интегрально-сорбционный метод контроля состава сточных вод
реклама

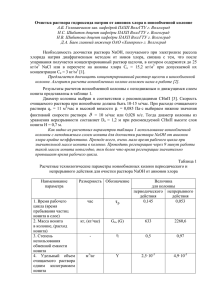
УДК 628.3:658.56 ИНТЕГРАЛЬНО-СОРБЦИОННЫЙ МЕТОД КОНТРОЛЯ СОСТАВА СТОЧНЫХ ВОД (ЭКОЛОГИЧЕСКИЙ СТОРОЖ) Е.В. Веницианов, Е.Н. Тарасова Институт водных проблем РАН, г. Москва, Россия Р.Х. Хамизов Институт геохимии и аналитической химии им. В.И. Вернадского РАН, г. Москва, Россия Скрытые залповые сбросы токсичных веществ в природные водоемы, к которым нередко прибегают промышленные предприятия, представляют серьезную экологическую опасность [4]. В связи с этим контроль качества сточных вод, отводимых в водоемы, имеет большое природоохранное значение. Хотя основное внимание уделяется разработке автоматизированных инструментальных систем мониторинга [3], проблема создания таких систем далека от решения вследствие их чрезвычайной дороговизны, отсутствия чувствительных сенсорных датчиков-анализаторов и надежных средств автоматизации анализа. Предложенный интегрально-сорбционный способ определения состава сточных вод не является альтернативой системам оперативного контроля, однако в сочетании с соответствующим математическим обеспечением позволяет с достаточной для практики точностью восстанавливать качество сточной воды за определенный расчетный период работы сорбционной колонки. При сорбции загрязняющего вещества его распределение по слою сорбента отражает изменение концентрации этого вещества в сточной воде по времени. Это распределение определяется разными способами: напрямую спектрофотометрически, изготовив колонку из стекла или сделав ее разборной, состоящей из нескольких секций, и затем осуществляя регенерацию в каждой секции отдельно, определяя содержание сорбированного компонента в регенерате. Выбор сорбента и форма его исходного состояния зависят от состава сточной воды. Сточная вода подается на вход колонки по байпасной схеме, а время сорбции выбирается экспериментально или на основе математической модели процесса. Если известны физико-химические условия сорбции, то возникает сугубо математическая проблема: восстановить переменную входную концентрацию загрязняющих компонентов по известному итоговому распределению их концентраций по слою сорбента. Скорость продвижения по колонне («фазовая» скорость) точек концентрационного профиля, соответствующих постоянным концентрациям некоторого i-го компонента в растворе или в сорбенте ai x, t = Const или с( x, t ) Const , выражается соотношением [7] v (1) Q c const , ai ci где v – линейная скорость раствора, отнесенная к сечению колонны. Пусть некоторый раствор, например, часть потока сточной воды предприятия, постоянно со средней скоростью v пропускается через сорбционное устройство. Если в результате какого-либо процесса, например, скрытого залпового сброса загрязняющего компонента i, в слое сорбента, равновесном с макросоставом сточной воды, например, находящегося преимущественно в форме иона Me z , сформирована зона (пик) микрокомпонента i, то для определения фазовой скорости переноса этого пика можно воспользоваться соотношением ai a i , (2) Г i Me z ci c где Г i - коэффициент Генри для линейной изотермы, i Me z - коэффициент однократного разделения компонентов, c – суммарная a – емкость ионита, концентрация всех компонентов в растворе. При известном значении фазовой скорости, определяемой по формулам (1) и (2), и с использованием экспериментальных данных о перемещении середины пика микрокомпонента в колонке x , определяемом с помощью послойного анализа состава ионита, всегда можно рассчитать время t x Q , то есть восстановить время залпового сброса. По данным о количестве вещества и ширине пика можно восстановить локальную концентрацию и протяженность скрытого сброса. Изложенная идея остается в силе для случая нескольких микрокомпонентов, а также для нескольких последовательных сбросов. Возможность использования описанного метода в реальных условиях обусловлена разрешением двух самостоятельных проблем. Первая из них связана с обратной задачей динамики ионного обмена, относящейся к классу некорректных задач, решение которых требует привлечения современных методов решения некорректных задач математической физики [1]. Другой проблемой, с которой мы сталкиваемся при описании динамики ионного обмена из растворов с малыми концентрациями целевых компонентов (соизмеримых с ПДК в природных водах), является повышенная селективность сорбентов к этим компонентам, значительно превосходящая оценки, которые могут быть сделаны из известных литературных данных по равновесиям обмена на ионитах [8]. Такое нарушение линейности изотерм обмена при малых и сверхмалых концентрациях может быть связано со многими факторами, в том числе с неидеальностью (полифункциональностью) обычных ионитов. Отметим, что эффект инверсии селективности ионитов при обмене ионов одинакового заряда в литературе обсуждался 2,6 . Ранее был предложен и экспериментально апробирован подход к решению этой проблемы, обеспечивающий принципиальную возможность формирования и перемещения в слое ионита зоны целевого загрязняющего компонента с помощью специального ввода в состав исходного ионита микродобавок («зарядка сорбента») определяемых компонентов или близких к ним по свойствам «сильноудерживаемых» компонентов. В работе использовали катионит КУ-2х8 и раствор, моделирующий катионный состав реки Москвы: Na+- 1,74; K+-0,179; Ca2+- 2,0; Mg2+- 0,494 мг-экв/л. В качестве примеров загрязняющих микрокомпонентов были взяты ионы Cu2+ и Ni2+ в концентрациях 5…10 мг/л, каждый в виде соответствующих сульфатных растворов. Согласно [5], использовали исходную Li-форму катионита, так как катионит КУ-2 менее селективен к иону Li+, чем к иону Na+ – наименее сорбируемому из макрокомпонентов исследуемого раствора. Это позволяет по скорости переноса по колонне переднего фронта обмена Na+–Li+ уточнять среднее значение линейной скорости пропускания раствора в случаях, когда последняя может неконтролируемым образом измениться. Образцы катионита Li-КУ-2 с микродобавками компонентов Cu2+ или Fe3+ на уровне 0,0046 мг-экв/г (~ 0,5% обменной емкости) готовили в статических условиях. Эксперименты проводили на ионообменной колонке L = 100 cм S = 0,65 см2 со съемным фильтрующим дном при длине слоя катионита l = 45…71 см. Объемная скорость пропускания раствора в пределах одного опыта оставалась постоянной и варьировалась от опыта к опыту в пределах 300…600 мл/ч. В заданные моменты времени прерывали подачу модельного раствора и в течение определенного времени через колонки пропускали раствор того же макросостава с добавкой Cu2+ (или смеси Cu2+ + Ni2+) в концентрации 5…10 мг/л. Этим имитировали «залповый выброс» контролируемого токсичного микрокомпонента с превышением ПДК в 5…10 раз. Отработанный катионит по специально разработанной методике равномерно выгружали порциями по 0,5…2 см слоя катионита в стеклянные бюксы, а затем с помощью различных известных аналитических методик определяли содержание элементов в пробах ионита. По полученным результатам строили кривые распределения концентраций компонентов по слою ионита. С ,м г-э к в /г i 4 12345- 3 6- 2 1 0 0 5 10 15 20 25 L, см Распределение компонентов в слое катионита КУ-2 – Li+ после пропускания модельного раствора в течение 36 ч v = 292 см/ч: 1 – Na+, 2 – Ca2+; 3 – Li+, 4 – Mg2+; 5 – K+; 6 – Cu2+ (СCuх50). В промежутке от t 1= 2ч до t2=4 ч раствор содержал ион-ЗВ (5 мг/л Cu2+) На рисунке представлены экспериментальные данные по распределению компонентов в слое катионита КУ-2 в Li+-форме после пропускания через него исследуемого модельного раствора в течение 36 ч. Ввод микрокомпонента«загрязнителя» (Cu2+ в концентрации 6,5 мг/л) соответствует интервалу времени от t1=2 ч до t2=4 ч. Распределение катионов макросостава модельного раствора соответствует обычным закономерностям и известным значениям ионообменных параметров для рассматриваемой системы. Для зоны Cu2+, находящейся преимущественно на фоне ионов Сa2+, наблюдается практически полное удерживание в начальном (лобовом) слое катионита, что для исследованной продолжительности процесса (t = 32 ч) соответствует Cu значению коэффициента разделения Сa 41,2, или константы равновесия ионного ~ ~ обмена K = 6,42. При сравнении этой величины с известными из литературы ~ ~ Cu значениями K Ca = 0,5…0,8 [8] очевидно весьма существенное расхождение. Этот эффект может быть обусловлен неизбежным наличием в ионите, наряду с сульфоновыми группами, микропримесей групп с повышенной селективностью к ионам переходных металлов. В этом случае введение в ионит до начала процесса микроколичеств «сильноудерживаемых» ионов, в частности, Cu2+, должно приводить к устранению или ослаблению влияния примесей этих дополнительных функциональных групп. В присутствии микродобавки целевого компонента имеет место заметное смещение его концентрационной зоны вдоль слоя катионита за реальное время эксперимента, а равновесные характеристики ионообменников приближаются к известным из литературы [8], что позволяет использовать эти ионообменники для создания устройств непрерывного контроля динамики загрязнения природных вод. В настоящее время ведется работа по адаптации этого метода к реальным сточным водам. Библиографический список 1. Веницианов Е.В., Тихонов Н.А., Трубецков Н.К. Интегрально-сорбционный способ контроля состава сточных вод. //Водные ресурсы. 1996. Т.23. № 5.С.575-577 2. Иванов В.А., Малых Я.Н., Горшков В.И. Равновесие обмена щелочных металлов и водорода на ионите КУ-2x8 при малых содержаниях H+// Физ. химии. 1986. Т.60. № 3. С. 715-718. 3. Израэль Ю.А. Экология и контроль состояния природной среды. Л.: Гидрометеоиздат. 1979. 376 с. 4. О состоянии окружающей природной среды Российской Федерации в 1995 году.- Гос. Доклад Минприроды РФ 1996 г. М., 1996. 451 с. 5. Патент РФ № 2121673. Способ сорбционного контроля загрязнения водных объектов /Зеленская О.Б., Крачак А.Н., Веницианов Е.В., Хамизов Р.Х., Аргин М.А. - Опубл. 10.11.98, Бюл. № 31. 6. Райхенберг Д. Селективность ионного обмена. // В кн. Ионный обмен. /Под. ред. Я.Маринского. М.: Мир.1968. С. 104-173. 7. Сенявин М.М. Ионный обмен в технологии и анализе неорганических веществ. М.: Химия.1980. 272 с. 8. Тремийон Б. Разделение на ионообменных смолах. М.: Мир.1967.432 с.