***** 1 - clinicpharm
реклама

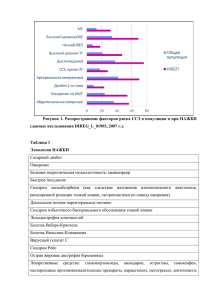
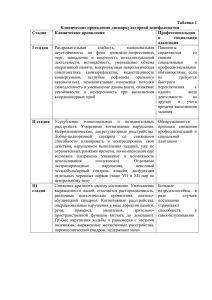
ПОЛЯКОВА С.И. 2016 Холестатический гепатит Синдром Алажилля, ПСВПХ 13 типа, несиндромальная дуктулярная гипоплазия , цитрина недостаточность, ARC синдром, галактоземия, фруктоземия, нарушение синтеза желчных кислот Гепатоцеллюлярная карцинома Тирозинемия А1АТ недостаточность Porphyria cutanea tarda Гликогеновая болезнь Болезнь Вильсона гемохроматоз Жировая дистрофия Болезни цикла образования мочевины, болезни β-ОЖК, абеталипопротеинемия, липодистрофия ДЛКЛ (б. Вольмана, накопление эфиров холестерина) лизосомальных субстратов (Гоше, Помпе, Данон, Фабри, Нимана-Пика) Narkowitz et al 2009 % 100 80 60 40 20 0 Коаг/ патия Энцеф/п Желтуха Гепато мегалия Сплено мегалия Асцит желтуха +/- коагулопатия ПВ> 50 “, ПТИ <70% энцефалопатия Рвота Инверсия сна Нарушение вскармливания Агрессивное или заторможенное поведение % 100 Гипогликемия и печеночная ЭП 80 60 все 40 20 В-ОЖК ПСВПХ БЦОМ С.Алажилля ГБ С.Кароли Б. Гл/Неогенеза А-1-АТН Митохондриальные Тирозинемия Органические B-ОЖК Ацидурии (ПА, ММА) ГБ болезни накопления Порт. Тромбофилия гипертензия Бадда-Киари 0 Коаг/п Энцеф/п Желтуха ЛБН Гепато мег Сплено мег Асцит, Mts декомпенсация Коагулопатия (II, VII, IX,X) • Кровотечения, ВЖК Энцефалопатия (недостаточность детоксикационной функции, системы глютатиона), аммониогенеза • Судороги, метаболическая эпилепсия, рвота, летаргия, Reye подобный синдром Билиарная недостаточность • Мальабсорбция, стеаторея, дефицит ЖР витаминов, атаксия, ахоличный стул недостаточность белковосинтетической функции, гликогенолиза и синтеза гликогена, βОЖК (глюконеогенеза) • Дефицит альбумина (отеки), трансферрина (СПЖ), церулоплазмина, гипогликемия, нарушение аминокислотного обмена Тирозинемия 1 типа Вероятность выживания % Возраст первых симптомов (г) наследственное заболевание аминокислотного обмена (аминоацидопатия), обусловленное недостаточностью фермента фумарилацетоацетатгидролазы (FAH)(ОМИМ +276700) До появления нитизинона До 1992 г Умирали Трансплантация Тансплантация и лечение нитизиноном Скрининг НТ1 и лечение до ОПН Эра Орфадина с 1992 г. Скрининг НТ и раннее лечение Диагноз установлен в возрасте 5 лет 3 года Альфафетопротеин на фоне лечения орфадином Алина С. Никита М. 1000000 Дима К. Дима Б. 100000 Лена К. Толя А. 10000 Надя Б. Таня Б. 1000 Саша К. Алеша Ч. 100 Маммед Э 10 Алла П. Раббия И 1 Кристина Д 0 1 2 3 4 5 0-1 через 2 недели, 1…5 интервал 0,5 года Гликогеновая болезнь = гепатомегалия + гипогликемия + кукольный habitus 1Б Гирке 4 тип Андерсена 1а тип Болезни β –окисления жирных кислот: Миопатия Кардиомиопатия Гипераммонемия Гиперурикемия Повышение КФК Дикарбоновая ацидурия Повышенные октаноил и гексаноил карнитина Дефекты среднецепочечной ацилКоАдегидрогеназы Детей без лечения, как правило, умирают от сердечной или дыхательной недостаточности в возрасте до трех лет. Существуют три формы TFP дефицита: “ранняя”, “инфантильная” и “мягкая”. Неонатальная форма первые симптомы метаболического кризиса: гипогликемия экстремальных сонливость изменения в поведении раздражительном настроении мышечная слабость плохой аппетит лихорадка тошнота понос рвота метаболический ацидоз > 20 нозологических форм ГИПОГЛИКЕМИЯ + ОСТРАЯ ПЕЧЕНОЧНАЯ НЕДОСТАТОЧНОСТЬ КАРДИОМИОПАТИЯ CT карнитин транспортер CPTI карнитин пальмитоил трансфераза CACT карнитин / ацилкарнитин транслоказа CPTII карнитин пальмитоил трансфераза + + + + + + + + VLCAD очень дл/цеп. ацКоА ДГ MCAD ср/цепочечная ацКоА ДГ + + + + + + LCHAD/MTP дл/цепоч. - 3 гидроксиацил– SCHAD короткоцеп.3- гидроксил КоА ДГ + + MAD множественная ацКоА ДГ + Дефицитный энзим КоА ДГ /михондр. трифункциональный белок ОСТРЫЙ РАБДОМ ИОЛИЗ СЛАБО СТЬ ДРУГИЕ НАРУШЕНИЯ + + + ПКА + + РЕТИНОПАТИЯ, НЕЙРОПАТИЯ, МАЛЬФОРМАЦИИ ГИПЕРИНСУЛИНИ ЗМ + + МАЛЬФОРМАЦИИ * - часть больных могут иметь все признаки или никакие при высокой остаточной активности фермента Органические кислоты в моче Ацилкарнитины Дефицитный энзим CT карнитин транспортер Плазма Моча НЕТ СВОБ. КАРНИТИНА, С0 (+ двукарбоновые кислоты) С6-С12 насыщ. и ненасыщ. ДКК CPTI карнитин пальмитоил трансфераза CACT карнитин / ацилкарнитин транслоказа CPTII карнитин пальмитоил трансфераза С18:1; C18:2; C16; C16-DC; C18:1-DC С6-С12 насыщ. и ненасыщ. ДКК ТЯЖЕЛЫЕ CPTII карнитин пальмитоил трансфераза ↑(С16+С18)/С2** СРЕДНЕЙ ТЯЖЕСТИ VLCAD очень дл/цеп. ацКоА ДГ С16:1; C14:2; C14:1;C18:1** MCAD ср/цепочечная ацКоА ДГ С10:1; C8; C6 Гексаноил-, суберил-, фенилпропиноил- ДКК [suberic>adipic], КТ SCAD короткоцеп АцКоА-ДГ C4 Бутирил- Этилмалоновая, метилянтарная, КТ LCHAD/MTP дл/цепоч. - 3 гидроксиацил–КоА ДГ 3-гидрокси-двукарбоновые кислоты /михондр. трифункциональный белок C18:1-OH; C18-OH C16:1-OH; C16:-OH** SCHAD короткоцеп.3- гидроксил КоА ДГ С4-OH 3-гидроксимасляная, 3-гидроксиглютаровая ДКК MAD множественная ацКоА ДГ:ТЯЖЕЛАЯ C4;C5;C5-DC; C6; C8; C10; C12; C14:1; C16;C18:1 СРЕДНЕ ТЯЖЕЛАЯ C6;C8,C10; C12 С6-С12 насыщ. и ненасыщ. ДКК Изобутирил-, изовалерил-, гексаноил-, суберил- Этилмалоновая, глютаровая, 2гидроксиглютаровая Этилмалоновая, адиповая, КТ * при среднетяжелых формах абс. кол-во в норме, соотношение меняется в катаболическую фазу Предполагаемая продолжительность голодания в разных возрастных группах возраст <6 мес 6-8 мес. 8-12 мес. 1-7 л >7 л Начало голодания 4 утра полночь 19 ч 17 ч 15 ч Продолжительность 8ч 15 ч 20 ч 24 ч 24 ч Прекращать при достижении гипогликемии!!! Жалобы: тошнота, боли в животе, слабость, потливость Анамнез болезни: в 3 года потеря сознания, судороги по утрам, грудное вскармливание до 2 лет, свободное, на фоне фебр. Температуры в 2 г 3 мес. первый эпизод тонич. Судорог – впервые зафиксирована гипогликемия 1,5 ммоль/л. Рецидивы судорог на фоне перерывов в еде более 4 часов. Жировая дистрофия печени. Поступила с Ds/ Гликогеновая болезнь? Анамнез жизни: близкородственный брак, токсикоз 1-3 триместр, роды оперативные x 4 мес. Энцефалопатия АЦИДОЗ КЕТОНУРИЯ ГИПОГЛИКЕМИЯ ↑ЛАКТАТ ↑ АЛТ И АСТ ↑ КФК ↑ МОЧЕВАЯ КИСЛОТА ↓ ЦИТОПЕНИЯ ПОТЕРЯ ВЕСА БЦОМ Органич. ацидурии Болезни βокисления жирных кислот Синдром Пируват гиперинсулинизм карбоксилазная а с ГА недостаточность +/(+) - + + +/+ + +/+ +/+ + + + - + ++ + + +/- - + + - - + deficiency<1 :2,000,000 CPS1 deficiency 1:1,300,000 OTC deficiency 1:56,500 ASS1 deficiency 1:250,000 ASL deficiency 1:218,750 ARG deficiency 1:950,000 NAGS Суммарно 1:8000 Аргинин (п) Цитруллин (п) Гомоцитруллин (моча) Глютамин (смж, п) Орнитин (п) Оротовая (моча) кислота Аргинина сукцинат (моча) 1 NAGS ↓ ↓n n ↑-↑↑ n ↓-n Нет дан. 2 СPS1 ↓ ↓n n ↑-↑↑ n ↓-n Нет дан. 3 OTC ↓ ↓n n-↑ ↑-↑↑ n ↑↑-(n- у новор.) Нет дан. 4 ↓ ↑↑↑ n ↑↑↑ n ↑-n n 5 ASS (CTLN1) ASL ↓ ↓ n ↑-n n ↑-n ↑↑↑ 6 ARG1 ↑ n ↑-n n ↑-n Нет дан. 7 HHH n-↑↑ новор ↑↑↑-> 1 мес n n ↑↑ ↑-↑↑ ↑-n ↑-n 8 9 Citrin def. GS def. ↑ N ↑ n n n n ↓↓ ↑↑, но не у новор. n n n n n n 10 THAN n-↓ n-↑ n n n Нет сведений Нет дан. 11 P5CS Def. n-↓ ↓-n n ↑-n ↓-n n n УЗИ , МРХПГ, холангиография расширение желчных протоков? Расширены протоки Не расширены протоки внепеченочные причины ГГТП сыворотки крови нормальная ГГТП зуд? нет нарушение синтеза первичных ЖК есть ПСВПХ 1,2 типы гепатит А Лекарст. гепатит (токсический) ТМС мочи на первичные и аномальные желчные кислоты повышенная ГГТП измененные протоки склерозирующий холангит молекулярно-генетическое исследование синдром Алажилля α1 антитрипсиновая недостаточность муковисцидоз лекарственный гепатит ПСВПХ 3 Аутоимм. холангит биопсия печени, холангиография холангит 85% вода, 15% сухой остаток: • на 50-67% из солей желчных кислот (ХДХК на 45% и ХК 31%) Конъюгированные желчные кислоты являются растворителями. ЖК обеспечивают поддержание холестерина желчи в растворенном виде, препятствуя его кристаллизации и образованию холестериновых камней 5 6 схема каналикулярного транспорта желчи из гепатоцита, гены и белки, ответственные за синтез и трансмембранный перенос желчи [5]: 1. ген ATP8B1(перенос фосфотидилсерина, PS) – ПСВПХ 1 тип 2. ген ABCB11(BSEP, bile salt export pomp, АТФ зависимый транспорт таурин- и глицинконьюгированных ЖК через каналикулярную мембрану) – ПСВПХ 2 тип 3. ген ABCC2 (MRP2) – Дубина-Джонсона синдром 4. ген ABCB4 (MDR3, multidrug resistence protein) нарушение экскреции фосфотидилхолина (PC) ПСВПХ 3 тип 5. OATP1B1, OATP1B3 – Ротора синдром 6. дефект образования первичных желчных кислот Синдром Алажилля Перегрузка железом Билиарная атрезия Несиндромальная дуктулярная гипоплазия Habitus = 50% диагноза Habitus + anamnesis = 80% диагноза Синдром внутрипеченочного холестаза с установленными мутациями ВНУТРИКЛЕТОЧНЫЙ ДУКТАЛЬНЫЙ Муковисцидоз С. Алажилля семейный Неонатальный внутрипеченочный холестаз -1 (с.Байлера) синусоидальный ПСВПХ 2-3 типов холестаз с ихтиозом Дефект синтеза желчных MDR-3 deficiency Прогрессирующий кислот Протопорфирия Методы визуализации PFIC-1 PFIC-2 MDR3 deficiency (PFIC-3) ЗУД да да средний ГГТП норма норма высокий Желчные кислоты в крови повышены повышены повышены Генная диагностика + + + Гистология Порт. Тракта N N фиброз Гистология каналикулярный холестаз гигантоклеточный гепатит неспецифический холангит Клинические Ассоциации диарея, мальабсорбци, панкреатит нет нет прогноз вариабельный плохой плохой (цирроз + порт. гипертензия) Дуктулярная гипоплазия при СА До 6 мес. у 60% больных, старше - у 95% Синдром исчезающих желчных протоков Синдром Алажиля (×10), возраст 3 месяца (×20), иммуногистохимическая реакция с цитокератином (CK19) Заместительная терапия дефицита жирорастворимых витаминов Лечение витаминного дефицита Препарат/доза цель Вит. К (кровоточивость) Фитоменадион, 1 мг/день (осторожно анафилакися!) Показатели времени свертывания Вит. Д (гипокальцемия, 10% глюконат кальция 0,1тетания, карпо-педальный 0,3 мл/кг, ВВ медленно спазм, рахит) 1,25-дигидрохолекальциферол, внутрь Нормализация ионизированного кальция 0,25-1,00 мкг/день Вит Е. (демиелинизация, атаксия) альфа-токоферол ацетат. внутрь 50 мг Восстановление содержания в сыворотке Вит. А (ночная слепота, кератиновая недостаточность) Ретинола ацетат, внутрь 2,5 тыс. Ед Холестатический гепатит Синдром Алажилля, ПСВПХ 13 типа, несиндромальная дуктулярная гипоплазия , цитриновая недостаточность, ARC синдром, галактоземия, фруктоземия, нарушение синтеза желчных кислот Гепатоцеллюлярная карцинома Тирозинемия А1АТ недостаточность Porphyria cutanea tarda Гликогеновая болезнь Болезнь Вильсона гемохроматоз Жировая дистрофия Болезни цикла образования мочевины, болезни β-ОЖК, абеталипопротеинемия, липодистрофия Накопление липидов (б. Вольмана, накопление эфиров холестерина) лизосомальных субстратов (Гоше, Помпе, Данон, Фабри, Нимана-Пика) Polyakova1963@list.ru 8-916-226-40-20 Полякова Светлана Игоревна