую энергию и распадается на альдегидную форму витамина А (транс-ретиналь) и белок (опсин). В темноте при участии витамина А (цис-ретиналя) и опсина родопсин восстанавливается, что способствует восприятию черно-белого изображения. При дефиците витамина А
родопсин в темноте восстанавливаться не может, поэтому черно-белое
изображение не воспринимается. В результате нарушается зрение в сумерках, хотя больные днем видят нормально.
• Ксерофтальмия (сухость конъюнктивы и роговой оболочки глаза) - связана с гиперкератозом эпителия слезных протоков, их закупоркой, высыханием конъюнктивы и роговой оболочки глаза.
• Усиление процессов кератинизации (переход цилиндрического эпителия
в плоский в коже и слизистых оболочках) - при гиповитаминозе А обусловлено тем, что витамин А принимает участие в окислительновосстановительных процессах и, следовательно, при его дефиците эти
процессы угнетаются. Возникает кислородная недостаточность. В условиях гипоксии изменяется трофика тканей, усиливается процесс кератинизации слезного канала - глазное яблоко не омывается слезой, обладающей бактерицидным действием. Все это ведет к сухости роговой оболочки, а под действием микробов - к ее размягчению, изъязвлению (кератомаляции). Усиление кератинизации приводит к интенсивному слущиванию эпидермиса кожи, образованию трещин, ороговению эпителия дыхательного и пищеварительного трактов, мочеполовых путей. В протоках
многих желез (подчелюстная, околоушная, поджелудочная) отмечаются
процессы гиалинизации. В результате возникает функциональная недостаточность железистого аппарата, органов дыхания, пищеварения, стерильность.
Гипервитаминоз А.
Под влиянием больших доз витамина А возникают дистрофические изменения в печени, почках, сердце, диафрагме, трубчатых костях и других органах.
267
Гипервитаминоз А может развиваться у детей в результате приема больших количеств рыбьего жира, препаратов витамина А. Гипервитаминоз развивается после приема 1000000-6000000 МЕ витамина А. Много свободного витамина А содержится в печени белого медведя, тюленя, моржа, поэтому жители
Севера ограничивают использование печени этих животных и рыб.
Гиповитаминоз К
Витамин К (филлохинон) — важнейший антигеморрагический
витамин. Гиповитаминоз К может быть эндогенного и экзогенного
происхождения. Эндогенный гиповитаминоз возникает вследствие нарушения
всасывания витамина К в кишечнике (дефицит желчи, панкреатического сока),
при поражении кишечника (поносы), остром и хроническом поражении печени,
приеме медикаментозных средств,
блокирующих синтез факторов свертывания крови. При недостаточности поступления в организм витамина К возникает кровоточивость, кровоизлияния в
кожу даже при самой незначительной травме. Наблюдаются также кровоизлияния в суставы, сетчатку глаза, носовые кровотечения, кровоточивость десен при
жевании твердой пищи и чистке зубов.
Основное проявление гиповитаминоза К — нарушение свертывания крови. При этом в печени понижается образование активных форм ряда факторов
свертывания крови: протромбина (фактор П), проконвертина (фактор VII), фактора Кристмаса (фактор IX), фактора Стюарта-Пауэра (фактор X). Витамин К
осуществляет коферментные функции в реакции карбоксилирования остатков
глютаминовой кислоты в молекуле протромбина и других белков, превращая
их в активные формы.
Витамин К участвует в переносе электронов при биологическом окислении в
клетках микроорганизмов и при осуществлении процесса фотосинтеза в растениях.
Гиповитаминоз Е.
Витамин Е (токоферол) — фактор размножения. Дефицит витамина Е вызывает мышечную дистрофию, склонность к гемолитической анемии (гемолиз
эритроцитов у взрослых, нарушение биосинтеза гема у недоношенных детей),
тормозит синтез коэнзима А, который имеет хиноновую структуру. Этот комплекс участвует в переносе водорода с желтых ферментов на цитохромы. Витамин Е принимает участие в процессе фосфорилирования в митохондриях клеток. Токоферол является сильным биологическим антиоксидантом, тормозящим процессы окисления ненасыщенных липидов.
При дефиците токоферола и усилении процессов перекисного окисления
ненасыщенных липидов повреждается структура липопротеидных мембран
клеток, субклеточных структур, ингибируется синтез простагландинов. При
дефиците витамина Е нарушается аккумуляция жирорастворимых витаминов во
внутренних органах, особенно ретинола. При гиповитаминозе Е понижается
тканевое дыхание, активируется процесс перекисного окисления липидов,
268
нарушаются практически все виды обмена веществ, возникают дистрофические
изменения.
Водорастворимые витамины.
Авитаминоз С (скорбут, цинга).
Витамин С (аскорбиновая кислота, противоцинготный фактор).
Одним из проявлений гиповитаминоза С является геморрагический диатез (цинга). Наиболее ранний признак заболевания - разрыхленность десен,
кровоточивость при легкой травме, развитие гингивита, зубы начинают шататься и могут выпадать. Возникают также кровоизлияния в волосяные фолликулы
голеней, бедер и предплечий. Кожа становится шершавой и сухой. При тяжелой
форме гиповитаминоза мышцы при пальпации болезненны вследствие кровоизлияний, в том числе под надкостницу и периартикулярные ткани коленных и
голеностопных суставов, что приводит к ограничению подвижности в суставах
нижних конечностей. В наиболее тяжелых случаях могут развиваться кровоизлияния в перикард,
плевру, желудочно-кишечные, маточные кровотечения, а также гипохромная
анемия.
Патогенез цинги представлен следующими механизмами:
• Нарушением окислительно-восстановительных процессов в пентозном
цикле - аскорбиновая кислота и ее дегидроформа образуют окислительновосстановительную систему. В пентозном цикле аскорбиновая кислота
служит переносчиком водорода. Она легко окисляется, отдавая два атома
водорода. Образующаяся дегидроаскорбиновая кислота легко восстанавливается за счет окисления других соединений.
• Гипофункцией надпочечников. Аскорбиновая кислота связана непосредственно с обменными процессами в клетках надпочечников. Понижение
ее содержания в данной железе свидетельствует о низкой функциональной способности надпочечников.
• Падением активности ряда ферментов, в частности гексокиназы, приводит к задержке всасывания углеводов из кишечника, затруднению отложения гликогена в печени, что может привести вначале к гипергликемии,
а затем и к гипогликемии. Вместо гликогена в печени может отложиться
жир, и тогда развивается жировая инфильтрация печени.
• Торможением синтеза белков и усилением их распада. Это вызывает:
возникновение дистрофических изменений в организме, слабость сердечной мышцы; торможение активности остеобластов; в результате задерживается образование белковой костной матрицы и, следовательно нарушаются процессы окостенения; уменьшение выработки антител и как следствие — снижение устойчивости организма к инфекциям; нарушение образования коллагена из проколлагена, что ведет к снижению эластичности сосудистых стенок, увеличению их проницаемости, кровотечениям и
отекам, заживление ран затягивается.
269
Клиническая картина цинги связана с нарушениями иммунной и эндокринной систем. Происходят сдвиги во всех видах обмена веществ.
Обычно наблюдается легкое течение заболевания, ограничивающееся разрыхленностью и кровоточивостью десен, кровоизлияниями в фолликулы, быстрой
утомляемостью и снижением работоспособности.
Авитаминоз В1 (болезнь бери-бери).
Витамин В1 (тиамин, аневрин) — противоневритный фактор.
Бери-бери относится к заболеваниям, возникающим вследствие
недостаточного поступления в организм или нарушения утилизации
витамина В1. Для бери-бери характерна классическая триада: полиневриты,
мышечная атрофия и поражение сердечно-сосудистой системы. Заболевание
встречается преимущественно в Юго-Восточной Азии среди населения, употребляющего в пищу полированный рис, бедный витаминами группы В).
Различают следующие формы заболевания:
• Экзогенная форма бери-бери встречается среди населения, питающегося
полированным рисом, а также при несбалансированном питании, при исключении из рациона хлеба из муки грубого помола (ржаной, серый,
пшеничный), бобов, крупы и других продуктов.
• Эндогенная форма возникает при нарушении всасывания, усвоения или
повышенном потреблении витамина В1, что наблюдается в результате
применения некоторых антибиотиков и сульфаниламидов, вливания щелочных растворов, употребления продуктов, содержащих тиаминазу.
Клиническая картина - резкое истощение, отеки, сердце расширено за счет
правых отделов, увеличиваются гипофиз, надпочечники, щитовидная железа.
При микроскопии выявляются жировая дистрофия миокарда, его серозный
отек, дегенеративные "изменения в ЦНС с распадом аксонов нервных волокон
и их миелиновых оболочек.
Гиповитаминоз витамина РР (пеллагра).
Основное значение никотиновой кислоты для организма заключается в
том, что она входит в состав адениннуклеотидов, которые необходимы в процессах переноса водорода с субстрата на флавиновые ферменты.
Причины пеллагры:
1. Алиментарная недостаточность витамина РР. У людей пеллагра
чаще встречается при однообразном питании кукурузой, полированным
рисом, вареным горохом, сухарями и другими продуктами с низким
содержанием триптофана, из которого синтезируется никотиновая кислота.
2. Нарушение синтеза никотиновой кислоты в организме человека и животного.
Возникновение эндогенного РР-гиповитаминоза обычно связывают с дефицитом фосфопиридоксаля (нарушается обмен триптофана и тормозится синтез
никотиновой кислоты), отсутствием субстратов общего фермента (катализирует
синтез никотиновой кислоты).
При дефиците витамина РР тормозится первый этап в окислительновосстановительных процессах — образование АТФ. Нарушение процессов
270
окислительного фосфорилирования ведет к трофическим расстройствам организма - появляются функциональные сдвиги в нервной системе, пеллагроидные
изменения кожи, поражается слизистая оболочка пищеварительного тракта,
возникает диарея.
Гиповитаминоз В6.
Витамин В6 — пиридоксаль, пиридоксин. Фосфопиридоксаль коферментная форма витамина В6.
Витамин В6 участвует в обмене веществ в виде фосфопиридоксаля в следующих процессах: переаминировании (фосфопиридоксаль является активной
группой трансаминаз), декарбоксилировании (эта реакция осуществляется
только с участием фосфопиридоксаля).
Вследствие отсутствия фосфопиридоксаля процессы переаминиования и
декарбоксилирования нарушаются. Это приводит к изменению обмена многих
аминокислот, например глутаминовой, триптофана, серина.
Дефицит витамина В6 приводит к изменениям функции нервной системы
(повышенная возбудимость, эпилепсия) и пеллагроидным изменениям кожи изза нарушения синтеза никотиновой кислоты из триптофана.
Гиповитаминоз В12.
Витамин В 12 участвует в синтезе нуклеиновых кислот, в углеводном обмене. Он восстанавливает рибонуклеозиды до соответствующих дезоксирибонуклеотидов.
Дефицит витамина В12 (цианокобаламина) возникает как вследствие недостаточного поступления в организм продуктов животного происхождения,
содержащих этот витамин, так и в результате нарушений образования фактора
Кастла, продуцируемого обкладочными клетками желудка, при заболевании
желудка.
При гиповитаминозе В12 наблюдается анемия, которая характеризуется
резким снижением числа эритроцитов в периферической крови, мегалобластическим типом кроветворения.
Гиповитаминоз В9 (фолиевой кислоты).
Фолиевая кислота активизирует синтез нуклеиновых кислот. Она способствует восстановлению холина и метионина в организме.
При недостаточности витамина В9 нарушается образование эритроцитов,
возникает мегалоцитарная анемия. Развитие мегалоцитарной анемии объясняется тем, что синтез гемоглобина при недостаточности фолиевой кислоты менее заторможен, чем процесс образования эритроцитов. В связи с этим в крови
появляются эритроциты с высоким содержанием гемоглобина. В результате
цветовой показатель становится выше единицы. В крови появляются мегалоцитарные формы эритроцитов. При дефиците фолиевой кислоты отмечается также
лейкопения.
271
Гиповитаминоз В3 (пантотеновая кислота).
Пантотеновая кислота входит в состав ацетилкоэнзима А, который осуществляет реакцию метилирования в окислительном превращении пировиноградной кислоты.
Гиповитаминоз В3 возникает вследствие нарушения всасывания пантотеновой кислоты в кишечнике при чрезмерном разрушении ее протеолитическимй ферментами.
При дефиците пантотеновой кислоты происходит распад белков, жиров и
углеводов. Вследствие снижения анаболических процессов возникают трофические расстройства.
Проявляется синдромом "жжения в стопах", апатией, парестезией в руках,
нарушением секреторной функции желудка.
Гиповитаминоз Н.
Витамин Н (биотин) — антисеборейный фактор.
Дефицит биотина возникает вследствие приема с пищей большого количества сырых яиц (в сыром яйце имеется белок авидин, который прочно связывает биотин пищи и мешает его всасыванию).
Патогенез биотиновой недостаточности: снижение активности фермента
карбоксилазы ацил-КоА, блокирующее переход малонил-КоА в продукты его
метаболизма - снижение синтеза жиров; торможение включения пировиноградной кислоты в цикл Кребса, что приводит к нарушению жирового и углеводного обмена, образованию АТФ.
При недостатке у человека отмечаются себорея, дерматит.
ПАТОФИЗИОЛОГИЯ КИСЛОТНО-ЩЕЛОЧНОГО
РАВНОВЕСИЯ.ПАТОФИЗИОЛОГИЯ ФОСФОРНО-КАЛЬЦИЕВОГО
ОБМЕНА
Нарушения КЩР.
Кислотно-щелочное равновесие это баланс между кислыми и щелочными
продуктами, которые поступают в организм, образуются в процессе метаболизма и выводятся из организма . Кислотно-щелочное равновесие – одно из важнейших гомеостатических свойств внутренней среды организма, характеризующееся относительным постоянством соотношения водородных и гидроксильных ионов и определяющее оптимальный характер обменных процессов и физиологических функций.
Кислотами называют вещества, способные при диссоциации в растворах
выделять ион водорода (донаторы протона), а щелочами – вещества, способные
связывать ион водорода.
В организм с пищей поступает множество разнообразных веществ – вода,
белки, жиры, углеводы, минеральные соединения, витамины. В результате метаболических реакций из них образуются эндогенные кислоты (нелетучие и летучие).
272
Нелетучие кислоты – кислоты, которые не способны превращаться в газ и
удаляться легкими (серная, молочная, пировиноградная, β-оксимасляная, ацетоуксусная).
Летучие кислота – угольная кислота, легко расщепляющаяся на воду и
углекислый газ, который выводится легкими.
Для оценки КЩР используется отрицательный десятичный логарифм
концентрации ионов водорода, который обозначается рН и называется водородным показателем. рН внутриклеточной, тканевой жидкости и крови относится к числу жестких гомеостатических констант.
Показатели рН.
рН крови 7,35 – 7,45
рН цереброспинальной жидкости 7,4 – 7,5
рН сока поджелудочной железы 7,8 – 8,4
рН печеночной желчи 7,3 – 8,0
рН слюны 5,8 – 7,8
рН желудочного сока 1,4 – 1,8
рН сока тонкой кишки 7,5 – 8,6
рН сока толстой кишки 8,0 – 9,0
При сдвиге рН на 0,1 единицы наблюдаются нарушения со стороны дыхательной, сердечно-сосудистой системы, изменяется состав мочи; сдвиг на 0,3
единицы приводит к коматозному состоянию; на 0,4 единицы, если он продолжителен – несовместим с жизнью.
Классификация нарушений КЩР.
К нарушениям КЩР относятся:
Ацидоз – сдвиг КЩР в кислую сторону,обусловленный аномальным
накоплением кислот в организме или потерей оснований.
Алкалоз – сдвиг КЩР в щелочную сторону,обусловленный аномальным
накоплением бикарбонат-аниона в организме или потерей кислот.
Ацидемия – снижение рН артериальной крови ниже 7,40.
Алкалемия – повышение рН артериальной крови выше 7,40.
Ацидозы и алкалозы делятся на экзогенные (вызванные отравлениями
кислотой или щелочью) и эндогенные (связанные с нарушениями механизмов
КЩР).
По интенсивности сдвигов КЩР:
ацидоз
компенсированный
7,40-7,35
субкомпенсированный 7,34-7,20
декомпенсированный 7,19-6,80
По патогенезу:
рН в норме
7,35-7,45
алкалоз
7,40-7,45
7,46-7,55
7,56-7,80
273
• Газовый (дыхательный) ацидоз (алкалоз) – нарушение КЩР связано с первичным изменением уровня парциального давления СО2 в артериальной
крови.
• Метаболический (негазовый) ацидоз (алкалоз) – нарушение КЩР вследствие
первичных изменений уровня нелетучих кислот и оснований.
Существуют также смешанные (газовый и метаболический, газовый и
выделительный) и комбинированные (метаболический и выделительный ацидозы) виды ацидоза и алкалоза.
Способы оценки и параметры КЩР.
Параметры КЩР определяются путем анализа газов крови в пробах артериальной крови микроионометрическим методом.
КЩР характеризуется с помощью:
• Актуальный (истинный) рН – значение рН артериальной крови, определенное без доступа воздуха при 38 0С.
• Актуальное (истинное) парциальное напряжение углекислого газа (рСО2) –
значение рСО2 артериальной крови, определенное без доступа воздуха при
38 0С.
• Стандартный бикарбонат (SB) – содержание бикарбонатов в плазме крови
(ммоль/л) при полном насыщении ее кислородом и при рСО2, равном 40 мм
рт. ст., определяемое при 38 0С.
• Актуальный (истинный) бикарбонат (АВ) – концентрация бикарбоната в
плазме крови при истинном рСО2, определяемое при 38 0С.
Если SВ больше АВ – имеет место газовый алкалоз, если АВ больше SВ – имеет место газовый ацидоз.
• Буферные основания (ВВ) – сумма всех анионов цельной крови, принадлежащих к буферным системам.
• Избыток (недостаток) буферных оснований (ВЕ) – разность между средним
нормальным содержанием буферных оснований и найденным значением
концентрации буферных оснований.
Избыток ВЕ свидетельствует о метаболическом алкалозе, недостаток ВЕ – о
метаболическом ацидозе.
Для оценки характера биохимических изменений при метаболическом
ацидозе введено понятие – анионный просвет (англ.- anion gap).
Анионный просвет (дефицит, интервал, зазор, А-) – искусственно выводимое соотношение между концентрацией катиона натрия и суммой концентраций анионов хлорида и бикарбоната в плазме, удобное для оценки происхождения метаболического ацидоза в клинике. Он определяется по формуле:
А- = [Na+] - { [Cl-] + [HCO3-] } =12±4 мэкв/л
Количество анионов и катионов в плазме эквивалентно, поэтому анионный просвет отражает концентрацию неизмеряемых анионов (анионы органических кислот, сульфаты, анионные эквиваленты белка).
Увеличение показателя анионного просвета косвенно свидетельствует об
увеличении в крови неизмеряемых анионов, возникшем в ответ на падение
274
концентрации HCO3- и характеризует тяжесть ацидоза. Если падение HCO3- сопровождается повышением уровня Cl-, который может быть измерен, наблюдается нормальная величина анионного просвета.
Механизмы компенсаций сдвигов КЩР.
К компенсаторным механизмам относятся химические механизмы
нейтрализации кислот и оснований (буферные системы внутренней среды, буферные функции клеток, компенсаторные изменения метаболизма) и функциональные механизмы (изменения в работе легких, почек, печени, ЖКТ, скелета).
Буферные системы – системы, которые состоят из двух и более компонентов один из которых является кислотой, другой – щелочью. Буферные системы могут связать и нейтрализовать избыток кислот и щелочей.
• Карбонатная буферная система – основная буферная система организма,
определяется постоянством соотношения угольной кислоты и ее кислой соли (H2CO3/NaHCO3). Это соотношение постоянно поддерживается в пропорции 1/20.
• Фосфатная буферная система действует за счет поддержания постоянства
соотношений одно- и двуметаллической соли фосфорной кислоты
(NaH2PO4/Na2HPO4).
• Белковая буферная система – главный внутриклеточный буфер, способна
проявлять свои свойства за счет амфотерности белков, которые в одном случае реагируют с щелочами как кислоты (образуются щелочные альбуминаты), а в другом – с кислотами как щелочи (образуются кислые альбуминаты).
• Гемоглобиновая буферная система обеспечивает буферную емкость крови, в
связи с тем, что оксигемоглобин является гораздо более сильной кислотой,
чем восстановленный гемоглобин.
Натрий-водородный противопереносчик (белок NHE) – является одним из
универсальных белков организма, осуществляет обмен катионов между клетками и внеклеточной жидкостью, принимает участие в процессах ацидогенеза в
почках.
При ацидозе клетки поглощают Н+ и взамен отдают во внутреннюю среду
Na+, Са+, К+. При алкалозе ионы движутся в противоположных направлениях.
Изменения метаболизма. При ацидозе внутренняя среда освобождается
от кислот, происходит их усиленное сжигание и связывание (молочная кислота
усиленно поглощается гепатоцитами). При алкалозе включение кислот в обменные процессы наоборот тормозится (гепатоциты «оставляют» молочную
кислоту в крови).
Функциональные механизмы компенсации.
Система внешнего дыхания. Углекислый газ выводится из организма через легкие. Дыхательный центр активируется при избытке СО2 и при любом
ацидозе, следовательно интенсивность вентиляции зависит от содержания в
крови СО2 и Н+. При возникновении в организме ацидоза, первично, независимо от состояния легких, они включаются в компенсацию этого состояния и развивается вторичная гипервентиляция и СО2 выводится из организма в больших
275
количествах, что способствует нормализации рН внутренней среды. При возникновении в организме алкалоза, первично, независимо от состояния легких,
они также включаются в компенсацию и развивается вторичная гиповентиляция и СО2 задерживается в крови, что способствует нормализации рН внутренней среды.
Мочевыделительная система. Участвуют в регуляции КОС за счет процессов осуществляемых в почечных канальцах:
• Ацидогенез – активный транспорт (секреция) в первичную мочу Н+, освобождающихся при диссоциации в канальцевых эпителиоцитах угольной
кислоты, которая образуется в эпителиоцитах из СО2, захваченного из крови.
В результате внутренняя среда освобождается от излишка ионов водорода.
• Аммониогенез – активный транспорт (секреция) в первичную мочу ионов
аммония, образующихся из аминогрупп при дезаминировании в эпителиоцитах глютамина, аспарагина.
• Реабсорбция бикарбоната – активный транспорт этого соединения из первичной мочи обратно в кровь. В результате предотвращается потеря буферных оснований организма.
При первичном ацидозе любого происхождения почки активно включаются в компенсаторные реакции организма – усиливается ацидогенез, аммониогенез и реабсорбция бикарбоната. В результате этого организм избавляется
от избытка нелетучих кислот и предотвращает потерю с мочой больших количеств бикарбоната – рН смещается в щелочную сторону.
При первичном алкалозе функциональная активность канальцев снижается – уменьшается ацидогенез и реабсорбция бикарбоната. В результате этого
ионы водорода задерживаются в организме, а бикарбонат остается в первичной
моче (снижение реабсорбции) и выводится из организма – рН смещается в щелочную сторону.
Печень вырабатывает глютамин, используемый почками для аммониогенеза и метаболизирует лактат и кетокислоты, закисляющие внеклеточную жидкость.
ЖКТ поддерживает КЩР за счет секреции слизистой желудка протонов
водорода и поступления при этом в экстрацеллюлярное пространство эквивалентного количества бикарбоната, секреции поджелудочной железой и энтероцитами бикарбоната, который нейтрализует протоны, не связанный бикарбонат
всасывается в кишечнике. Компенсаторные механизмы – при потерях желудочного содержимого усиливается продукция и всасывание бикарбоната, при потерях кишечного содержимого всасывание бикарбоната из ЖКТ снижается.
Скелет является депо кальция и фосфатов организма и под влиянием парат-гормона и кальцитонина может компенсировать отклонения КЩР в кислую
сторону. При этом кальций и фосфаты выходят из костей во внеклеточную
жидкость и первичную мочу, что приводит к остеолизу, возникает деминерализация костей и зубов. Кальций и фосфат реагируют во внеклеточной жидкости с угольной кислотой и нейтрализуют катионы кислот. Кальций и кислый
фосфат теряются с мочой, натрий и бикарбонат реабсорбируются.
276
Газовый ацидоз – первичное увеличение содержания в организме углекислого газа.
Причинами являются:
Заболевания органов дыхательной системы (пневмония, бронхит, эмфизема, ларингоспазм, бронхоспазм, пневмоторакс, гидроторакс и гемоторакс, аспирация, асфиксия, муковисцидоз).
Заболевания, связанные с угнетением ЦНС (дыхательного центра) или с
периферическими нервно-мышечными расстройствами (травмы, отеки и опухоли головного мозга; отравления, нарушающие центральную стимуляцию вентиляции – барбитуровое и опиатное; токсическое действие лекарств, нарушающие
периферические и центральные механизмы вентиляции – анестетики, седативные препараты, наркотики, алкоголь, миорелаксанты).
Гемодинамическая недостаточность различного генеза, препятствующая
доставке СО2 в легкие.
Это все приводит к недостаточному выведению СО2 из организма.
Тяжелая мышечная работа приводит к чрезмерному образованию СО2
организме.
Аварийные ситуации на производстве, замкнутые помещения, ошибки
при лечении газовыми смесями, приводящие к повышению содержания СО2 во
вдыхаемом воздухе.
Патогенез: возникает как следствие гиповентиляции, что приводит к гиперкапнии (повышение рСО2 в крови), а следовательно и к увеличению концентрации ионов водорода и снижению рН.
• Включение в работу всех буферных систем крови и клеточных буферов.
• Активация работы почек – усиление ацидогенеза, аммониогенеза и реабсорбции бикарбоната.
• Активация β-адренорецепторов, приводящая к вазодилятации и возникновение симптомов интракраниальной (внутричерепной) гипертензии (сильные
головные боли), отек мозга, сопор с переходом в кому.
• Активация симпатической нервной системы и выделение катехоламинов
приводит к спазму резистивных сосудов, централизации кровообращения,
повышение АД, МОК, ЦВД, ОЦК, что увеличивает нагрузку на сердце. Может привести к возникновению сердечной недостаточности. Возникает ишемия периферических органов.
• Значительная концентрация СО2 в крови повышает возбудимость блуждающего нерва – остановка сердца, бронхоспазм, усиление секреции слизи, что
дополнительно затрудняет дыхание.
• Активация симпатической нервной приводит к спазму резистивных сосудов
почек, закислению мочи и снижение её количества.
• Увеличение выхода из клеток ионов калия в обмен на ионы водорода приводит к развитию гиперкалиемии, что приводит к сердечным аритмиям.
277
Патогенетическая терапия зависит от причины, вызвавшей газовый ацидоз,
направлена на уменьшение степени или ликвидацию дыхательной недостаточности, симптоматическое лечение.
Метаболический (негазовый) ацидоз – характеризуется нарушениями
метаболизма, приводящих к накоплению нелетучих кислот и веществ с кислыми свойствами.
Развивается вследствие первичной гипобикарбонатемии:
• При поступлении кислых продуктов извне (передозировка ацетилсалициловой кислоты, отравления этиленгликолем, метиловым спиртом).
• При нарушении выделительной функции почек (острая и хроническая почечная недостаточность, гипоальдостеронизм).
• При избыточной эндогенной продукции ионов водорода (кетоацидоз при сахарном диабете, при голодании, при избытке жира в пище; лактоацидоз при
циркуляторной и гемической гипоксии) и при избыточной потере бикарбонатов (болезни ЖКТ – гиперсаливация, поносы, свищи).
Патогенез: накопление нелетучих кислот или снижение концентрации
бикарбоната, повышение содержания ионов водорода.
• Включение буферных систем крови и клеточных буферов.
• Стимуляция инспираторных нейронов дыхательного центра при накоплении
кислых продуктов приводит к компенсаторному развитию гипервентиляции
и углекислый газ в больших количествах выводится из организма, что способствует нормализации рН внутренней среды.
• Активация работы почек – усиливается ацидогенез, аммониогенез и реабсорбция бикарбоната. рН организма возвращается к норме за счет уменьшения избытка нелетучих кислот и предотвращения потери с мочой бикарбоната.
• Циркуляторная гипоксия вследствие снижения кровоснабжения головного
мозга приводит к нарушению энергетического обеспечения нейронов и развитию гипоксической энцефалопатии вплоть до развития коматозного состояния.
• Гиперкалиемия вследствие обмена ионов калия на ионы водорода клетками
приводит к аритмиям.
• Увеличение концентрации водородных ионов приводит к вторичной гипервентиляции – дыхание Куссмауля.
• Гипокапния возникает вследствие гипервентиляции и приводит к снижению
сосудистого тонуса – артериальная гипотензия, сердечно-сосудистая недостаточность.
• Снижения кровотока в почках приводит к развитию олигурии.
• Потери ионов кальция костной тканью в обмен на ионы водорода приводит
к развитию остеопороза.
• Повышение проницаемости стенок артериол и прекапилляров, снижение реабсорбции жидкости в связи с венозным застоем, гиперосмия и гиперонкия
тканей приводят к отекам.
278
Патогенетическая терапия: устранение непосредственной причины, вызвавшей метаболический ацидоз; удаление из организма и нейтрализация избытка кислот – улучшение легочной вентиляции, коррекция электролитного
обмена (вливание бикарбонатов); симптоматическое лечение.
Газовый алкалоз – характеризуется увеличением рН и гипокапнией.
Причины возникновения:
• Острая гипоксия различного генеза (горная болезнь, тромбоэмболия легочной артерии).
• Хронические заболевания, вызывающие гипервентиляцию (пневмофиброз,
бронхиальная астма, застойная сердечная недостаточность).
• Возбуждение дыхательного центра (стресс, лихорадка, токсико-септический
шок, отравление салицилатами, опухоли мозга, черепно-мозговая травма,
энцефалит, эмоциональные состояния – истерия).
• Неверно выбранный режим искусственной вентиляции легких.
Патогенез. Эти причины приводят к увеличению эффективной альвеолярной
вентиляции – гипервентиляции, что приводит к увеличенному выделению углекислого газа – возникает гипокапния.
• Включение буферных систем – освобождают протоны и концентрация
бикарбоната в плазме снижается.
• Клетки поглощают ионы натрия и калия и выделяют в кровь ион водорода,
что приводит к гипокалиемии, также калий теряется вместе с мочой из-за его
усиленной секреции вместе с Н+. Возникают мышечная слабость, парез кишечника, параличи скелетной мускулатуры, нарушения сердечного ритма.
• Снижение ацидогенеза, снижение секреции калия и реабсорбции бикарбоната в почках.
• Вследствие снижения объемного мозгового кровотока в результате повышения тонуса мозговых сосудов и компенсаторного перехода ионов кальция в
клетки в обмен на ионы водорода отмечаются парестезии кожи конечностей
и вокруг рта, мышечные спазмы в конечностях, легкая или выраженная сонливость, головные боли, нарушения сознания, кома.
• Вследствие компенсаторного снижения реабсорбции натрия вместе с
бикарбонатом возможно развитие обезвоживания.
Патогенетическая терапия: коррекция дыхания (ликвидация причины гипервентиляции), устранение дефицита углекислого газа в организме – применение
дыхательных смесей богатых СО2 (карбоген: 95% СО и 5% О2), симптоматическое лечение.
Метаболический (негазовый) алкалоз характеризуется повышением рН
крови и увеличением концентрации бикарбоната.
Причины возникновения:
• Связанные с потерей ионов водорода:
-Потеря желудочного содержимого при рвоте (токсикоз беременности, пилороспазм, высокая кишечная непроходимость) и отсасывании через зонд.
279
-Потери протонов почками при действии петлевых диуретиков и тиазидов.
-Первичный и вторичный гиперальдостеронизм.
• Связанные с избытком поступления бикарбоната:
-Поступление с пищей больших количеств щелочи.
-Ятрогенное назначение больших доз бикарбоната или его предшественников.
-При массивной гемотрансфузии
Патогенез. Повышение уровня бикарбоната и снижение концентрации катиона водорода и хлорид-аниона во внеклеточной жидкости.
• Буферные системы крови и клеток как при газовом алкалозе.
• Снижение ацидогенеза и реабсорбции бикарбоната в почках (кроме гиперальдостеронизма).
• Гиповентиляция – возникает при снижении концентрации водородных
ионов, задерживает СО2 в организме для понижения рН и вызывает гипоксию.
• При гиперальдостеронизме возникает гипокалиемия (увеличивается выведение ионов калия).
• Клинические проявления как при газовом алколозе.
Патогенетическая терапия: устранение причины метаболического алкалоза,
восстановление нормального уровня, симптоматическое лечение.
Смешанные формы нарушений КЩР – возникают при сочетании у больного двух и более форм простых нарушений КЩР.
Если смешанное нарушение КЩР обусловлено наличием двух типов ацидоза, то следует ожидать большее снижение рН.
Если одновременно развиваются два противоположных нарушения КЩР
(ацидоз и алкалоз), то величину рН будет определять преобладающее нарушение. Иногда рН может оставаться нормальным.
Могут наблюдаться следующие сочетания:
• Метаболический ацидоз и метаболический алкалоз, например – диабетический кетоацидоз и рвота.
• Метаболический ацидоз и дыхательный ацидоз, например – лактоацидоз,
остановка дыхания.
• Метаболический ацидоз и дыхательный алкалоз, например – отравление
этиленгликолем, пневмония.
• Метаболический алкалоз и дыхательный ацидоз, например – дренирование содержимого желудка, передозировка седативных препаратов.
• Метаболический алкалоз и дыхательный алкалоз, например – применение
диуретиков, печеночная недостаточность.
Связь нарушений КЩР с нарушениями вводно-электролитного обмена.
Довольно часто нарушения водно-электролитного обмена приводят к
нарушениям КЩР в связи с потерей или задержкой в организме веществ, обладающих кислыми или щелочными свойствами.
280
При потере содержимого желудка наблюдается гипогидратация, которая
приводит к метаболическому алкалозу – потеря жидкости с высоким содержанием хлора и водорода.
При потере содержимого кишечника наблюдается гипогидратация, которая приводит к метаболическому ацидозу – потеря жидкости с высоким содержанием бикарбонатов.
Кальций играет важную роль во многих метаболических и физиологических процессах организма человека – главный составной элемент кости, передача внутриклеточных сигналов, участие в различных ферментативных реакциях, коагуляция крови, нервно-мышечная проводимость. 99% кальция содержится в костях, нормальное содержание кальция в плазме крови составляет 2,2 –
2,6 мМ/л, ионизированного кальция 1,1 – 1,4 мМ/л. Количество ионизированного кальция зависит от величины рН: алкалоз уменьшает концентрацию ионизированного кальция, ацидоз – повышает.
Фосфор содержится в плазме крови в количестве 1,0 – 1,5 мМ/л, необходим
для прочности костей и является их существенным элементом, является буферной системой.
Тесная связь обмена кальция и фосфора обусловлена тем, что они образуют
нерастворимые соединения типа оксиапатита, составляющие основу кристаллической структуры обызвествленных тканей (костей и твердых тканей зубов).
Содержание кальция в плазме крови регулируется гормоном паращитовидной железы паратирином (паратгормон), витамином D и гормоном щитовидной
железы кальцитонином.
Паратирин стимулирует активность остеокластов и резорбцию костной ткани, благодаря чему Са++ из костей поступает в кровь, повышает реабсорбцию
Са++ в почечных канальцах, усиливает всасывание Са++ в кишечнике ( при
наличии достаточного количества витамина D). Все это приводит к повышению
содержания Са++ в жидкой части крови.
Тиреокальцитонин тормозит резорбцию костной ткани и тем самым препятствует мобилизации Са++ и фосфатов из костей, уровень Са++ в крови под влиянием этого гормона снижается. Уровень тиреокльцитонина повышается у беременных и кормящих женщин, что предотвращает остеопороз в связи с затратой
Са++ при формировании костной ткани плода и потерей этого иона с молоком.
Нарушения кальций-фосфорного обмена может проявляться расстройством
всасывания кальция и фосфатов в кишечнике, обызвествлением скелета, а также отложением фосфатно-кальциевых солей в мягких тканях.
Расстройство всасывания кальция и фосфата наблюдается в случаях изменения нормального соотношения этих элементов в диете (2 : 1), употребления
пищи, богатой оксалатами и инозитфосфатной (фитиновой) кислотой, упорного
поноса, а также при рахите.
Гипокальциемия – понижение концентрации кальция в плазме крови. Причинами могут быть нарушения высвобождения или синтеза паратирина паращитовидными железами, при злокачественных новообразованиях, острой и
281
хронической почечной недостаточности, остром панкреатите, дефиците витамина D, применение лекарственных средств (противосудорожные, фенобарбитал).
Механизмы, обуславливающие гипокальциемию при разных заболеваниях,
неодинаковы. При злокачественных новообразованиях – в результате влияния
медиаторов, высвобождаемых опухолевыми клетками, стимулируется функция
остеобластов и усиливается образование костей. При острой и хронической почечной недостаточности гипокальциемия обусловлена нарушением продукции
1,25-дигидроксивитамина D3 клетками проксимальных канальцев, приводящей
к дефициту витамина D, а также задержкой фосфатов.
Проявления гипокальциемии. Повышенная возбудимость нервной ткани
может способствовать гиперактивным рефлексам, судорогам, тетании – спонтанное сокращение мышц, особенно кистей и стоп. При быстром снижении
кальция возможен ларингоспазм. Тетанию могут провоцировать эмоциональное
напряжение, физическая нагрузка, беременность, тошнота и рвота. Изменения
на ЭКГ – удлинение интервалов QT, ST, инверсия зубца Т, возможна желудочковая тахикардия.
Рахит. Этиология. Одним из основных факторов является гиповитаминоз D,
дефицит витамина D экзогенного или эндогенного происхождения. Помимо неправильного вскармливания и алиментарной недостаточности витамина D к
нему может приводить и нарушение образования его активных форм в организме при недостатке ультрафиолетовых лучей (зимой и осенью, в городах),
заболевания печени и почек (в них происходит образование активных форм витамина).
Патогенез. Витамин D представляет собой стероидное соединение и известен в виде витамина D2 (эргокальциферол) и витамина D3 (холекальциферол),
которые очень близки по строению, физико-химическим свойствам и влиянию
на организм человека. Поступающий с пищей витамин D подвергается преобразованию в печени и почках, в результате чего образуется 1,25-дигидроксивитамин D, обладающий гормоноподобным действием. Это соединение влияет
на генетический аппарат клеток кишечника, благодаря чему повышается синтез
белка, специфически связывающего кальций и обеспечивающего его транспорт
в организме. При недостатке витамина D нарушается всасывание и обмен кальция, его концентрация в крови падает, что вызывает реакцию паращитовидных
желез и повышение секреции паратирина, регулирующего обмен кальция и
фосфора. Избыточная секреция паратирина ведет к мобилизации кальция из
костной ткани, подавлению реабсорбции фосфатов в почечных канальцах, в
связи с чем содержание неорганических фосфатов в крови падает. В то же время резко увеличивается активность щелочной фосфатазы. Нарушения фосфатно-кальциевого обмена приводят к развитию ацидоза, что сопровождается
нарушением возбудимости нервной системы.
Нарушение минерализации костей из-за местных и системных причин, не
связанных с дефицитом активного витамина D приводит к картине «Dрезистентного рахита». Примером является наследственная аутосомнорецессивная гипофосфатазия – при дефекте щелочной фосфатазы у больных
282
замедляется гидролиз пирофосфата, который в костях является ингибитором
минерализации.
Клинические проявления. Начальными признаками рахита являются изменения со стороны нервной системы. Ребенок становится раздражительным, часто плачет, потеет. Характерными признаками рахита служат изменения в скелете и трубчатых костях. У ребенка долго не зарастают роднички, наблюдается
размягчение костей черепа. Серьезные нарушения возникают в грудной клетке:
размягчаются ребра, грудина выступает вперед, на местах соединения ребер с
реберными хрящами появляются рахитические четки. Грудная клетка деформируется, теряет способность расширяться в переднезаднем и фронтальном
направлениях, что ведет к ограничению акта дыхания – возникает недостаточность функции дыхания, возможно развитие ателектазов, пневмонии. Деформации грудной клетки приводят к нарушению кровообращения: отмечаются застойные явления в печени и воротной вене, которые приводят к недостаточности функций органов брюшной полости (ухудшение всасывания в кишечнике,
развитие метеоризма, энтероколита, наблюдается увеличение живота).
Лечение. Рациональное питание, массаж, гимнастика, препараты витамина
D2,ультрафиолетовая терапия.
Профилактика. Рациональное питание, достаточная инсоляция, санитарногигиенический режим, закаливание.
Гипервитаминоз D.
При поступлении в организм больших доз витамина D у детей прекращается рост скелета, кости утолщаются, особенно в области эпифизов, приостанавливается рост; костей в длину. Наблюдается раннее зарастание родничков,
что приводит к возникновению микроцефалии. Из-за отсутствия разрушения
костной ткани рост длинных трубчатых костей в длину нарушается и развивается деформация скелета. При гипервитаминозе D нередко отмечается обызвествление мягких тканей.
Гиперкальциемия – повышение концентрации кальция в плазме крови.
Причинами могут быть гиперпаратиреоз (аденома паращитовидных желез, гиперплазия), первичный альдостеронизм, злокачественные новообразования (рак легких, молочной железы, предстательной железы, миелома, лимфома), саркоидоз.
Механизмы развития гиперкальциемии при гиперпаратиреозе обусловлены повышением концентрации в плазме крови паратирина и 1,25дигидроксивитамина D3. При раке легких опухоль продуцирует гуморальный
фактор, способствующий резорбции кости и увеличению реабсорбции кальция
канальцами почек. При миеломе – продукция лимфокинов, способных стимулировать остеокласты. При саркоидозе – избыточная продукция гранулематозной тканью 1,25-дигидроксивитамина D3.
При гиперкальциемии подавляется секреция паратирина и увеличивается высвобождение кальцитонина.
283
Клинические проявления: слабость, тошнота, рвота, боли в костях, переломы, боли в пояснице (отложение солей кальция в почках), депрессия, спутанность сознания, парестезии, ступор, кома. На ЭКГ: укорочение сегмента ST и
интервала QT.
Обызвествление мягких тканей.
Если гиперкальциемия сопровождается нормальным или повышенным
уровня фосфора сыворотки, образуются кристаллы фосфата кальция, которые
откладываются во всех органах. Кальцификация наблюдается когда содержание
комплексов кальция и фосфора в сыворотке превышает 70 мг/л.
Патогенез. Усилением всасывания кальция в кишечнике приводит к увеличению его содержание в крови. Гиперкальциемия по принципу обратной связи приводит к торможению функций паращитовидных желез. Вследствие возникшей гипосекреции паратгормона усиливается процесс реабсорбции фосфора
в канальцах почек, развивается гиперфосфатемия. При появлении гиперкальциемии и гиперфосфатемии в условиях щелочной среды образуются труднорастворимые соединения фосфорнокислого кальция, которые, проходя через почечный фильтр, задерживаются в почках и приводят к их обызвествлению.
Также фосфорнокислый кальций откладывается в тканях (стенки сосудов,
внутренние органы). В результате такого отложения возникают кальцинозы,
служащие благоприятной почвой для развития склеротических изменений.
Гипофосфатемия – снижение концентрации неорганических фосфатов в
плазме.
Причины – увеличение потери фосфатов с мочой (гиперпаратиреоз, дефицит витамина D, остеомаляция при онкологических заболеваниях, алкогольная интоксикация, ацидоз, увеличение объема внутриклеточной жидкости, лекарственные средства – кальцитонин, диуретики, глюко- и минералокортикоиды), уменьшение всасывания фосфатов кишечником (применение антацидов,
дефицит витамина D, злоупотребление алкоголем, голодание), приток фосфатов
в клетки (снижение массы тела, респираторный алкалоз, интоксикация салицилатами, введение инсулина, глюкозы, фруктозы).
Гипофосфатемия приводит к снижению 2,3-дифосфоглицерата в эритроцитах, что приводит к повышенному поглащению кислорода гемоглобином и
тканевой гипоксии, падению в тканях содержания аденозинфосфата, нарушению хемотаксиса и бактерицидных свойств лейкоцитов, тромбоцитопении,
склонности к геморрагиям. Повреждение мышц развивается при сочетании
уменьшения фосфатов со снижением содержания внутриклеточного фосфора и
увеличением содержания в клетках воды, натрия и хлоридов. Дефекты скелета
могут развиваться при гиперфосфатемии в связи с увеличением синтеза 1,25дигидроксивитамина D3 и уменьшении выделения паратирина.
Гиперфосфатемия – увеличение содержания фосфатов в плазме крови.
Причинами являются снижение экскреции фосфатов вследствие развития почечной недостаточности, гиперпаратиреоз, повышенное поступление фосфатов
из внеклеточной жидкости, выход фосфора из клеток при усиленных катаболи-
284
ческих процессах (инфекции, синдром длительного сдавливания, перегревание,
применение цитостатиков, гемолитические анемии, острая лейкемия).
ЛИТЕРАТУРА:
1. Патологическая физиология: Учебник / Н.Н. Зайко, Ю.В. Быць, А.В. Атаман
и др.; Под. ред. проф. Н.Н. Зайко, проф Ю.В. Быця. – 3-е изд. – М.: МЕДпресс-информ, 2006. – 640 с.
2. Патологическая физиология / Под ред. проф. Адо А.Д. Москва "Триада-Х"
2002. – 616 с.
3. Атаман А.В. Патологическая физиология в вопросах и ответах.Учебное пособие.-Винница: Новая книга, 2008.-544с.
4. А.Ш. Зайчик, Л.П. Чурилов. Основы общей патологии. Учебное пособие для
медицинских вузов.-ЭЛБИ-СПБ.-1999.
5. Патофизиология / Под ред. проф. П.Ф. Литвицкого. – М.: Гэотар-Медицина,
2003.
6. Внутренние болезни. В 10 книгах. Книга 2. Перевод с английского. / Под редакцией Е. Браунвальда и др. / - М.: Медицина, 1993.
7. Общая патология человека: Руководство для врачей/ Под ред. А.И.Струкова,
В.В.Серова, Д.С.Саркисова. В 2Т.-2-е изд., перераб. и доп.- М.: Медицина,
1990.
8. Ажаев А. Н., Брязгунов И. П. Перегревание организма // БМЭ / Под ред. Б.
В. Петровского. -3-е изд. - М., 1982. -т. 18.
9. Романцев Е. Ф., Блохина В. Д. Жуланов З. И. Молекулярные механизмы лучевой болезни. -М.: Медицина, 1984.
10. В.П.Щипков, Г.Н.Кривошеина Общая и медицинская генетика. М:
ACADEMIA, 2003.
11. В.Ю.Шанин. Клиническая патофизиология. Учебник для медицинских вузов. – СПб: Специальная литература, 1998.
12. Перспективы медицинской генетики/ Под ред. Н.П. Бочкова, М.: Медицина,
1982.
13. Симонов П.В. Темперамент, характер, личность. - М.: Медицина, 1985.
14. Форель Ф., Мотульски А. Генетика человека: В 3Т.: Пер. с англ.-М.:Мир
1990.
15. Маянский А.Н., Маянский Д.Н. "Очерки о нейтрофиле и макрофаге", Новосибирск Наука. 1989.
16. Ройт А., Бростофф Дж.,Мейл Д, Иммунология: Пер. с англ.-М.:Мир,2000.
17. Иммунопатология. В 3-х томах./ Под ред. У. Пола, М., ”Медицина”, 19871989гг.
18. Першин Б.Б. Стресс, вторичные иммунодефициты и заболеваемость, М., АО
ЗиС, 1994г.
19. Хаитов Р.М. и др. Экологическая иммунология, М., изд. ВНИРО, 1995г.
20. Константинов Н.А. Иммунные комплексы и повреждение тканей, ”Медицина”, 1996г.
21. Ярилин А.А. Основы иммунологии. Москва Медицина 1999г.
285
22. Хаитов Р.М. и др. Иммунология.М: Медицина ,2000.
23. Сиротинин М.Ф., "Состояние капиллярного русла при некоторых видах сосудистой патологии", Киев.-1981.
24. Проценко В.А., Шпак С.И., Доценко С.М. "Тканевые базофилы и базофильные гранулоциты крови". М.,-1987.
25. Джексон Н.П. "Периферическое кровообращение", М:,"Медицина"-1982.
26. Чернух А.М. и др. " Микроциркуляция ", "Медицина".-1984.
27. Сверчкова В.С. "Гипоксия - гиперкапния и функциональные возможности
организма". Изд. "Наука" Казахской ССР. Алма-Ата, 1985г.
28. Агаджанян Н.А., Гневушев В.В., Катков А.Ю.”Адаптация к гипоксии и биоэкономика дыхания” Москва, изд.УДН, 1987г.
29. Агаджанян Н.А., Елфимов А.И.”Функции организма в условиях гипоксии и
гиперкапнии”, М., ”Медицина”, 1986г.
30. Механизмы развития и компенсации гемической гипоксии. Под общей ред.
М.М.Середенко, Киев,”Наукова думка”, 1987г.
31. Зиновьев Ю.В., Козлов С.А., Савельев О.Н. ”Резистентность и гипоксия”,
Изд. Красноярского университета, 1988г.
32. Мусил Я. Основы биохимии патологических процессов: Пер.с чешского.-М.:
”Медицина”, 1985.
33. Лишко В.К., .Шевченко М.И ”Мембраны и жизнь клетки.” Киев, ”Наукова
думка”.- 1987.
34. Браин Д.Д, МоженокТ.П. Неспецифический адаптационный синдром клеточной системы.// Изд. ”Наука”.- Ленинград.- 1987.
35. Физиология гормональной рецепции./ Под ред. В.Г.Шляпиной Л.: Наука.1986.
36. Проценко В.А., Шпак С.И. Ингибиторы протеолитических ферментов - протекторы клеточный повреждений.// Физиол.ж. СССР.-1985.
37. Хорст А. Молекулярные основы патогенеза болезней: Пер. с польск.- М.:
”Медицина”, 1982.
38. Кавешникова Б.Ф. "Современные направления развития коагулологии".//
"Гематология и трансфузиология", 1985г.
39. "Воспаление" Руководство для врачей /Под ред. В.В. Серова, В.С. Паукова,
М., "Медицина",1995г.
40. Пекарский Д.Е. "Гипертермические реакции", "Здоровье", Киев, 1981г.
41. Лоурин М.И. "Лихорадка у детей", М., 1985г.
42. Максимович В.А. "Эрготермическая устойчивость человека". Киев, 1985г.
43. Веселкин П.И. Лихорадка. /БМЭ/, под ред. Б.В.Петровского, М., 1980.
44. Гомеостаз /под ред. А.Д. Горизонтова/, М., Медицина, 1981, 257с.
45. Андрющенко Е.В., Алиферова В.Ф. Субфебрилитет, К., ”Здоров,я”,1986г.
46. Дворецкий Р.И. Дифференциальный диагноз при лихорадках в клинике
внутренних болезней, М., Медицина, 1993г.
47. А.Ш. Зайчик, Л.П. Чурилов.Механизмы развития болезней и синдромов.
Книга 1 «Патофизиологические основы гематологии и онкологии» Учебник
для медицинских вузов.-ЭЛБИ-СПб.-2002.
286
48. М.Э.Вуд, П.А.Банн Секреты гематологии и онкологии. :СПб: Невский диалект, 2001.
49. Внутренние болезни в 10 книгах. Книга 8. Онкология и эндокринология.
Ред. Т.Р. Харрисон, Е. Браунвальд и др., М., Медицина, 1996г.
50. Колосов А.Е. Современные классификации опухолей. Кишинев, 1990г.
51. Коган А.Х. Наследственность и развитие опухолей. Моск. мед. акад. им.
И.М.Сеченова, М., 1991г.
52. Онкология.- М: ВИНИТИ.- (Итоги науки и техники)/ Сер. "Онкология",
Т.19: Макрофаги и противоопухолевый иммунитет, 1990г.
53. Сейц И.Ф., Князев П.Г.Молекулярная онкология.-Л.:Медицина, 1986.
54. А.Ш. Зайчик, Л.П. Чурилов.Основы патохимии .Учебное пособие для медицинских вузов.-ЭЛБИ-СПБ.-1999.
55. В.Дж.Маршал. Клиническая биохимия М.-СПб.: «Издательство БИНОМ» «Невский диалект», 2000.
56. Дж. Теппермен, Х. Теппермен. Физиология обмена веществ и эндокринной
системы. Москва, "Мир", 1989г.
57. С.М.Лейтес. Проблемы регуляции обмена веществ в норме и патологии, М.,
1978 г.
58. Врожденные и приобретенные энзимопатии / Под ред. Т. Ташева. М., "Медицина". 1981г.
59. Хорст А. Молекулярные основы патогенеза болезней / Пер. с англ.М.:Медицина, 1982
60. Кон Р.М.,Рот К.С. Ранняя диагностика болезней обмена веществ:Пер. с
англ.-М.:Медицина, 1986.
61. Эндокринология / Под ред. Н.Левина.-М.:Практика,1999.
62. Потемкин В.В.Эндокринология.-М.:Медицина,1993.
63. Калинин А.П. Первичный гиперпаратиреоз.-Бишкек:Илим,1992.
64. Щитовидная железа: фундаментальные аспекты / Под ред. А.И.Кубарко и
S.Yamashita.-Минск,1998.
65. Холодова Е.А. и соавт. Справочник по клинической эндокринологии.Минск:Белорусь, 1998.
66. Руководство по клинической эндокринологии/Под ред. Н.Т.Старковой.СПб.:Питер,1996.
67. Васильев В.Н. Здоровье и стресс.- М.: Знание, 1991.
СОДЕРЖАНИЕ
1.Предмет, задачи и методы патофизиологии.Нозология,
этиология и патогенез. ………………………………… ….
7
287
2. Общая нозология …………………………………………..
3.Патогенное действие факторов внешней среды………………
3. Роль наследственности и конституции в патологии………….
4. Роль реактивности в патологии.Патология иммунологической реактивности. ………………………………………
5.Аллергия………………………………………………………
6.Типовые нарушения периферического кровообращения…….
7. Патофизиология клетки………………………………………
8. Гипоксия…………….…………………………………………
9. Патофизиология воспаления……………………………………
10. Лихорадка……………………………………….. ……………
11. Патофизиология тканевого роста.Опухоли…………………...
12. Патофизиология обмена веществ ……………………………..
-Патофизиология энергетического обмена …………………
-Патофизиология углеводного обменов…………………….
-Патофизиология азотистого и липидногообменов…………...
-Патофизиология водно-электролитного обмена…………...
-Патофизиология обмена макро-, микроэлеметов,витаминов..
-Патофизиология кислотно-щелочного равновесия…………
14
32
47
58
92
105
121
146
163
187
194
221
225
229
241
252
260
271
288