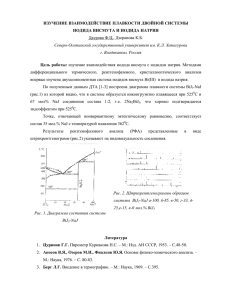
Министерство образования и науки Российской Федерации Федеральное государственное бюджетное образовательное учреждение
реклама

Министерство образования и науки Российской Федерации Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования «Кузбасский государственный технический университет имени Т.Ф. Горбачева» На правах рукописи СУРОВАЯ Виктория Эдуардовна ТЕРМО – ФОТО – И ГАЗОСТИМУЛИРОВАННЫЕ ПРЕВРАЩЕНИЯ НАНОРАЗМЕРНЫХ ПЛЕНОК ВИСМУТА, ОКСИДА МОЛИБДЕНА (VI) И СИСТЕМЫ НА ИХ ОСНОВЕ специальность 02.00.04 – физическая химия диссертация на соискание ученой степени кандидата химических наук Научный руководитель: д.х.н., профессор Черкасова Т.Г. Омск 2015 2 ОГЛАВЛЕНИЕ ВВЕДЕНИЕ…………………………………………………………………. 5 ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ…………………………………………. 12 1.1 Свойства висмута…………………………………………………… 12 1.2 Свойства оксида висмута (III)……………………………………… 25 1.3 Термостимулированные превращения наноразмерных слоев оксида молибдена (VI)…………………………………………………. 31 1.4 Термодинамика и кинетика химического окисления металлов…………………………………………………………………. 34 1.4.1 Условия сплошности пленок………………………………… 35 1.4.2 Кинетические закономерности окисления висмута….......... 38 ГЛАВА 2. МЕТОДИКА ЭКСПЕРИМЕНТА………………………………. 39 2.1 Приготовление образцов для исследований………………………. 39 2.2 Методы определения толщины пленок……………………………. 40 2.3 Метод оптической спектроскопии…………………………………. 42 2.4 Метод кварцевого микровзвешивания…………………………….. 43 2.5 Методика измерения контактной разности потенциалов………. 48 2.6 Электрофизические методы исследования……………………….. 50 2.7 Актинометрия источника излучения……………………………… 52 ГЛАВА 3. ИССЛЕДОВАНИЕ ТЕРМО - ФОТО – И ГАЗОСТИМУЛИРОВАННЫХ ПРЕВРАЩЕНИЙ НАНОРАЗМЕРНЫХ ПЛЕНОК ВИСМУТА, ОКСИДА МОЛИБДЕНА (VI) И СИСТЕМЫ НА ИХ ОСНОВЕ……………………………………… 54 3.1 Закономерности формирования оксида висмута (III) на поверхности наноразмерной пленки висмута……..…………………... 54 3.1.1 Изменение оптических свойств наноразмерных пленок висмута под действием температуры….………………………….. 55 3.1.2 Кинетические особенности окисления наноразмерных пленок висмута……………………………………………………… 61 3 3.1.3 Определение термоэлектронной работы выхода наноразмерных пленок висмута и оксида висмута (III) методом контактной разности потенциалов………………………………… 3.1.4 Модельные представления процесса окисления наноразмерных пленок висмута…………………………………… 3.2 Фотохимическое окисление наноразмерных пленок висмута…. 3.2.1 Спектры отражения оптического наноразмерных поглощения пленок висмута и 72 79 82 зеркального до и после облучения светом…………………………………………………… 82 3.2.2 Кинетические закономерности стимулированных светом превращений наноразмерных пленок висмута………………….. 84 3.2.3 Фотоэлектрические свойства систем Bi - Bi2O3…………… 89 3.2.4 Модельные представления стимулированного светом процесса окисления наноразмерных пленок висмута…………… 91 3.3 Модификация наноразмерных пленок висмута в атмосфере аммиака………………………………………………………………….. 95 3.3.1 Влияние газообразного аммиака на оптические свойства наноразмерных пленок висмута………………………………….. 3.3.2 Кинетические закономерности 95 взаимодействия наноразмерных пленок висмута с газообразным аммиаком…… 98 3.3.3 Контактная разность потенциалов для пленок Bi и BiN…. 103 3.3.4 Модельные представления процесса превращения наноразмерных пленок висмута в атмосфере газообразного аммиака при Т = 293 К……………………………………………. 3.4 Исследование термо- фото- и 105 газостимулированных превращений наноразмерных пленок висмута методом кварцевого микровзвешивания………………………………………………………. 108 3.5 Термостимулированные превращения в наноразмерной системе Bi – MoO3…………………………………………………........................ 113 3.5.1 Изменение оптической плотности и отражательной 4 способности наноразмерной системы Bi – MoO3 под действием температуры………………………………………………………… 113 3.5.2 Кинетические закономерности степени термического превращения наноразмерных пленок MoO3 в процессе термической обработки системы Bi – MoO3……………………… 119 3.5.3 Контактная разность потенциалов для наноразмерных пленок Bi и MoO3…………………………………………………… 122 3.5.4 Модельные представления термостимулированных превращений наноразмерных пленок оксида молибдена (VI) в системе Bi – MoO3…………………………………………………. 123 3.6 Образование центров окраски в наноразмерных пленках оксида молибдена (VI) под действием света………………………………….. 3.6.1 Влияние облучения на оптические 127 свойства наноразмерных пленок оксида молибдена(VI)………………….. 128 3.6.2 Кинетические зависимости степени фотохимического превращения центров окраски наноразмерных пленок MoO3…. 3.6.3 Модельные представления 130 фотостимулированных процессов в наноразмерных пленках оксида молибдена (VI)…. 133 ЗАКЛЮЧЕНИЕ…………………………………………………………….. 135 ОСНОВНЫЕ РЕЗУЛЬТАТЫ И ВЫВОДЫ…………………………….. 138 СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ…………………….. 140 ПРИЛОЖЕНИЕ…………………………………………………………….. 157 5 ВВЕДЕНИЕ Актуальность темы. В последнее время тонкопленочные покрытия на основе металлов, оксидов металлов, гетеросистем металл-оксид значительно улучшают характеристики транзисторов и интегральных схем, служат основой для создания эффективных лазеров, светоизлучающих и поглощающих элементов, электрохромных и фотохромных дисплеев, электрохромных зеркал, молекулярных фильтров, осуществляющих эффективную очистку и опреснения воды [1 – 5]. Одним из важнейших направлений применения висмута является производство полупроводниковых материалов для термоэлектрических приборов, самозаряжающихся гальванических и высокоэнергетических элементов, способных использоваться при высоких рабочих температурах [6, 7]. Оксид висмута (III) используют для окрашивания стекла и в производстве покрытий, поглощающих ультрафиолетовое и инфракрасное излучение [6 9]. Оксид молибдена (VI) используется в производстве металлического молибдена, который служит компонентом жаропрочных и коррозионностойких сплавов, а также применяется в высокотемпературных вакуумных печах сопротивления в качестве нагревательных элементов и теплоизоляционных материалов [10 – 11]. Выяснение закономерностей термо-, фото-, газо- стимулированных превращений наноразмерных пленок висмута, оксида молибдена (VI) и системы Bi-MoO3, представляет интерес в связи с созданием новых функциональных материалов, обладающих полезными для практического использования свойствами востребованных в ряде областей новой техники, в том числе нано-, сенсорной. Обозначенные выше аспекты определяют актуальность выполненных в настоящей работе исследований. Целью работы является изучение природы и закономерностей процессов, протекающих в индивидуальных наноразмерных пленках висмута, оксида 6 молибдена (VI) различной толщины и двухслойной системе на их основе в зависимости от температуры обработки, интенсивности облучения и времени воздействия газообразного аммиака. Для достижения поставленной цели решались следующие задачи: 1. Выяснить основные закономерности изменения оптических свойств наноразмерных пленок висмута, в результате воздействия газообразного аммиака при Т = 293 К и термической обработки в диапазоне длин волн = 190 - 1100 нм методом оптической спектроскопии. 2. Методом оптической спектроскопии в диапазоне длин волн = 190 1100 нм исследовать влияние интенсивности падающего света и времени облучения на спектры поглощения и зеркального отражения наноразмерных пленок висмута и оксида молибдена (VI). 3. Установить влияние размерных факторов на процессы термо-, фото- и газостимулированных превращений наноразмерных пленок висмута. 4. Установить фотостимулированных влияние размерных превращений факторов наноразмерных на процессы пленок оксида молибдена (VI). 5. Исследовать зависимость степени термического превращения наноразмерных пленок MoO3 в процессе термической обработки системы Bi MoO3 в интервале температур (Т = 373 – 673 К). 6. Определить методом контактной разности потенциалов значения термоэлектронных работ выхода наноразмерных пленок висмута, оксида висмута (III), нитрида висмута, оксида молибдена (VI). 7. Установить качественный состав продуктов, образующихся в процессе термической обработки, облучения светом, а также взаимодействия пленок висмута с газообразным аммиаком. Связь темы работы с планами НИР. Работа проводилась в соответствии с темпланом ФГБОУ ВПО «Кузбасский государственный технический университет имени Т.Ф. Горбачева» по государственному заданию Министерства образования и 7 науки Российской Федерации на выполнение научно-исследовательских работ на 2011-2014 г. № Г36-2012/3.1216.2011 «Полифункциональные материалы энергосберегающих и энергоэффективных технологий», а также в рамках базовой части ГЗ № 3478: «Проведение исследований и разработка технических решений обогатительного инновационных и утилизации техногенных металлургического продуктов, магнитной отходов горно- производств с фракции редкоземельных и получением элементов» Минобрнауки РФ на 2014 г. Научная новизна. 1. Впервые систематически исследованы закономерности изменения оптических свойств наноразмерных пленок висмута, в зависимости от времени воздействия газообразного аммиака при Т = 293 К и температуры в интервале длин волн = 190 - 1100 нм. 2. Впервые обнаружено влияние интенсивности падающего света и времени облучения на оптическую плотность и отражательную способность наноразмерных пленок висмута и оксида молибдена (VI). Впервые 3. установлено влияние размерных факторов термостимулированных превращений наноразмерной системы Bi - MoO3, а также наноразмерных пленок MoO3 в процессе термообработки системы Bi MoO3. 4. Впервые зарегистрировано уменьшение отражательной способности практически до нулевого значения – «эффект просветления» для наноразмерных системы Bi - MoO3 с толщиной индивидуальных подслоев Bi (d = 45 - 92 нм), MoO3 (d = 33 - 8 нм), в интервале длин волн (λ = 410 610 нм). 5. Определены значения контактных потенциалов (относительно электрода сравнения из платины) наноразмерных пленок висмута, оксида висмута (III), нитрида висмута, оксида молибдена (VI). Практическая значимость работы. Результаты исследований являются продуктивной основой для создания новых тонкослойных 8 регистрирующих сред, с управляемым уровнем термо- и фоточувствительности, светоотражающих и поглощающих покрытий, высоко селективных датчиков, принципиально новых материалов, стабильных в условиях коррозионного воздействия окружающей среды, а так же позволят прогнозировать и направленно изменять поведение наноразмерных пленок висмута и оксида молибдена (VI) за счет образования двухслойных объектов на их основе. Методы исследования и результаты работы используются в курсе лекций и лабораторном практикуме «Строение и свойства полифункциональных материалов и нанокомпозитов», «Наноматериалы и нанотехнологии», «Актуальные направления химической технологии» для студентов, обучающихся по направлению подготовки 240100 «Химическая технология», профиль «Химическая технология неорганических веществ» Института химических и нефтегазовых технологий Кузбасского государственного технического университета имени Т.Ф. Горбачева (акт внедрения ФГБОУ ВПО КузГТУ им. Т.Ф. Горбачева, г. Кемерово). Основными положениями, выносимыми на защиту являются: 1. Установленные закономерности в изменении оптических свойств наноразмерных пленок висмута в результате термо-, фото- и газостимулированных превращений, с толщиной слоев висмута (d = 3 - 120 нм), температурой (Т = 373 - 673 К), интенсивностью облучении светом ( = 360 нм, I = (1,8 - 7,0)·1015 квант·см-2·с-1), временем воздействия газообразного аммиака ( = 1 мин - 5400 час), взаимосвязь между ними. 2. Продуктом термо- и фотостимулированных превращений наноразмерных пленок висмута в атмосферных условиях является оксид висмута (III). 3. Продуктом взаимодействия наноразмерных пленок висмута в атмосферных условиях с газообразным аммиаком при Т = 293 К, является нитрид висмута. 9 4. Обоснование возможности направленно изменять оптические свойства наноразмерных пленок оксида молибдена (VI), висмутом в интервале температур (Т = 373 - 673 К), путем создания гетеросистемы Bi - MoO3 с различной толщиной индивидуальных подслоев, в атмосферных условиях. 5. Кинетические кривые степени термо- и фотостимулированного окисления наноразмерных пленок висмута удовлетворительно описываются в рамках линейного, обратного логарифмического, кубического и логарифмического законов. 6. Кинетические кривые степени превращения наноразмерных пленок висмута в атмосфере удовлетворительно газообразного описываются в аммиака рамках при Т = 293 К линейного, обратного логарифмического, параболического и логарифмического законов. Личный вклад автора. Результаты исследований, представленные в защищаемых положениях и выводах, получены лично автором. Идея исследования, постановка задач, анализ результатов обсуждались совместно с научным руководителем. Апробация работы. Результаты диссертационной работы опубликованы в 63 научных трудах, из которых 10 статей в ведущих рецензируемых журналах, входящих в перечень ВАК РФ, остальные в материалах Международных и Всероссийских конференций. Основные положения диссертации докладывались и обсуждались на конференциях различного масштаба, а именно: XV Всероссийской научной конференции студентов-физиков и молодых ученых «ВНКСФ – 15» (Кемерово, 2009г.); Международной научной студенческой конференции «Студент и научно-технический прогресс» (Новосибирск, 2009, 2010, 2011г.); I, VI Всероссийской, 54, 59 научно-практической конференции «Россия молодая» (Кемерово, 2009, 2014г.); XII Международной научно- практической конференции «Химия – XXI век: Новые технологии, новые продукты» (Кемерово, 2009г.); V, VI, VII Всероссийской конференции «Исследования и достижения в области теоретической и прикладной химии» 10 (Барнаул, 2009, 2011, 2013, 2014 г.); Всероссийской научно-практической конференции «Научное творчество молодежи». (Анжеро-Судженск, 2010, 2011, 2012 г.); XVII, XVIII, XIX, XX Международной конференции студентов, аспирантов и молодых ученых «Ломоносов» (Москва: МГУ, 2010, 2011, 2012, 2013, 2014 г.); 10th International Conference on Modification of Materials with Particle Beams and Plasma Flows (Tomsk, 2010), Международной Российско-Казахстанской конференции по химии и химической технологии (Томск, 2011 г., Караганда, Казахстан, 2012 г.); VI, VII, VIII Международной конференции молодых ученых: «Теоретическая и экспериментальная химия жидкофазных систем» (Иваново, 2011, 2012, 2013 г.); IV Всероссийской конференции по химической технологии с международным участием ХТ’12 (Москва, 2012 г.); Общероссийской с международным участием научной конференции «Полифункциональные химические материалы и технологии» (Томск, 2012 г.); V Школе-семинар сети ЦКП научным оборудованием «Исследования и метрология функциональных материалов» (Томск, 2012 г.); I, II Всероссийской конференции «Химия и химическая технология: достижения и перспективы» (Кемерово, 2012, 2014 г.); Всероссийской молодежной научной школы «Химия и технология полимерных и композиционных материалов» (Москва, ИМЕТ РАН, 2012 г.); Всероссийской молодежной научной конференции «Инновации а материаловедении» (Москва, ИМЕТ РАН, 2013 г.); II Всероссийской научно – технической конференции «Высокие технологии в современной науке и технике» (Томск, 2013 г.), Всероссийской научно-практической конференции молодых ученых, аспирантов и студентов «Современное состояние и проблемы естественных наук» (Юрга, 2014 г.); III Международной научно-практической конференции «Современные тенденции и инновации в науке и производстве» (Междуреченск, 2014 г.); XI Российской конференции молодых научных сотрудников и аспирантов «Физико-химия и технология неорганических материалов» (Москва, 2014 г.). Полный перечень публикаций включен в список литературы. 11 Объем и структура работы: Диссертация состоит из введения, 3 глав, заключения и выводов, списка цитируемой литературы из 162 наименований, приложения и содержит 156 страницы машинописного текста, включая 65 рисунков и 29 таблиц. Автор выражает глубокую благодарность и признательность научному руководителю, директору Института химических и нефтегазовых технологий Кузбасского государственного технического университета им. Т.Ф. Горбачева доктору химических наук, профессору Черкасовой Т.Г. за предоставленную тему, внимание, поддержку и консультации на всех этапах выполнения работы; доктору химических наук, профессору Кемеровского государственного университета Суровому Э.П., за дельные советы и полезные обсуждения результатов; кандидату химических наук, доценту Кемеровского государственного университета Бугерко Л.Н за проявленный интерес к работе, ценные указания и дискуссии; а также, кандидату химических наук, доценту КемГУ Борисовой Н.В., кандидату химических наук, ведущему инженеру КемГУ Бину С.В., кандидату химических наук, ассистенту КемГУ Рамазановой Г.О. за помощь экспериментальных исследований и моральную поддержку. в проведении 12 ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ 1.1 Свойства висмута Общие сведенья о висмуте. Висмут (Bismuthum), Bi – химический элемент 15-й группы (по устаревшей классификации — главной подгруппы V группы) шестого периода периодической системы химических элементов Д. И. Менделеева. Порядковый номер 83, атомная масса 208,98040 а.е. Распределение электронов в атоме висмута - 1s22s22p63s23p63d104s24p64d104f145d106s26p3. Степени окисления +3, +5 и -3, очень редко +1 и +2. Сродство к электрону 0,7 эВ. Атомный радиус 0,182 нм, ионные радиусы (в скобках указаны координационные числа) 0,110 нм (5), 0,117 нм (6), 0,131 нм (8) для Bi3+, 0,090 нм (6) для Bi5+, 0,213 нм для Bi3- [12]. Энергия ионизации (эВ) при последовательном переходе от Bi0 к Bi3+ 7,289; 16,74; 25,57; 45,3; 56,0. Поперечное сечение захвата тепловых нейтронов для висмута 3,4∙10-30м2. Электроотрицательность, эВ: 1,9 (по Полингу), 1,67 (по Оллреду), 4,69 (абсолютная). Эффективный заряд ядра: 6,30 (по Слейтеру), 13,34 (по Клименте), 16,90 (по Фрезе—Фишеру)[6]. Природный висмут имеет всего один изотоп — 209 Bi. Изотоп радиоактивен с периодом полураспада 1,9±0,2∙1019 лет. Кроме 209 209 Bi α- Bi известны и другие изотопы, большинство из которых имеет изомерные состояния. Долгоживущими считаются (0,368∙106 лет), 207 Bi (период полураспада 31,55 года), 208 Bi 210 Bi (3,04∙106 лет) — одно из изомерных состояний. Все остальные радиоактивны и короткоживущие: периоды их полураспада не превышают нескольких суток. Изотопы висмута с массовыми числами от 184 до 208 и от 215 до 218 получены искусственным путём (самый тяжелый из изотопов висмута — 210 Bi, 211 Bi, 212 Bi, 213 215 Bi, а не Bi и 214 209 Bi, как предполагали ранее), остальные — Bi — образуются в природе, входя в цепочки радиоактивного распада ядер урана-238, урана-235 и тория-232 [13]. Первые сведения о висмуте встречаются в трудах минералога и металлурга начала XVI в. Георга Агриколы. В 1739 г. немецкий химик И. Потт установил, что висмут является самостоятельным элементом, а не 13 разновидностью сурьмы, свиица или олова, как это полагали ранее. Символ элемента Bi впервые введен в химическую номенклатуру в 1819 г. шведским химиком Й.Я. Берцелиусом [14]. Содержание висмута в земной коре 2∙10-5 % по массе, в морской воде — 2∙10-5 мг/л. Мировая добыча/потребление едва превышает 6000 тонн в год (от 5800 до 6400 тонн в год). В рудах находится как в форме собственных минералов, так и в виде примеси в некоторых сульфидах и сульфосолях других металлов. Обладая высокой степенью изоморфизма с мышьяком и сурьмой, висмут часто входит в состав арсенидов и антимонидов никеля, кобальта, железа. Кроме того, для этого элемента характерно образование сульфовисмутитов свинца, серебра и меди [14]. В мировой практике около 90 % всего добываемого висмута извлекается попутно при металлургической переработке свинцово-цинковых, медных, оловянных руд и концентратов, содержащих сотые и иногда десятые доли процента висмута. Висмутовые месторождения встречаются крайне редко (Германия, Монголия, Боливия, Австралия, Перу, Россия, а также и др. странах). Известно более 70 минералов висмута. Из них промышленный интерес представляют: висмут самородный (содержит 98,5—99 % Bi), висмутин Bi2S3 (81,30 % Bi), тетрадимит Bi2Te2S (56,3—59,3 % Bi), козалит Pb2Bi2S5 (42 % Bi), бисмит Bi2O3 (89,7 % Bi), бисмутит Bi2CO3(OH)4 (88,5—91,5 % Bi), виттихенит Cu3BiS3, галеновисмутит PbBi2S4, айкинит CuPbBiS3[12,14]. Структура висмута. При обычных условиях висмут имеет ромбоэдрическую кристаллическую решетку (α-модификация), с периодом а=0,47457 нм и углом а=57°14'13". Энергия кристаллической решетки 208 мкДж/кмоль. При высоких давлениях, как следует из диаграммы состояния (рис. 1.1), образуется еще ряд модификаций (табл. 1.1). 14 Рис. 1.1. Диаграмма состояния висмута при высоких давлениях. Штриховые линии — приблизительные границы областей существования фаз [6,12]. Так, α-модификация (I) при давлении 2,57 ГПа и 25 °С переходит в моноклинную (II), при 2,72 ГПа — в (III), при 4,31—в (IV), около 5 — в (V), при 7,74 — в кубическую (VI) и при 30 ГПа —в (IX) [6,12]. Таблица 1.1 Характеристика некоторых кристаллических модификаций висмута [6,12] Показатель Кристаллическая решетка Пространственная группа Параметры: a, нм b, нм c, нм угол, град Число формульных единиц в ячейке Модификация I II VI VII Ромбоэдричес кая Моноклин ная Кубическая Тетрагона льная R3m C2m Im3m ___ 0,4746 0,3800 0,657 57,23 (α) 0,6674 0,6117 0,3304 110,33 (β) 2 4 2 0,568 8 15 Физические свойства. Висмут - блестящий серебристый металл с розоватым оттенком. Температура плавления 544,5 К, температура кипения 1833 ± 5 К. Энтальпия плавления ΔHпл = 10,48 кДж/моль. Энтальпия испарения ΔHисп = 179,1 кДж/моль, молярная теплоемкость С = 26,0 Дж/Кмоль [6]. В изломе висмут имеет грубозернистое строение, но при температуре 225— 250 °С он может быть подвергнут пластической деформации. Висмут является самым диамагнитным металлом, наиболее плохо проводит тепло, а при температуре ниже 7 К обладает свойствами сверхпроводника. При плавлении резко возрастают электропроводность и плотность висмута, последняя достигает 10,55 г/см, что связывают с наличием ковалентных связей в кристаллическом висмуте и их отсутствием в расплаве [6]. Плотность висмута при н.у. ρ = 9,79 мг/м3, по данным [14] при комнатной температуре, определенная пикнометрическим методом, ρ = 9,840 мг/м3, рентгеновским, ρ = 9,807 мг/м3. С повышением температуры плотность висмута возрастает, достигая максимума при температуре плавления, а при дальнейшем температуры вновь уменьшается (рис. 1.2) [14]. Рис.1.2 Зависимость плотности висмута от температуры росте 16 Удивительно, но в отличие от других металлов, которым свойственно при понижении температуры уменьшаться в объеме, висмут при затвердевании увеличивается в объеме на 3,3 %, что связано с наличием ковалентных связей в твердом висмуте и отсутствием их в расплавленном [6, 14, 15]. Тепловые и термодинамические. Температура плавления 544,5 К, температура кипения 1833 ± 5 К, характеристическая температура θD =117К [6,12,14]. Зависимость температуры плавления висмута от давления приведено в таблице 1.2 [14]. Таблица 1.2 Зависимость температуры плавления висмута от давления Р, МПа 0,1013 101,3 2026 405,2 810,4 1013 tпл, °С 271 267,5 263,5 256,6 238,0 288,8 Удельная теплота плавления висмута при 298 К ΔHпл = 54,69 кДж/кг. Удельная теплота сублимации при 1193 К в вакууме ΔHсубл. = 789,63 кДж/кг, при температуре кипения и обычном давлении 897,23 кДж/кг, при 298 К 947,6 кДж/кг, а при температуре плавления 1041,47 кДж/кг. Удельная теплота испарения ΔHисп. =857,49 кДж/кг (при температуре кипения) [14]. В таблице 1.3 представлена зависимость удельной теплоемкости cp висмута от температуры [14]. Таблица 1.3 Зависимость удельной теплоемкости висмута от температуры Т, К 10 100 293 544(тв) 544(ж) 873 1273 ср, Дж/(кгК) 9,8 110,2 128,95 141,51 142,35 157,42 175,42 Удельная электронная теплоемкость сэлр = [0,021 мДж/(мольК2)Т. Висмут является одним из худших проводников тепла среди металлов, его теплопроводность при 273 К составляет около 2 % теплопроводности серебра. Теплопроводность висмута в зависимости от температуры приведена в таблице 1.4 [14]. 17 Таблица 1.4 Зависимость теплопроводности висмута от температуры Поликристалл Т, К , Вт/(мК) 70 293 540 870 1070 12,6 8,41 7,29 16,4 15,1 Монокристалл Т, К 200 800 900 , Вт/(мК) 1,3 2,0 6,0 Теплопроводность висмута сильно анизотропна. При 291 К IIc = 6,65 Вт/(мК), а ┴c = 9,25 Вт/(мК) [14]. Температурный коэффициент линейного расширения висмута при 273 К α = 13,3710-6 К-1 при 293 К α = 13,410-6 К-1. При давлении 101,3 МПа температурный коэффициент линейного расширения составляет 12,9410-6 К-1 (293 К); 12,9810-6 К-1 (90 – 288 К); 14,610-6 К-1 (273 – 540 К) [14]. В таблице 1.5 представлена зависимость температурного коэффициента объемного расширения β висмута от температуры [14]. Таблица 1.5 Зависимость температурного коэффициента объемного расширения висмута от температуры Т, К 2 5 20 28 58 75 283 β106, K-1 0,0236 0,28 11,85 18,42 31,5 34,13 39,65 Поверхностное натяжение висмута в вакууме = 300 мН/м при 365°С и =343 мН/м при 600°С. Поверхностное натяжение жидкого висмута в зависимости от температуры указано в таблице 1.6 [14]. Таблица 1.6 Температурная зависимость поверхностного натяжения жидкого висмута t, °С 270 300 500 780 , мН/м 390 376 363 344 18 Динамическая вязкость жидкого висмута = 1,6610-3 Пас при T=549 K, кинематическая вязкость висмута = 1,95105 м2/спри T=548 K [14]. Электрические и магнитные. Отличительной особенностью висмута является более высокая удельная электрическая проводимость в жидком состоянии по сравнению с твердым; удельное электрическое сопротивление твердого висмута при 542 К ρ = 2,67 мкОмм, а жидкого при 545 К ρ= 1,27 мкОмм. Электросопротивление висмута при 278 К ρIIc= 1,27 мкОмм, a ρ┴c=0,99 мкОмм. Отношение удельного электросопротивления жидкого висмута к таковому для твердого составляет параллельно (II) оси с ρж/ρтв = 0,35, перпендикулярно (┴) оси с ρж/ρтв = 0,47 [14]. В таблице 1.7 представлена зависимость удельного электрического сопротивления ρ и удельной электрической проводимости висмута от температуры [14]. Таблица 1.7 Зависимость удельного электрического сопротивления ρ и удельной электрической проводимости висмута от температуры: T, K , МСи/м ρ, мкОмм 73 2,46 0,348 173 ____ 0,756 273 0,92 1,068 290,5 ____ 1,200 373 0,625 1,565 473 ____ 2,145 573 ____ 1,289 673 ____ 1,342 773 0,61 1,399 873 ____ 1,4525 1023 ____ 1,5355 1273 0,598 1,675 19 Интересной особенностью висмута является увеличение (на ~200 %) его электросопротивления (106,810-8 Омм при Т= 273 К) под влиянием магнитного поля, что используется для измерения индукции сильных магнитных полей [6,14]. По данным [14] температурный коэффициент электрического сопротивления висмута α = 4,210-3 К-1 (273 – 373 К). Температура перехода в сверхпроводящее состояние Тс 7 К, а при давлении 2,47 ГПа Тс = 4,2 К. Абсолютный температурный коэффициент ЭДС висмута при 273 К, е = 70 мкВ/К, при этом параллельно оси с, е = 100 мкВ/К, а перпендикулярно е = -53 мкВ/К. Температурный коэффициент ЭДС Е висмута по отношению к платине при температуре холодного спая 273 К и горячего 173,16 К и 373,16 К равна +7540 и – 7340 мкВ соответственно [14]. Коэффициент вторичной электронной max= l,15 эмиссии при ускоряющем напряжении первичных электронов 0,550 кэВ [14]. Висмут — самый диамагнитный металл, его магнитная восприимчивость , м3 /кг: -1,68410-8 (тв.), а при 293 К, = -1,3410-9 [6,14]. Оптические. Интегральный коэффициент излучения при T=298 К T = 0,048, при T=378 К T = 0,061. Отражательная способность (коэффициент отражения n D) пленок висмута представлена в таблице 1.8 [14]. Таблица 1.8 Коэффициент отражения (nD) пленок висмута толщиной 0,04—0,1 мкм , нм 103 – 73 103 – 59 nD, % 15 – 0,2 65 – 70 20 80 Угол падения Показатель преломления n для пленок висмута толщиной 0,04 – 0,1 мкм указан в таблице 1.9 [14]. 20 Таблица 1.9 Показатель преломления пленок висмута толщиной 0,04—0,1 мкм , нм 52 400 450 500 550 n 1,2 1,1 1,46 1,53 1,58 На рисунках 1.3, 1.4 приведены зависимости показателей преломления и поглощения от длины волны, для пленок висмута толщиной 0,3 мкм и 0,1 мкм, соответственно [14]. Рис. 1.3 Зависимость показателя преломления пленок висмута толщиной 0,3 мкм от длины волны Рис. 1.4 Зависимость показателя поглощения пленок висмута толщиной 0,1 мкм от длины волны 21 Механические свойства. Модуль нормальной упругости висмута при растяжении, по различным данным, колеблется в интервале £=32,0-=-34,0 ГПа. Для отожженного висмута значения модуля упругости в зависимости от температуры представлены в таблице 1.10 [14]: Таблица 1.10 Зависимость модуля упругости от температуры Е, ГПа 36,8 34,1 25,5 t,°C 187 19 230 Модуль сдвига, кг/мм: 1260. Поверхностное натяжение, Н/м: 0,376 (Т=573 К), 0,363 (Т=773 К), 0,344 (Т=1053 К)[6]. Механические свойства висмута чистотой 99,999 % в зависимости от температуры испытания приведены в таблице 1.11 [14]. Таблица 1.11 Механические свойства висмута чистотой 99,999 % tисп, °C В, МПа 0 14/15 50 , % φ, % ___ /0 1/0 24/26 24/26 33/2 100 17/19 67/16 95/11 160 14/16 45/65 88/99 210 ___ ___ ___ /12 /52 /96 Примечание. В числителе — свойства при скорости испытания 210-2 с-1, в знаменателе – при 210-1 с-1. Зависимость ударной вязкости KCU висмута от температуры указаны в таблице 1.12 [14]. Таблица 1.12 Ударная вязкость KCU висмута t, °C 18 66 93 149 204 232 KCU, кДж/м2 2 3 5 9 7 7 22 Временное сопротивление висмута чистотой 99,999 % при более низких температурах и скорости испытания 210-2 с-1 даны [14]. Таблица 1.13 Работа удара для образцов висмута без надреза В, МПа 13,7 9,8 3,9 tисп, °C -45 -100 -200 Температурная зависимость работы удара для образцов висмута без надреза представлена в таблице 1.14 [14]. Таблица 1.14 Работа удара для образцов висмута без надреза t, °C КС, Дж 18 66 93 198 204 232 1,32 2,00 4,00 6,99 5,99 5,99 В таблице 1.15 приведена зависимость предела выносливости висмута при испытаниях на круговой изгиб от условий испытания [14]. Таблица 1.15 Предел выносливости висмута при испытаниях на круговой изгиб Скорость вращения образца, об/мин 2800 60 2 Число циклов t, °C R, МПа 103 -197 7,8 104 -196 6,9 105 20 14,7 106 20 10,8 107 20 7,1 105 20 14,7 106 20 8,8 107 20 4,7 105 20 5,9 106 20 2,9 23 Ползучесть при испытаниях на изгиб [14]: t, °C . . . . . . . . . . . . . . . 66 Нагрузка , МПа . . . . . . . . . 4,20 121 177 204 3,15 2,45 2,10 2,8 2,8 2,8 Скорость установившейся ползучести 105 %/с . . . . . . . 2,8 Твердость по Бринеллю отожженного висмута НВ = 94,2 МПа, микротвердость H=177 МПа [14]. Коэффициент Пуассона = 0,330, сжимаемости = 2,8610-11 Па-1[14]. Химические свойства. Висмут почти не окисляется при обычной температуре в сухом воздухе, длительное время, сохраняя серебристо-белый цвет, а во влажном воздухе покрывается тончайшим слоем оксида [14]. Во влажном воздухе постепенно покрывается буроватой пленкой оксидов. Заметное окисление начинается около 500°С. Выше 1000°С горит голубоватым пламенем с образованием Bi2O3 [12]. Оксид висмута (IV) Bi2O4 получается при окислении оксида висмута (III) ферроцианидом калия в концентрированном растворе едкого кали или персульфата аммония в разбавленном растворе едкого натра [14]. Оксид висмута (V) Bi2O5 — ангидрид не выделенной в свободном состоянии висмутовой кислоты НВiO3. Соли висмута (III) легко гидролизуются, переходя в основные соли, малорастворимые в воде [14]. Соединение висмута с водородом — висмутин, или гидрид висмута BiH3,— очень нестойкий ядовитый газ, разлагающийся уже при комнатной температуре; получается при действии НСl на сплав висмута с магнием [14]. Не реагирует с соляной кислотой и разбавленной H2SO4. С азотной кислотой образует нитрат [6], с концентрированной серной кислотой при нагревании – гидросульфат (BiH(SO4)2). Осаждение гидроксисолей висмута начинается при pH 1,6, полное осаждение достигается при pH = 4,8. Исключением являются перхлоратные растворы растворимости гидроксиперхлората Bi(OH)2ClO4 [12]. из-за высокой 24 С азотом висмут дает нитраты. При нагревании кристаллогидрата Bi(NO3)35H2O до 30°С начинает выделяться азотная кислота, при 75,5 °С кристаллогидрат распадается на жидкую фазу и основную соль состава Bi2O5N2O5H2O. Водой гидролизуется с образованием основных солей [14]. Со многими металлами (Na, К, Ru, Cs, Ca и др.) образует тугоплавкие интерметаллические соединения – висмутиды (Na3Bi, Mg3Bi), с сурьмой — непрерывный ряд твердых растворов, с легкоплавкими тяжелыми металлами (Pb, Sn, Cd и др.) — эвтектики с температурой плавления от 33 до 156°С. Электрохимический эквивалент для висмута со степенью окисления +5 равен 0,43316 мг/Кл, со степенью окисления +3 – 0,72193 мг/Кл [14]. Жидкий висмут незначительно растворяет фосфор. При воздействии сероводорода на оксид висмута образуется моносульфид висмута BiS — вещество, неустойчивое при нагревании на воздухе и в парах воды. Сульфид висмута Bi2S3 получается в виде черно-бурого осадка при действии сероводорода на растворы солей висмута или при сплавлении висмута с серой. Трехйодистый висмут BiI3 получают нагреванием висмута в парах йода или смеси йода и висмута в углекислоте или водороде [14]. Висмут легко образует галогениды. Однохлористый висмут BiCl— неустойчивое твердое вещество черного цвета, получается растворением металлического висмута в расплавленном BiCl. Двуххлористый висмут BiCl2 получается при медленном действии хлора на висмут или при восстановлении треххлористого висмута в процессе нагревания с фосфром, серебром, цинком, ртутью, оловом. Треххлористый висмут ВiС13 получают хлорированием металлического висмута или растворением Bi2O3 в соляной кислоте или висмута в царской водке [14]. Необходимо отметить, что сплавы на основе висмута характеризуются ферромагнитными свойствами ввиду чего, идут на изготовление мощных постоянных магнитов. Кроме того висмутовые покрытия имеют большое значение для производства так называемых «автоматных сталей», особенно нержавеющих, и очень облегчает их обработку резанием на станках- 25 автоматах [12,14,15]. Сплав 88% Bi и 12% Sb (сурьма) из этого сплава изготовляют быстродействующие усилители и выключатели. Соли висмута применяются при изготовлении красок для дорожных знаков, "вспыхивающих", когда на них падает луч автомобильной фары. Известные с давних пор косметические наклонности висмута проявляются сегодня в создании с помощью его солей перламутровой губной помады [6 – 9]. Расширение областей применения висмута выдвигает новые научнотехнические задачи, в частности, изучение изменения свойств изделий на основе наноразмерных пленок висмута в условиях агрессивного воздействия окружающей среды. 1.2 Свойства оксида висмута (III) Сесквиоксид висмута (оксид висмута (III)) Bi2O3 — лимонно-желтого цвета при обычной температуре. При нагревании становится оранжевым, а при охлаждении приобретает первоначальный цвет. В природе встречается в виде минерала бисмита или висмутовой охры (89,7 % Bi) [6,13]. Структура желтовато-белые оксида висмута кристаллы, (III). которые Оксид имеют висмута(III) четыре образует полиморфные модификации: стабильные — моноклинной α-Вi2O3 и гранецентрированной высокотемпературной -Вi2O3, а также метастабильные — тетрагональной βВi2O3 и кубической объемно-центрированной γ-Вi2O3 [6,13]. При нагревании моноклинная модификация α-Вi2O3 превращается при 730 °С в высокотемпературную -модификацию и при 825 °С оксид висмута плавится. В случае охлаждения из расплава отмечаются экзотермические эффекты при 824 °С, обусловленные кристаллизацией фазы β-Вi2O3 [13]. При охлаждении до 650 и 540 °С имеют место последовательные превращения -фазы в β и α-фазу или при 640 и 540 °С превращения > γ > α. При 640 °С в некоторых случаях имеет место непосредственное превращение -фазы в α, минуя стадию образования метастабильных модификаций, и в редких случаях возможно превращение β-фазы в γ с сохранением модификации γ-Вi2O3 до обычной температуры. В случае 26 нагревания метастабильной γ-Вi2O3 от комнатной температуры при 340 °С имеет место превращение γ-фазы в α [6]. На рисунке 1.5 представлена схема температурных областей существования стабильных и метастабильных фаз Вi2O3 [6]. Рис. 1.5 Схема температурных областей существования стабильных и метастабильных фаз Вi2O3 Необходимо отметить, что наличие у оксида висмута (III) полиморфных форм с различной кристаллической структурой объясняет причины различий физико-химических свойств материалов на основе оксида висмута [6]. Низкотемпературная α-Вi2O3 кристаллизуется в моноклинной сингонии пр. гр. Р21с [13, 16, 17]. Кристаллическая структура α-Вi2O3 впервые была исследована Силленом [16]. Позднее на монокристалле и порошках, было установлено, что структура α-Вi2O3 имеет слоистый характер и образована из слоев двух типов атомов висмута, расположенных параллельно плоскости, которые разделены слоями из атомов кислорода (рис. 1.6) [6]. 27 Рис. 1.6 Проекция элементарной ячейки α-Вi2O3 на плоскость XZ: а) по данным работы Малмроса, б) предложенная Силленом. 1 – Bi (1), 2 – Bi (2), 3 – O (1-3) Координационный полиэдр атомов Bi (1) содержит пять атомов кислорода в вершинах искаженного октаэдра, шестая вершина которого занята неподеленной электронной парой Bi+3. Наиболее короткая связь Bi-O (0,2133 нм) расположена в транс-положении к 6s2 -неподеленной паре, а остальные четыре расстояния равны 0,2210, 0,2218, 0,2546 и 0,2629 нм. Атомы Bi (2) окружены шестью атомами кислорода, лежащими в вершинах сильно искаженного октаэдра. Координационные полиэдры атомов висмута, объединяясь вершинами и ребрами, образуют трехмерную сетку с туннелями, минимальный диаметр которых составляет 0,25 нм и в которые направлены неподеленные электронные пары ионов Bi +3. Расстояния трех коротких связей Bi-О составляют 0,2130, 0,2201, 0,2283 нм и трех более длинных — 0,2422, 0,2559, 0,2787 нм [6]. Высокотемпературная модификация -Вi2O3 кристаллизуется в кубической сингонии (структурный тип CaF2) при 729 °С из моноклинной модификации α-Вi2O3, и она устойчива до температуры плавления оксида (825 °С). Гранецентрированная кубическая структура -Вi2O3 относится к пр. гр. Fm3m. Атомы висмута статистически размещаются в вершинах тетраэдра, расположенного асимметрично тетраэдру из атомов висмута с фактором занятости 3/16. Кислородная субъячейка -Вi2O3 сильно деформирована, 28 атомы кислорода в структуре распределены статистически, занимая 75 % позиций, и разупорядоченность структуры определяет высокую ионную проводимость -Вi2O3 (рис. 1.7) [6]. Рис. 1.7 Структурные модели флюоритоподобной ячейки для -Вi2O3: а) Модель Силлена, б) модель Гаттова, в) модель Виллиса со статистическим смещением атома кислорода из центра тетраэдра. Тетрагональная модификация β-Вi2O3 представляет собой искаженную флюоритовую структуру с вакансиями в кислородной субъячейке. Данная модификация относится к пр. гр. P4b2 [16]. На рис. 1.8 представлена проекция структуры β-Вi2O3 на плоскость [6]. Цифрами позиционные параметры атомов в направлении плоскости [17]. указаны 29 Рис. 1.8 Проекция структуры β-Вi2O3 на плоскость. а) по данным Силлена, б) Ауривиллиуса и Малмроса. Метастабильная кубическая модификация γ-Вi2O3, образуется при охлаждении расплава или высокотемпературной модификации β-Вi2O3, предварительно выдержанной в кислороде, имеет сходство со структурой силленита [6]. Физические свойства. Физические свойства оксида висмута (III) исследованы в основном для α-Вi2O3. Основные физические свойства оксида висмута (III) приведены в таблице 1.16 [6,13]. Таблица 1.16 Основные физические свойства оксида висмута (III) Температура плавления, tпл 825 °С Температура кипения, tкип 1890 °С Плотность, ρ 8,90 г/см3 Теплоемкость, С0р 114 ж/(мольК) Теплота образования, ΔH0обр — 578 кДж/моль Энтропия в стандартном состоянии, S0298 -151,6 Дж/(мольК) Теплопроводность, 0,8 Вт/мК 30 В результате исследования зависимости электропроводности от полиморфного состояния Вi2O3 Харвигом и Герардсом установлено [18], что проводимость β, γ и -фаз является преимущественно ионной и обусловлена подвижными ионами кислорода. Электропроводность α-Вi2O3 в зависимости от парциального давления кислорода описывается уравнением G =P0,25(O2). Последнее свидетельствует о электронном характере проводимости, и дырки являются основными носителями заряда. В области 650—730 °С в α-фазе имеет место смешанная ионно-электронная проводимость [6]. На рис. 1.9 приведена обычная зависимость электропроводности висмута (III) оксида от температуры на образцах с платиновыми электродами для цикла нагревание—охлаждение [6]. Измерения в которых проявляется β-фаза представлены прерывистой линией. Рис. 1.9 Зависимость электропроводности оксида висмута (III) от температуры. 1- исходные измерения, α, β, γ и - структурные модификации оксида висмута (III) Из рисунка видно, что проводимость возрастает более чем на три порядка при переходе от α к -модификации при 729 °С. При охлаждении на 80 или 90 °С кривая гистерезиса свидетельствует об образовании промежуточных β– или γ-фаз соответственно [6]. 31 Химические свойства. Оксид висмута(III) — типичный основной оксид, бинарное неорганическое соединение металла висмута и кислорода с формулой Bi2O3, не растворимые в воде. Cесквиоксид висмута легко растворяется в концентрированных горячих кислотах (T=343-353K), а также в разбавленных растворах минеральных кислот с образованием соответствующих солей, что широко используется в производстве его соединений. С плавиковой кислотой образует фторид висмутила (BiOF). Молекулярный водород восстанавливает оксид висмута (III) до металла при T=543K. Под действием окислителей при высоких температурах (T= 673 – 873K) образуются висмутаты (МехBiO3, МеxBiO4) [13]. В растворах щелочей растворимость Вi2O3 очень низка. В расплавах щелочей Вi2O3 проявляет кислотные свойства и вступает в реакции с оксидами, гидроксидами и карбонатами металлов, образуя двойные и более сложные оксидные соединения. Многие из данных соединений обнаруживают полезные физические свойства и используются в качестве сегнетоэлектрических, сцинтилляционных, акустооптических, сверхпроводящих и других материалов [6]. На основе оксида висмута (III) изготавливают ряд противоопухолевых препаратов для лечения онкологических заболеваний, антисептических и заживляющих средств, в том числе лекарств от многих желудочно-кишечных заболеваний [6 - 9]. 1.3 Термостимулированные превращения наноразмерных слоев оксида молибдена (VI) Структура, физико-химические свойства оксида молибдена (VI) подробно рассмотрены в работах [10, 14, 19 - 22]. Оптические свойства наноразмерных пленок MoO3 привлекают внимание исследователей по всему миру [19 - 26]. В работах [22,23] приведена зависимость оптической плотности, отражательной способности MoO3 от 32 первоначальной толщины, температуры, времени термической обработки образцов, а также влияние [20] освещения УФ-светом, на спектры поглощения MoO3. Кроме того, ряд исследователей [22 - 25] выделяют на спектрах поглощения характерные для пленок и монокристаллов MoO3 две спектральные области поглощения и отражения – коротковолновую λ 330 нм и длинноволновую λ 330 нм. Однако, спектры оптического поглощения, измеренные [19, 20, 22 - 25] на образцах MoO3 не дают возможности определить оптическую ширину запрещенной зоны слоев MoO3 из-за наложения на него полосы поглощения в интервале = 330-400 нм с максимумом при λ 350 нм. В процессе термической обработки на спектрах поглощения слоев MoO3 исследователями [22, 23] наблюдается уменьшение оптической плотности образца в интервале = 330-400 нм с максимумом = 350 нм (центр 1) (что приводит к смещению края полосы поглощения в коротковолновую область спектра) и увеличение в интервале = 400-1000 нм с максимумом = 870 нм (центр 2). Необходимо отметить, что в длинноволновой области спектра для образцов толщиной d 20 нм наблюдается увеличение, а для препаратов толщиной d 20 нм уменьшение оптической плотности [23]. Кроме того, по мере увеличения толщины слоев MoO3 (вплоть до 130 нм) при постоянной температуре и времени термической обработки наблюдается последовательное уменьшение эффектов изменения оптической плотности образцов во всем исследованном спектральном диапазоне [22, 23]. Полоса поглощения с максимумом при = 350 нм для монокристаллов MoO3 связана со стехиометрическим недостатком кислорода и обусловлена вакансиями кислорода с одним захваченным электроном [(Vа)++ е] (аналог F-центра) [19]. Авторами [22, 23], было установлено, что, степень превращения центра 2, во всем исследованном интервале температур, по мере уменьшения толщины слоев MoO3 (при постоянном времени термообработки) – возрастает, а увеличение температуры термообработки (при постоянной толщине пленок 33 MoO3) приводит к возрастанию скорости термического превращения. Кроме того, [(Vа)++ е]-центр образуется в процессе приготовления (т.е. термического испарения и осаждения на стеклянные подложки), термической обработки в интервале температур Т = 373-573 К пленок МоО3 за счет перехода электронов из валентной зоны с образованием дырок (р) на уровни анионной вакансии (Vа)++ + е [(Vа)++ е]. (1.1) Дырки, могут захватываться собственными (Vк6-) и примесными (Т-) дефектами с выделением кислорода и освобождением анионных вакансий: р + Vк6- [Vк6- р] + р [Vк6- р] О2 + 2Va++ + Vк6-, (1.2) где Vк6- и Va++ – катионная и анионная вакансии, р + Т- Т0 + р Т+ О2 + 2Va++ + Т [22,23]. (1.3) Глубина залегания [(Vа)++ е]-центра, по данным [22, 23] составляет EF1 = 3,54 эВ. Уменьшение максимума поглощения при = 350 нм, а также формирование максимума поглощения при = 870 нм в процессе термической обработки слоев MoO3 – взаимосвязанные процессы и являются результатом преобразования центра [(Vа)++ е]. Известно [22, 23, 27], для того, чтобы обеспечить при термическом возбуждении электронной подсистемы твердого тела переход электрона с нижнего заполненного уровня на верхний незаполненный и обеспечить достаточную скорость этого процесса необходимо, чтобы средняя энергия фонона (kT) соответствовала величине преодолеваемого энергетического барьера. Преобразование [(Vа)++ е]-центра авторы [22, 23] осуществляют двумя путями: а) перевод электрона с уровня залегания центра на дно зоны проводимости: [(Vа)++ е] (Vа)++ + е (1.4) (для обеспечения этого процесса потребуется энергия EF1) б) перевод электрона с потолка валентной зоны с образованием дырок (p) на уровень центра: 34 е + [(Vа)++ е] [е (Vа)++ е] (1.5) для обеспечения этого процесса потребуется энергия [22, 23]: E = Eшзз - EF1, (1.6) где Eшзз – термическая ширина запрещенной зоны MoO3; EF1 – глубина залегания [(Vа)++ е]-центра (Eшзз = 3,54 эВ – меньше на 0,2-0,3 эВ, чем оптическая ширина запрещенной зоны [19]). 1.4 Термодинамика и кинетика химического окисления металлов В природных условиях большинство металлов находятся в связанном состоянии в виде оксидов или солей. Следовательно, для них это состояние является термодинамически наиболее устойчивым. Для того, чтобы из природных соединений получить металлы или сплавы, которые используются как конструкционные материалы, нужно затратить энергию. Таким образом, в промышленных условиях большинство металлов и сплавов находятся в термодинамически неустойчивом состоянии [28]. Стремление металлов перейти из металлического в ионное состояние характеризуется величиной уменьшения свободной энергии (ΔG0) и составляет сущность процессов химической коррозии. Наибольшее значение в практических условиях имеет химическая коррозия при повышенных температурах на границе металла с газовой фазой, так называемая газовая коррозия. Продуктами газовой коррозии обычно являются оксиды металлов, за исключением особых случаев эксплуатации металлических изделий, когда могут получаться и другие соединения, например сернистые металлы [29]. Термодинамическая вероятность образования продуктов окисления на поверхности металлов определяется по величине изменения свободной энергии системы при протекании коррозионной реакции. Химическая реакция окисления: Ме + ½О2 ↔ МеО будет находиться в равновесии, если парциальное давление кислорода (Ро2) и упругость диссоциации оксида (РМеО) станут равны. При Ро2 > РМеО 35 реакция будет протекать в сторону образования окисла. Наоборот, при РМеО > Рo2 реакция будет протекать в обратном направлении. Оксид будет диссоциировать (разлагаться) на чистый металл и кислород. Следовательно, если известны значения упругостей диссоциации оксидов металлов для разных температур, то, полагая парциальное давление кислорода равным постоянной величине (для воздуха при атмосферном давлении Рo2 равно около 0,2 атм.), можно легко определить температурные границы термодинамической вероятности процесса окисления [29]. Образование продуктов коррозии осуществляется в результате протекания ряда последовательных и параллельных реакций. На рис. 1.10 представлена схема образования сплошной оксидной пленки при окислении металла кислородом из газовой фазы. Рис. 1.10 Схема процесса образования оксидной пленки на металле. Отличительной особенностью газовой коррозии металла является в отдельных случаях затухание процесса во времени. Это происходит тогда, когда на поверхности металла образуется защитная пленка. Если эта пленка является сплошной и имеет хорошую адгезию с поверхностью, то она изолирует металл от контакта с агрессивной средой и коррозия прекращается [28]. 1.4.1 Условия сплошности пленок Поверхностная пленка, которая образуется на металле, определяет его коррозионную устойчивость в агрессивной среде. Продукты коррозионной реакции, которые образуются непосредственно на тех участках 36 металлической поверхности, которые вступают в реакцию, могут тормозить дальнейший процесс коррозии [28]. При таком механизме коррозии дальнейший рост поверхностной пленки будет зависеть от возможности проникновения через эту пленку коррозионных агентов. Согласно [29] в одних случаях пленки могут расти до заметных толщин, в других они могут быть чрезвычайно тонкими, порядка нескольких молекулярных слоев. Одним из основных условий, характеризующих способность образованного первичного слоя продуктов коррозии тормозить дальнейшее окисление металла, будет сплошность получаемой пленки. Критерий сплошности впервые был сформулирован Пиллингом и Бедвортсом. Условие сплошности выполняется тогда, когда молекулярный объем химического поверхностного соединения (оксида, VОк), больше объема металла (VМе), израсходованного на образование молекулы оксида. В противном случае можно ожидать получения рыхлой, пористой пленки, неспособной покрыть плотным слоем весь металл и как следствие обладающей слабыми защитными свойствами. Соотношение объемов образованного оксида и исходного металла может быть легко подсчитано. При окислении 1 моля металла, его объем: V Ме A , d (1.7) где А – атомный вес металла; d – удельный вес металла. Соответственно, объем полученного оксида будет равен: VОк M , n D (1.8) где М – молекулярный вес оксида; n – число атомов металла в молекуле оксида; D – удельный вес оксида. Соотношение между объемом оксида и объемом металла согласно [29]: VОк M d . VMe n D A (1.9) Для висмута, применяемого в исследованиях, величинa соотношения 37 объемов VОк составляет 1,23. V Ме Необходимо отметить, металлы, имеющие соотношение VОк <1, образуют V Ме рыхлые пленки со слабыми защитными свойствами и характеризуются VОк высокими скоростями окисления. Металлы с отношением >1 образуют, V Ме как правило, пленки, значительно тормозящие дальнейшее протекание процесса окисления, т.е. обладающие хорошими защитными свойствами. Соотношение VОк носит название «критерий Пиллинга-Бедвортса». V Ме Данный критерий можно считать основанием только для приближенного суждения о стойкости металлов в условиях высокотемпературного окисления. Устойчивость металла против окисления зависит не только от сплошности пленки, но и от ряда других ее свойств (пористости, химической инертности к агрессивной среде, твердости, износостойкости, адгезии и близким к металлу коэффициенту термического расширения). Как отмечалось выше, сплошность пленки является необходимым, но недостаточным условием для проявления защитных свойств. В случае, когда VОк 1 , может происходить такое возрастание внутренних напряжений, VМе которое приводит к вспучиванию и отслаиванию пленки, что снижает ее защитные свойства. Ориентировочно можно считать, что достаточно хорошими защитными свойствами обладают пленки на металлах, удовлетворяющие условию [28]: 2,5 VОк 1. VМе (1.10) Дополнительными условиями, обеспечивающими защитные свойства пленки, являются их хорошая адгезия к металлу и физико-химические свойства. При формировании и росте защитной оксидной пленки важным 38 является также и условие ее ориентационного соответствия металлу, т.е. максимального сходства кристаллическитх решеток металла и образующегося оксида. 1.4.2 Кинетические закономерности окисления висмута Кинетические закономерности окисления висмута мало изучены. В работах [30 - 33] приведены результаты исследований термодинамических характеристик висмутсодержащих систем, кинетики окисления расплавов на основе висмута. Авторами [31, 32] было установлено, что чистый висмут при температурах 773, 873 и 973 К окисляется по параболическому закону, т.е. скорость окисления лимитируется процессом массопереноса в образующемся оксидном слое, а при 1073 и 1123 К - по линейному, в данном случае скорость реакции лимитируется процессами, протекающими на поверхностях раздела. При этом при 1073 К пленка Bi2O3 остается в твердом состоянии, в то время как при 1123 К расплавляется (Тпл(Bi2O3) = 1098 К). Кинетика окисления жидкого висмута на воздухе при T = 1273 К приведена в работе [33], авторами установлено, что с уменьшением высоты расплава висмута относительно расстояния до края тигля из BeO, диффузионный процесс все меньше лимитирует процесс окисления висмута и, как следствие, скорость окисления жидкого висмута увеличивается, а кинетические кривые окисления висмута на воздухе при T = 1273 К описываются в рамках линейного закона. Из анализа результатов рентгенофазового анализа [31] было установлено, что после охлаждения все пленки, полученные в результате окисления висмута, имеют α-Bi2O3 модификацию. Однако в работе [30] указывает на то, что переход α-фаза → δ-фаза Bi2O3 сопровождается изменением закона окисления висмута с параболического на линейный, в то время как увеличение температуры, приводящее к плавлению пленки, не только не изменяет механизм процесса, но и практически не сказывается на значениях скорости. 39 ГЛАВА 2. МЕТОДИКА ЭКСПЕРИМЕНТА 2.1 Приготовление образцов для исследований Объекты исследований: Для приготовления тонкопленочных образцов использовались следующие вещества: 1. порошок висмута (марка «Ч»); 2. оксид молибдена (VI) (марка «ХЧ»). Структура нанесения образцов. Образцы для исследований готовили методом термического испарения в вакууме (2∙10-3 Па), используя вакуумный универсальный пост «ВУП-5М» [34 – 36]. Двухслойные системы Bi-MoO3, MoO3 - Bi готовили путем последовательного нанесения пленок MoO3 на пленки Bi (предварительно нанесенные на подложку из стекла) и путем нанесения пленок Bi на пленку MoO3, соответственно. Одновременно с гетеросистемами, на одном расстоянии от лодочки-испарителя, наносились пленки Bi и MoO3, соответствующие толщинам подпленок двухслойных объектов. На рисунках 2.1, 2.2 в качестве примера приведены структуры индивидуальных слоев Bi, MoO3 и двуслойных систем Bi - MoO3, MoO3 – Bi. Рис. 2.1 Структура индивидуальных слоев Bi и MoO3. Рис. 2.2 Получение двухслойных систем Bi - MoO3 и MoO3 – Bi В качестве испарителя использовали лодочки из молибденовой и танталовой жести толщиной d = 3∙10-4 м для приготовления индивидуальных слоев MoO3 и Bi, соответственно. 40 Подготовка подложки. Подложками служили стекла от фотопластинок (ГОСТ 9284 – 59) толщиной 1·10-3 м и площадью 2·10-4-4·10-4 м2. Решающим фактором для нанесения пленок, безусловно, является чистота поверхности подложки. Процедуру очистки проводили следующим образом. Стеклянные подложки подвергали предварительной обработке в концентрированной азотной кислоте, в растворе дихромата калия в концентрированной серной кислоте, в кипящей дистиллированной воде и сушили [34, 35]. Очистка подложек производилась непосредственно перед нанесением на них исследуемых объектов. Обработанные подложки оптически прозрачны в диапазоне 300-1100 нм. Получение аммиака. Аммиак получали термическим разложением концентрированного гидроксида аммония, сушили и напускали в экспериментальную ячейку из стекла. 2.2 Методы определение толщины пленок Гравиметрический метод кварцевого микровзвешивания основан на сравнении эталонного изменения частоты кварцевого резонатора при нанесении на его поверхность пленки эталонной толщины с изменением частоты кварцевого резонатора с пленкой, толщину которой требуется определить. В результате уравнение имеет вид [37]: Fх , (2.1) d эталон Fэталон где dх, dэталон – толщины исследуемой и эталонной пленок; Fх, Fэталон – dх изменения частот резонаторов с исследуемой и эталонной пленками [34, 35]. Следует отметить, изменение частоты резонатора прямо пропорционально величине наносимой на его поверхность массы. Спектрофотометрический метод. Согласно закону Бугера-Ламберта-Бера величина поглощения (оптическая плотность) слоя прямо пропорциональна его толщине (при 41 условии одинаковой концентрации слоя светопоглощающих частиц). Толщину пленок (dx) рассчитывали по формуле: dх d эталон Aх Aэталон , (2.2) где dэталон – толщина эталонного образца; Ах, Аэталон – оптические плотности определяемого и эталонного образцов при определенной длине волны [35, 37]. Регистрацию спектров поглощения осуществляли на двулучевом спектрофотометре «Shimadzu UV-1700». Эллипсометрический метод. В основу метода положено фиксирование угла отражения падающего пучка света, сдвига фаз между падающим и отражающим светом и т.д., позволяет измерять толщину пленок, начиная с нескольких атомных слоев вещества [37]: d 15,17 0 , [Å] n 2 0,883 (2.3) где δ0 – приращение разности оптического хода, изменяющегося от 0 до 180°; n – показатель преломления. В эллипсометрическом методе экспериментально определяется разность фаз между компонентами электрического вектора, лежащими в плоскости падения света на измеряемую пленку, и перпендикулярной ей плоскости Δ ┴, и отношения амплитуд этих компонентов ψа исследуемой пленки. На практике для связи между экспериментально определенными и вычисленными величинами используют номограммы. Из построенных номограмм легко связать между собой Δ┴ и ψа с фазовой толщиной (dф), показателем преломления (n) и углом падения света (φ). Выражение для определения толщины образцов имеет вид [38]: d dф 360 n 2 sin 2 . (2.4) Толщину и показатель преломления пленок MoO3 оценивали с помощью лазерного эллипсометра «ЛЭФ-3М» (λ = 632,8 нм). 42 Интерференционный метод основан на наблюдении в микроскоп интерференционных полос, возникающих при рассмотрении в монохроматическом свете двух поверхностей, расположенных под углом друг к другу. Перед измерением на образце получали ступеньку – резкую боковую границу пленки на подложке. Для этого при осаждении пленки маскировали часть подложки или химически удаляли часть осажденной пленки[37]. На рисунке 2.3 изображен сдвиг интерференционных полос. Видно, что чередующиеся светлые и темные интерференционные полосы с шагом L на поверхности, как пленки, так и подложки смещаются относительно друг друга у их границы на значение l. С помощью интерференционного микроскопа «МИИ-4» измеряли смещение какой-либо определенной Рис. 2.3 – Сдвиг интерференционных полос. полосы, а расчет толщин образцов проводили по уравнению: d 1 l c ( ) , 2 L (2.5) где λс – длина волны монохроматического света, равна 0,54 мкм; L 1 c –шаг между соседними интерференционными полосами; 2 l – смещение интерференционной полосы [29, 34, 37]. 2.3 Метод оптической спектроскопии Метод оптической спектроскопии основан на законе светопоглощения Бугера – Ламберта – Бера и характеризует взаимосвязь между количеством поглощенного излучения с концентрацией вещества в исследуемом образце и его толщиной. Он был установлен экспериментально П. Бугером в 1729 г., 43 математически сформулирован М. Ламбертом в 1760 г. и в отношении к концентрации подтвержден А. Бером в 1852 г [37]. A lg I0 lg T k cl , I (2.6) где A – оптическая плотность; λ – длина волны; с – концентрация; I0 – интенсивность падающего излучения; I – интенсивность прошедшего излучения; Т – пропускание; k – коэффициент поглощения; l – толщина слоя. Спектры оптического поглощения и зеркального отражения регистрировали на спектрофотометре «Shimadzu UV-1700» (используя приставку зеркального отражения № 200-63687). Двулучевой спектрофотометр «Shimadzu управляемый UV-1700», персональным компьютером, позволяет регистрировать спектры поглощения (отражения) в спектральном диапазоне 190-1100 нм. В качестве источников излучения используются дейтериевая (185 – 357 нм) и галогеновая (350 – 1100 нм) лампы [36]. 2.4 Метод кварцевого микровзвешивания В качестве чувствительного элемента (микровесов) в данном методе применяется кварцевый резонатор. Кварцевый резонатор представляет собой электромеханическое устройство, основой которого является пьезоэлектрический элемент, изготовленный из кристаллов кварца (SiO2). Кристаллы имеют три кристаллографические оси, вдоль которых электрические свойства кварца одинаковы: х – электрическая, у – механическая, z – оптическая. Кристаллы кварца обладают прямым пьезоэффектом (возникновение зарядов под действием механического напряжения) и обратным (появление механических напряжений при электрической поляризации) [36]. Резонатор состоит из пьезоэлемента, кварцедержателя и корпуса; пьезоэлемент состоит из кристаллического элемента и пленочных электродов. Кристаллический пьезоэлемент может совершать различные 44 механические колебания, которые определяются характером движения его элементарных частиц. В значительной степени параметры резонатора определяются срезом пьезоэлемента – его ориентацией относительно кристаллографических осей кварца. В зависимости от того, по каким осям ориентированы толщина, ширина и длина пьезоэлемента, срез может обозначаться буквами х, у, z с указанием угла поворота относительно осей. Более широко в литературе применяется обозначение среза двумя буквами, например АТ. В генераторе резонатор может возбуждаться на частотах, отличных от частот механических резонансов. Реальное значение частоты колебаний резонатора в генераторе называют рабочей частотой. Частота, на которой возбуждаются колебания в кварцевом резонаторе, также весьма значительно зависит от температуры кристалла, причем зависимость эта различна для различных срезов и меняется в диапазоне от 80·10-6 до + 80·10-6 [35, 35]. Температурная зависимость для так называемых температурно-независимых срезов АТ и ВТ показана на рис. 2.4. В кварцевых резонаторах на частоту до 1 МГц используются резонаторы ВТ-среза, а выше 1 МГц – АТ-среза. Рис. 2.4 – Температурная зависимость резонансной частоты кварцевых резонаторов срезов АТ и ВТ. Необходимо отметить, что старение резонатора является его основной 45 качественной характеристикой. Оно характеризуется измерением частоты резонатора во времени при эксплуатации и хранении. Это изменение частоты во времени тем больше, чем больше рассеиваемая резонатором мощность. Масс-чувствительные пьезорезонансные датчики выполняются из тонких пластин или линз кварца температурно-независимого АТ-среза. В резонаторах возбуждаются колебания сдвига по толщине. Присоединяемая масса может наноситься с одной или с двух сторон, как на электроды, так и на периферию резонатора. Наращивание массы, т.е. процесс сорбции вещества, может происходить по-разному и носить как необратимый, так и обратимый характер. Например, при отработке технологии процессов напыления в установке заподлицо с поверхностью, на которую производится напыление, помещается пьезорезонатор – толщиномер, позволяющий непрерывно контролировать процесс по изменению частоты пьезорезонатора в зависимости от толщины, нанесенной на него пленки/ Зависимость изменения резонансной частоты резонатора от присоединенной массы выражается уравнением Зауэрбрея [36]: mf 02 f N к S , (2.7) где N – частотный коэффициент резонатора; к – плотность кварца; S – площадь поверхности кристалла, на которую нанесено покрытие; f0 – исходная частота резонатора. Преобразовав уравнение Зауэрбрея можно установить связь частоты с толщиной h' и плотностью ' присоединяемого материала, которая определяется в первом приближении формулой: f h f h , где и h – плотность и толщина резонатора. (2.8) 46 Допустим, что исследуемые вещества сорбируются по всей поверхности дискового резонатора, тогда с учетом (2.8), уравнение имеет вид: f m f m , (2.9) где m – масса резонатора. Очевидно, что относительное приращение массы может регистрироваться с тем же разрешением, что и относительное изменение частоты, т.е. 10 -6-10-7. Для кварцевых резонаторов регистрируемые приращения толщиной массы на h = 0,1 мм минимальные единицу поверхности m = (10-6-10-7)h = (10-6-10-7)2,650,01 = 2,65(10-8-10-9) г/см2. Максимальная присоединяемая масса не должна превышать 210-3 г/см2, и толщина пленок должна быть не более 1-2 мкм, в противном случае резко падает добротность резонатора, что приводит к нестабильности и большой погрешности измерения [34 – 36]. Установка для измерения массы методом микровзвешивания. Для измерения относительных изменений массы тонких пленок висмута в процессе окисления при различных температурах использовали специально разработанную и сконструированную установку, термокамерой которой, являлся сушильный шкаф MLW WS 51 с двухступенчатым регулированием мощности нагревания. В качестве датчика температуры была применена термопара ТПК Непосредственно 021-0,5/1000 в двери (точность сушильного термостатирования шкафа были 1 K). вмонтированы герметичные электрические вводы и установлен специальный кронштейн для установки двух кварцевых резонаторов, на один из которых наносится изучаемая пленка, а другой служит резонатором сравнения для компенсации не связанного с изменением массы паразитного температурного изменения частоты [36]. 47 Источником питания служил стабилизированный блок питания с выходным напряжением 5 В, обеспечивающий при изменении напряжения питающей сети на 10 % изменение выходного напряжения не более чем на 1 %. В качестве регистрирующего прибора используется цифровой частотомер Ч3-34, ко входам А и Б которого подключались выходы сдвоенного генератора [35]. Регистрация изменения массы нанесенной на поверхность резонатора пленки состоит в следующем. Кварцевые резонаторы извлекаются из корпусов и подвергаются двум-трем циклам нагрева и охлаждения для стабилизации параметров. Максимальная температура прогрева берется равной (или чуть выше) той температуре, при которой предполагается проводить дальнейший эксперимент. После этого при температуре 298 К путем подключения к специально изготовленному генератору определяется точная частота того резонатора, на который будет наносится пленка исследуемого вещества. Затем на поверхность резонатора при помощи специального электродов держателя – трафарета, резонатора обеспечивающего предотвращающего проводящей заданную постоянную металлической площадь замыкание пленкой пленки, и наносится исследуемая пленка и вновь определяется частота при 298 К. Разность частот до нанесения пленки и после принимается относительной массой пленки, выраженной в единицах частоты [36]. Далее оба кварцевых резонатора устанавливаются в предварительно прогретую до необходимой температуры термокамеру. Через 1 мин. производится первый отсчет частот генераторов. Далее отсчеты частоты производятся с прогрессивно возрастающими в зависимости от степени превращения вещества интервалами от 1 до 10 мин. После прекращения изменения частоты резонатор с пленкой извлекается из камеры, охлаждается на воздухе, производится измерение частоты при температуре 298 К, определяя таким образом полную массу, прибывшую (убывшую) в процессе эксперимента. 48 Уравнение для определения значения частоты резонатора с исследуемой .ист. Fxобр имеет вид [36]: 1 пленкой с учетом частоты резонатора сравнения .ист. .ист. Fxобр Fxобр.ист Fxобр Fxобр Fxср Fxср1 1 1 Значение частоты F0 резонатора при температуре (2.10) проведения эксперимента в момент времени = 0 рассчитывали путем вычитания из величины полного изменения массы величину изменения частоты от F1 до Fx. Затем строили кинетические кривые в координатах «степень превращения – время» (при известном значении полного изменения частоты, соответствующем стопроцентному превращению). 2.5 Методика измерения контактной разности потенциалов Для удаления электрона из твердого тела необходимо затратить некоторую энергию, именуемую работой выхода электрона . Работа выхода электрона частично, а в ряде случаев полностью, определяется заряженным двойным электрическим слоем или диполем на поверхности твердого тела. Измерение работы выхода электрона является одним из классических методов изучения поверхностных явлений [39]. Большинство процессов, изменяющих потенциальный барьер на поверхности, могут быть непосредственно исследованы этим методом. Он особенно полезен при исследовании природы электронного взаимодействия между хемосорбированными атомами и адсорбирующей поверхностью. С его помощью можно получить сведения о процессах образования хемосорбционной связи. Работа выхода играет важную роль в процессах электронных переходов при адсорбции и катализе [39] (имеется прямая связь между работой выхода электрона с поверхности и ее каталитическими свойствами). И наконец, измерение работы выхода электрона дает важную информацию при изучении процессов, протекающих в гетеропереходах и переходах “металл – полупроводник” [40], так как состояние поверхности контактирующих партнеров в большинстве случаев играет определяющую 49 роль в процессах, протекающих в темноте и при облучении светом различного спектрального состава в подобных системах [39]. В пространстве между двумя проводящими телами имеется контактная разность потенциалов, обусловленная разностью работ выхода электронов двух поверхностей. Контактную разность потенциалов (КРП) можно измерить различными методами Одним [41,42]. из наиболее распространенных методов измерения КРП является метод Кельвина [39, 42 44]. В этом методе исследуемая поверхность и относительный электрод образует конденсатор, емкость которого модулируется колебанием относительного электрода. Модуляция емкости приводит к изменению заряда на обкладках конденсатора, в результате чего в цепи, соединяющей обкладки конденсатора, появляется сигнал переменного тока. Величина сигнала в общем случае определяется значением КРП. Включение в эту цепь постоянного внешнего напряжения позволяет компенсировать КРП. При равенстве величин внешнего напряжения и КРП сигнал переменного тока исчезает. Уравнение для определения работы выхода электрона методом контактной разности потенциалов имеет вид [36]: VКРП а к , (2.11) где VКРП – контактная разность потенциалов; а,, к – работа выхода для анода и катода соответственно. Блок-схема установки приведена на рисунке 2.5. Экспериментальный комплекс «КРП-2» позволяет проводить измерения КРП в интервале давлений Р = 1·10-6-5·105 Па, в атмосфере различных газов и паров [36, 39]. Обеспечивается возможность термостатирования и линейного разогрева образца с различными скоростями (0,2, 0,1, 0,05, 0,01 К/с ) в интервале температур Т = 77-400 К, а также экспонирования (до и в процессе измерения) монохроматическим светом в интервале 200-2000 нм. Точность измерения контактной разности потенциалов составляет 0.01В. 50 Рис. 2.5 – Вакуумная и оптическая система установки КРП – 2: 1 – измерительная ячейка; 2 – высоковакуумная колебательная система; 3 – крышка (на кронштейне которой закрепляется исследуемый образец); 4 – вакуумные краны; 5 – масс-спектрометрическая лампа типа РМО-4С (в комплекте с измерительным блоком ИПДО-1); 6 – термопарный (ПМТ-2) манометрический преобразователь; 7 – ионизационный (ПМИ-2) манометрический преобразователь; 8 – механический насос; 9 –цеолитовый насос; 10 – высоковакуумный насос типа НОРД-100 с блоком питания; 11 – система напуска с газовым баллоном. 2.6 Электрофизические методы исследования Электрофизические методы исследования хорошо известны и широко применяются в различных областях науки и техники. Применение этих методов для изучения электрофизических свойств материалов позволяет определить их основные физические параметры [36, 39]. Широкое применение эти методы нашли для исследования ряда электрофизических характеристик гетеропереходов и переходов “металл – полупроводник”[39, 40]. Измерение темновых и световых вольтамперных характеристик, фотоэлектрических и оптических свойств дают возможность определить физические параметры контактов и их моделей [39, 45]. Блок - схема высоковакуумного экспериментального комплекса «Электрофизика», предназначенного для исследования фотоэлектрических характеристик неорганических материалов, приведена на рис. 2.6 [36, 39]. 51 Рис. 2.6 – Схема установки «Электрофизика»: 1 – вольтметр универсальный В7-21; 2 – ЭВМ типа IBM PC; 3 – усилитель постоянного тока У5-11; 4 – источник питания постоянного тока Б5-43А; 5 – монохроматор МДР-2 (МСД-1, набор светофильтров); 6 –осветитель с лампой ДРТ-250 (ДКсШ-1100); 7 – электрозатвор; 8 –измерительная ячейка; 9, 10 – вентиль; 11 – преобразователь манометрический термопарный (ПМТ); 12 – преобразователь манометрический ионизационный (ПМИ); 13 – вакуумметр (ВИТ-2); 14 – насос охлаждаемый разрядный диодный (НОРД-100); 15 – цеолитовый насос; 16 – БП-1100; 17 – БП-100; 18 – форвакуумный насос; 19 – линза; 20 – автоматический потенциометр КВП-1-503. По функциональному назначению она подразделяется на: вакуумную, электроизмерительную и оптическую системы. Вакуумная система обеспечивает поддержание и контроль рабочего давления в измерительной ячейке и включает в себя: измерительную ячейку, насос магниторазрядный НОРД-100, блок питания БП-100, цеолитовый насос, вакууметр манометрический ионизационно-термопарный термопарный, ВИТ-2, преобразователь преобразователь манометрический ионизационный, соединительные трубопроводы и запорные вентили [36, 39]. Электроизмерительная система предназначена для регистрации и записи на ЭВМ темнового тока, фототока, фотоЭДС и состоит из: источника 52 постоянного тока Б5-43А; усилителя У5-11; вольтметра универсального В7-21; ЭВМ типа IBM PC [36, 46 - 50]. Оптическая система предназначена для экспонирования образца, помещенного в измерительную ячейку монохроматическим светом в интервале длин волн от 250 до 1300 нм. В ее состав входят следующие приборы и оборудование: монохроматор МДР-2 (МСД-1, набор светофильтров), осветитель, лампа ДРТ-250 (ДКсШ-1100), блок питания лампы, фотозатвор, реле времени, кварцевые конденсаторы [39, 48 - 50]. Необходимо отметить, для регистрации фото-ЭДС в двуслойных системах Bi – MoO3, Bi – Bi2O3, Bi – BiN использовали, «прижимной» вариант, представляющий собой две стеклянные подложки, плотно прижатые друг к другу, одна с нанесенными висмутовыми электродами, другая с наноразмерной пленкой исследуемого образца (MoO3, Bi2O3, BiN) [51]. ФотоЭДС исследуемых гетеросистем измеряли при тщательном экранировании одного из висмутовых электродов (рис. 2.7). Рис. 2.7 Геометрия измерения фото-ЭДС гетеросистем различного состава: 1,4 – подложка из кварца; 2 – электроды из висмута; 3 – пленка исследуемого образца (MoO3, Bi2O3, BiN); 5 – экран. 2.7 Актинометрия источника излучения Актинометрию источников излучения (в данной работе использовались ртутная (ДРТ - 250) и ксеноновая (ДКсШ - 1000) лампы) проводили с помощью радиационного термоэлемента RT-0589. Установка для измерения абсолютных интенсивностей световых потоков приведена на рис. 2.8. 53 Рис. 2.8 – Установка для измерения абсолютных интенсивностей световых потоков: 1 – источник света; 2 – водяной фильтр; 3 – светофильтр (монохроматор); 4 – фотозатворы; 5 – термостолбик; 6 – вольтметр. Свет от источника света (1) проходит через кварцевую кювету, наполненную водой с толщиной слоя 2 см (2), и для выделения требуемого участка спектра стеклянный полосовой светофильтр (монохроматор) (3) и попадает на приемник термостолбика (5). Световая энергия, превращаясь в тепло, создает разность потенциалов (термо-ЭДС), которая регистрируется вольтметром (6). Расчет абсолютных интенсивностей световых потоков проводили по формуле [52 - 54]: Nф P P , h h c (2.12) где Nф – поток фотонов, [фотон/см2·с]; Р – энергетическая освещенность, [Вт/см2]; h – постоянная Планка, [6,62·10-34 Дж·с]; ν – частота излучения; c – скорость света, [3·1010 см/с]. После преобразования получаем: P U S , (2.13) где U – разность потенциалов, регистрируемая гальванометром, [В]; S – площадь приемника термостолбика, [см2]; γ – чувствительность термостолбика, [В/Вт]. Для термостолбика марки RT-0589 (S· = 3,7·102). 54 ГЛАВА 3. ИССЛЕДОВАНИЕ ТЕРМО - ФОТО – И ГАЗОСТИМУЛИРОВАННЫХ ПРЕВРАЩЕНИЙ НАНОРАЗМЕРНЫХ ПЛЕНОК ВИСМУТА, ОКСИДА МОЛИБДЕНА (VI) И СИСТЕМЫ НА ИХ ОСНОВЕ Получение перспективных для науки и практики материалов, изучение их физико-химических свойств, выяснение корреляций между составом, структурой и свойствами соединений представляют значительный интерес для физики и химии твердого тела [8, 55 - 58]. Выяснение особенностей процессов, протекающих в наноразмерных слоях различных материалов, представляет интерес в связи с необходимостью создания прочного физикохимического фундамента наноструктурированного состояния вещества, который будет служить надежной научной базой для получения новых функциональных материалов, обладающих полезными для практического использования свойствами [8, 56, 58]. 3.1 Закономерности формирования оксида висмута (III) на поверхности наноразмерной пленки висмута Среди разнообразных неорганических материалов особое место занимает висмут [6, 7, 9, 59, 60]. Висмут благодаря комплексу положительных свойств (пластичность, сравнительно низкая температура плавления (544 К) и довольно значительная температура кипения (1833 К), малое сечение захвата тепловых нейтронов и значительная способность к растворению урана, невысокая агрессивность к конструкционным материалам в сочетании с химической стойкостью и пожарной безопасностью, наименьшая токсичность из всех тяжелых металлов и др.) широко применяется во многих областях науки, техники, промышленности [6, 7, 9, 59, 60]. Однако в атмосферных условиях при контакте с окружающей средой висмут подвергается атмосферной коррозии [6, 7, 29, 61 - 65]. В настоящем разделе представлены результаты исследований, направленные на выяснение природы и закономерностей процессов, 55 протекающих в условиях атмосферы в наноразмерных слоях висмута различной толщины (d = 3 – 120 нм) в зависимости от температуры (Т = 373 – 673 К) и времени ( = 0,05 – 2500 мин) теплового воздействия. 3.1.1 Изменение оптических свойств наноразмерных пленок висмута под действием температуры В результате исследований оптических свойств наноразмерных пленок висмута разной толщины (нанесенных на стеклянные подложки) до, в процессе и после термической обработки (Т = 373…673 К) в атмосферных условиях при Т = 298 К, прежде всего, было установлено [66 – 73], что спектры поглощения и отражения пленок висмута до термообработки существенно зависят от их толщины. На рис. 3.1 представлены спектры поглощения пленок висмута толщиной d = 3…120 нм. Видно, что в исследуемом диапазоне длин волн на спектрах поглощения образцов толщиной более 28 нм можно выделить характерные для висмута полосы поглощения (в частности – максимумы поглощения при λ 410 нм и 735 нм) [67, 68, 70 – 73, 74]. Рис. 3.1 Спектры поглощения пленок висмута толщиной: 1 – 120 нм, 2 – 92 нм, 3 – 77 нм, 4 – 68 нм, 5 – 63 нм, 6 – 51 нм, 7 – 39 нм, 8 – 27 нм, 9 – 17 нм, 10 – 13 нм, 11 – 9нм, 12 – 3нм. 56 По мере уменьшения толщины пленок висмута на спектрах поглощения и отражения постепенно перестают проявляться характерные для висмута полосы поглощения и отражения. Для пленок висмута толщиной d 28 нм наблюдается бесструктурное поглощение и отражение в диапазоне = 190…1100 нм [70 – 72, 75, 76]. На рис. 3.2 в качестве примера приведены спектры отражения пленок висмута толщиной d = 3…120 нм. Рис. 3.2 Спектры отражения пленок висмута толщиной: 1 – 120 нм, 2 – 92 нм, 3 – 77 нм, 4 – 55 нм, 5 – 49 нм, 6 – 39 нм, 7 – 30 нм, 8 – 17 нм, 9 – 3 нм. Коэффициент отражения (R) светового потока, падающего по нормали к плоской поверхности твердого тела из вакуума (воздуха), может быть представлен через коэффициенты преломления (n) и поглощения (k) твердого тела в следующем виде [46, 77, 78]: (n 1) 2 k 2 R . (n 1) 2 k 2 (3.1) Для химически чистого висмута при = 589 нм коэффициент преломления составляет 1,9, а коэффициент поглощения – 3,66 [46, 74]. Коэффициент отражения будет равен R = 0,65. Этому значению коэффициента отражения соответствуют пленки висмута толщиной более 120 нм. Из уравнения следует, что если в определенном спектральном 57 диапазоне твердое тело не поглощает свет, то коэффициент отражения будет зависеть только от значения показателя преломления. Основным продуктом при термической обработке пленок висмута является оксид висмута (III), коэффициент преломления которого составляет 2,63 [79], коэффициент отражения для Bi2O3 должен составить величину 0,2 (20%). При анализе спектров отражения пленок висмута (полученных методом термического испарения в вакууме) установлено, что по мере уменьшения толщины оптические свойства пленок висмута все в большей степени (при толщине пленки менее 28 нм практически полностью) определяются наличием пленок Bi2O3 на их поверхности [68, 70, 71 – 73, 75, 76]. В результате термической обработки пленок висмута разной толщины в интервале температур (Т = 373…673 К) в атмосферных условиях спектры поглощения, отражения и масса образцов претерпевают существенные изменения. На рисунках 3.3, 3.4 в качестве примера приведены спектры поглощения пленок висмута толщиной d = 25 нм до и после термической обработки при 373 К, и толщиной d = 41нм при 473 К соответственно. Рис. 3.3 Спектры поглощения пленок висмута толщиной (d = 25 нм) до (1) и после предварительной термической обработки при Т = 373 К: 2 – 3 мин, 3 – 20 мин, 4 – 45 мин, 5 – 90 мин, 6 – 140 мин, 7 – 190 мин, 8 – 270 мин, 9 – 360 мин, 10 – 480 мин, 11 – 620 мин, 12 – 710 мин, 13 – 860 мин. 58 Рис. 3.4 Спектры поглощения пленок висмута толщиной (d = 41 нм) до (1) и после предварительной термической обработки при Т = 473 К: 2 – 40 с, 3 – 3 мин, 4 – 5 мин, 5 – 8 мин, 6 – 12 мин, 7 – 18 мин, 8 – 25 мин, 9 – 35 мин, 10 – 50 мин, 11 – 90 мин, 12 – 180 мин, 13 – 360 мин. Причем, наблюдаемые изменения массы, спектров поглощения и отражения после термической обработки образцов в значительной степени зависят от первоначальной толщины пленок висмута, температуры и времени термообработки [66, 68 – 72, 76, 80, 81]. На рисунках 3.5, 3.6 представлены спектры поглощения пленок висмута толщиной d = 50 нм до и после термической обработки при 573 К и толщиной d = 39 нм при 673 К соответственно. 59 Рис. 3.5 Спектры поглощения пленок висмута толщиной (d = 50 нм) до (1) и после предварительной термической обработки при Т = 573 К: 2 – 1 с, 3 – 15 с, 4 – 40 с, 5 – 1,5 мин, 6 – 2 мин, 7 – 3 мин, 8 – 4 мин, 9 – 8 мин, 10 – 10 мин, 11 – 60 мин, 12 – 120 мин. Рис. 3.6 Спектры поглощения пленок висмута толщиной (d = 39 нм) до (1) и после предварительной термической обработки при Т = 673 К: 2 – 1 с, 3 – 3 с, 4 – 5 с, 5 – 10 с, 6 – 15 с, 7 – 30 с, 8 – 1 мин, 9 – 2 мин, 10 – 3 мин, 11 – 25 мин. 60 Видно, что термическая обработка приводит к существенным изменениям вида спектров поглощения образцов. Отметим, что наблюдаемые изменения не аддитивны в рассматриваемом спектральном диапазоне длин волн. Наряду с уменьшением в интервале = 330…1100 нм и увеличением в диапазоне = 300…330 нм значений оптической плотности образца формируется спектр поглощения нового вещества [66 – 73, 76, 80 – 82]. Оцененная по длинноволновому порогу поглощения, который находится при ≈ 387 нм, оптическая ширина запрещенной зоны образующегося вещества, составляет Е ≈ 3,2 эВ [66 – 73, 76, 80 – 82]. Край полосы поглощения пленок висмута оценивали по формулам (3.2), (3.3) [83], используя спектры поглощения образцов, подвергнутых термической обработке. а(hν) = А(hν – Еg)1/2, (3.2) а(hν) = А’(hν – Еg)3/2, (3.3) где α – коэффициент поглощения; h – постоянная Планка; ν – частота излучения; A, A – коэффициенты пропорциональности; Eg – ширина запрещенной зоны. Полученное значение ширины запрещенной зоны вещества удовлетворительно совпадает с шириной запрещенной зоны оксида висмута (III) [84]. Поэтому, было сделано предположение, что при термической обработке пленок висмута основным продуктом взаимодействия их с кислородом окружающей среды является оксид висмута (III) [66 – 73, 76, 80 – 82]. Закономерности изменения спектров поглощения пленок висмута по мере увеличения или уменьшения температуры термической обработки сохраняются (в коротковолновой области спектра – слева от изобестической точки наблюдается увеличение оптической плотности, в длинноволновой области спектра – справа от изобестической точки наблюдается уменьшение оптической плотности образцов) [68 – 73, 75, 82]. 61 При одинаковой исходной толщине пленок висмута с увеличением температуры имеет место возрастание эффектов изменения оптической плотности [68, 71 – 73]. По мере увеличения толщины пленок висмута (вплоть до 120 нм) при постоянной температуре термической обработки (в интервале 373…673 К), наблюдается последовательное уменьшение эффектов изменения оптической плотности образцов во всем исследованном спектральном диапазоне [66 – 73, 76, 80 – 82]. 3.1.2 Кинетические особенности окисления наноразмерных пленок висмута Для выяснения закономерностей протекания процесса взаимодействия пленок висмута с кислородом окружающей среды (используя результаты гравиметрических исследований и измерений спектров поглощения и отражения пленок висмута разной толщины до и в процессе термической обработки образцов при разных температурах) были рассчитаны и построены кинетические зависимости степени превращения (α = ()). Для построения кинетических кривых в координатах α = () (по результатам измерений спектров поглощения и отражения) был применен следующий подход [70 – 73, 80, 81]. Спектры поглощения пленок висмута, измеренные при различных временах термической обработки, пересекаются в одной (изобестической) точке, в которой оптическая плотность не зависит от времени термообработки, а интенсивность поглощения слоями висмута и оксида висмута (III) одинакова. Слева и справа от изобестической точки оптическая плотность (А) пленки висмута зависит от времени термической обработки, а при определенном времени термической обработки будет складываться из оптической плотности, связанной с наличием слоя висмута (АBi) и оксида висмута (III) (АBi2O3) [71 – 73]: А = АBi + АBi2O3………………………….(3.4) На основании анализа спектров поглощения и отражения пленок висмута и Bi2O3 для построения кинетических кривых α = () был выбран диапазон 62 длин волн = 400…900 нм, в котором пленки висмута имеют значительное поглощение, а поглощением Bi2O3 можно пренебречь [68, 69, 71, 72]. Если обозначить через α степень термического превращения пленок висмута в оксид висмута (III), то при длине волны (например, = 800 нм – рис. 3.3 – 3.6), соответствующей спектральной области, в пределах которой висмут поглощает, а оксид висмута (III) практически не поглощает свет [46, 74, 84], текущие оптические плотности пленок висмута (АBi) и оксида висмута (АBi2O3) можно представить в следующем виде [71 – 73]: АBi = А1Bi (1 – α), (3.5) АBi2O3 = А1Bi2O3·α, (3.6) где А1Bi, А1Bi2O3 – предельные значения оптической плотности пленок висмута и оксида висмута (III) при = 800 нм. В итоге получаем следующее выражение для степени термического превращения пленки висмута в оксид висмута (III) [69 – 72, 76, 82]: А = А1Bi (1 – α) + А1Bi2O3·α, (3.7) α = (А1Bi – А обр.) / (А1Bi – А1Bi2O3). (3.8) Известно [46, 77, 78], что падающая по нормали на поверхность какойлибо системы световая волна от источника излучения, претерпевает зеркальное отражение, рассеяние, поглощение и пропускание. При прохождении через границы нескольких сред (воздух – оксид висмута – висмут – стекло – воздух) с различными коэффициентами преломления (n) суммарная зеркально отраженная световая волна (R) будет складываться из нескольких составляющих R = R1 + R2 + R3+ R4, (3.9) где R1 – зеркально отраженная световая волна от границы воздух – оксид висмута (III), R2 – зеркально отраженная световая волна от границы оксид висмута (III) – висмут, R3 – зеркально отраженная световая волна от границы висмут – стекло, R4 – зеркально отраженная световая волна от границы стекло – воздух. Таким образом, измеряемое в реальных условиях на спектрофотометре 63 полное значение оптической плотности включает (как минимум) несколько составляющих A = Aобр. + Aотр. + Aрас., (3.10) где Aобр. – значение оптической плотности образца; Aотр. – значение оптической плотности, обусловленное потерями на зеркальное отражение света поверхностью образца; Aрас. – значение оптической плотности, обусловленное потерями на диффузное рассеяние света поверхностью образца. Специальными исследованиями [71 – 73] было установлено, что диффузное рассеяние поверхностью пленок висмута пренебрежимо мало по сравнению с зеркальным отражением и, как следствие, Aрас. можно считать 0. Тогда A = Aобр. + Aотр.. (3.11) После преобразований формула для расчета истинного (вызванного поглощением света в веществе) значения оптической плотности имеет вид: Aобр. = A + lg(1 – R). Степень термического превращения пленок (3.12) висмута зависит от первоначальной толщины, температуры и времени термической обработки [66, 68 – 73, 76, 81, 82]. В качестве примера на рисунках 3.7 – 3.10 приведены кинетические кривые степени превращения пленок висмута разной толщины при температурах (T = 373…673 K) соответственно. 64 Рис. 3.7 Зависимость степени превращения от толщины пленок висмута при Т = 373 К: 1 – 4 нм, 2 – 25 нм, 3 – 34 нм, 4 – 52 нм. Рис. 3.8 Зависимость степени превращения от толщины пленок висмута при Т = 473 К: 1 – 6 нм, 2 – 11 нм, 3 – 21 нм, 4 – 30 нм, 5 – 51 нм. Видно, что с уменьшением толщины пленок висмута наблюдается увеличение степени термического превращения во всем исследованном интервале температур (Т = 373…673 К) [66 – 72, 75, 80, 81]. 65 Рис. 3.9 Зависимость степени превращения от толщины пленок висмута при Т = 573 К: 1 – 5 мин, 2 – 1 мин. Рис. 3.10 Зависимость степени превращения от толщины пленок висмута при Т = 673 К: 1 – 9 нм, 2 – 27 нм, 3 – 39 нм, 4 – 55 нм, 5 – 77 нм. По мере увеличения температуры термообработки при постоянной толщине пленок висмута степень термического превращения возрастает [68, 71 – 73]. На рисунке 3.11 представлена температурная зависимость степени превращения пленок висмута толщиной 25 нм. 66 Рис. 3.11 Температурная зависимость степени превращения пленок висмута толщиной 25 нм: 1 – 6 мин, 2 – 3 мин, 3 – 1 мин. Кинетические кривые степени термического превращения пленок висмута разной толщины условно можно разбить на несколько участков (табл. 3.1 – 3.4) [66, 67, 71, 72, 75, 76, 80, 81]: линейный (α = Kτ + A), обратный логарифмический (K / α = B – lgτ), кубический (α3 = Kτ + B) и логарифмический (α = K lg(Bτ + 1)), где К – константа скорости формирования оксида висмута (III), A и B – постоянные интегрирования, τ – время взаимодействия. Таблица 3.1. Зависимость продолжительности линейного, обратного логарифмического, кубического, логарифмического участков от толщины пленок висмута в процессе термической обработки (T = 373 К). d,нм 4 25 34 52 α = Kτ + A ___ 0 – 6мин 0 – 50мин 0 – 80мин K/α = B – lgτ 0 – 6мин 6 – 20мин 50 – 110мин 80 – 135мин ___ 20 – 120мин 110 – 190мин 135 – 220мин 120 – 420мин 190 – 550мин 220 – 940мин α3 = Kτ + B 6 – 160мин 160 – 310мин 420 – 550мин 550 – 770мин 940 – 1480мин α = K lg(Bτ+1) 310 – 550мин 550 – 860мин 770 – 1100мин 1480 – 2560мин 67 Таблица 3.2 Зависимость продолжительности обратного логарифмического, кубического, логарифмического участков от толщины пленок висмута в процессе термической обработки (T = 473 К). d,нм 6 11 21 30 51 K/α = B – lgτ 0 – 20с 0 – 40с 0 – 1мин 0 – 1мин 0 – 1мин 1 – 5мин 1 – 8мин 1 – 12мин 5 – 15мин 8 – 25мин 12 – 40мин 20с – 1мин 40с – 3мин α3 = Kτ + B 1 – 6мин 3 – 12мин 6 – 15мин 12 – 25мин 15 – 40мин 25 – 60мин 40 – 90мин α = K lg(Bτ+1) 15 – 40мин 25 – 180мин 40 – 240мин 60 – 300мин 90 – 420мин Наличие соответствующих участков, а также их продолжительность определяется толщиной пленок висмута и температурой термообработки. По мере увеличения толщины пленок висмута и уменьшения температуры термообработки наблюдается увеличение продолжительности участков кинетических кривых степени превращения [67, 71, 72, 76, 80, 81]. Таблица 3.3. Зависимость продолжительности обратного логарифмического, кубического, логарифмического участков от толщины пленок висмута в процессе термической обработки (T = 573 К). d,нм 3 16 34 50 92 K/α = B – lgτ ___ ___ 0 – 5с 0 – 10с 0 – 15с ___ ___ 5с – 1мин 0 – 15с 0 – 1мин 1 – 4мин 2 – 8мин 5 – 15мин 15с – 2мин 1 – 5мин 4 – 10мин 8 – 15мин 15 – 40мин α3 = Kτ + B 10с – 2мин 15с – 5мин α = K lg(Bτ+1) 2 – 15мин 5 – 30мин 10 – 40мин 15 – 60мин 40 – 120мин 68 Таблица 3.4. Зависимость продолжительности обратного логарифмического, кубического, логарифмического участков от толщины пленок висмута в процессе термической обработки (T = 673 К). d,нм 9 27 39 55 77 K/α = B – lgτ ___ 0 – 4с 0 – 4с 0 – 5с 0 – 5с ___ ___ 4 – 30с 5 – 30с 5 – 1мин 0 – 5с 4 – 10с α3 = Kτ + B α = K lg(Bτ+1) 30с – 2мин 30с – 3мин 1 – 3,5мин 5с – 1мин 10с – 3мин 2 – 4мин 3 – 5мин 3,5 – 5мин 1 – 5мин 4 – 40мин 5 – 60мин 5 – 90мин 3 – 20мин На рисунках 3.12 – 3.15 представлены кинетические кривые степени превращения пленок висмута различной толщины при температурах (Т = 373, 473, 573, 673 К) соответственно. Линейный участок на кинетических кривых α = f (τ) удается зарегистрировать только при термической обработке пленок висмута толщиной d 25 нм при Т = 373 К (рис. 3.12, кривая 1) [71 – 73, 75]. 69 Рис. 3.12 Кинетические кривые термического превращения пленок висмута толщиной d = 52 нм при Т = 373 К: 1 – линейный; 2 – обратный логарифмический закон; 3 – кубический закон; 4 – логарифмический закон. 70 Рис. 3.13 Кинетические кривые термического превращения пленок висмута толщиной d = 21 нм при Т = 473 К: 1 – обратный логарифмический закон; 2 – кубический закон; 3 – логарифмический закон. Обратный логарифмический участок на кинетических кривых α = f (τ) проявляется при термической обработке при Т = 373 К и 473 К пленок висмута толщиной d = 3…120 нм (рис. 3.12, кривая 2), при Т = 573 К d 35 нм (рис. 3.14 кривая 1) и при Т = 673 К d 80нм (рис. 3.15 кривая 1) [66, 67, 71 – 73, 80, 81]. 71 Рис. 3.14 Кинетические кривые термического превращения пленок висмута толщиной d = 34 нм при Т = 573 К: 1 – обратный логарифмический закон; 2 – кубический закон; 3 – логарифмический закон. По мере увеличения времени термической обработки пленок висмута разной толщины в интервале температур Т = 373…673 К кинетические кривые α = f (τ) удовлетворительно описываются в рамках кубического (рис. 3.12 – 3.15, кривая 3) и логарифмического (рис. 3.12 – 3.15, кривая 4) законов [66, 67, 71, 72, 80, 81]. 72 Рис. 3.15 Кинетические кривые термического превращения пленок висмута толщиной d = 27 нм при Т = 673 К: 1 – обратный логарифмический закон; 2 – кубический закон; 3 – логарифмический закон. 3.1.3 Определение термоэлектронной работы выхода наноразмерных пленок висмута и оксида висмута (III) методом контактной разности потенциалов Для выяснения причин, вызывающих наблюдаемые изменения спектров поглощения и отражения, а также кинетических кривых степени превращения пленок висмута в процессе термообработки, при тщательном экранировании одного из электродов из висмута, были измерены U Ф систем Bi – Bi2O3 и КРП для пленок Bi, Bi2O3 [68 – 73, 76, 85]. Из анализа результатов измерений КРП было установлено, что в области контакта Bi – Bi2O3 (из-за несоответствия между работами выхода из контактирующих партнеров) возникает запорный электрический слой [68 – 73, 76, 85, 86]. 73 В таблице 3.5 приведены значения КРП (VКРП) для висмута и оксида висмута (III) в атмосферных условиях и в вакууме до и после тепловой обработки. Из таблицы видно, что КРП для образцов висмута и оксида висмута практически не зависит от изменения давления в экспериментальной ячейке [69 – 73, 85]. Таблица. 3.5 Контактная разность потенциалов между пленками висмута, оксида висмута (III) и относительным электродом из платины. КРП, В Образец Давление, Па 1∙105 1∙10-5 1∙10-5* Bi 1) +1,11 +1,11 +0,21 Bi 2) +1,12 +1,12 +0,20 Bi 3) +1,11 +1,11 +0,21 Bi2O34) +0,23 +0,22 +0,22 Bi2O35) +0,23 +0,22 +0,21 1) Пленки висмута толщиной (d = 92 нм) на стекле получены путем термического испарения в вакууме 2∙10-3 Па. 2) Пленки висмута толщиной (d = 87 нм) на стекле получены путем термического испарения в вакууме 2∙10-3 Па. 3) Пленки висмута толщиной (d = 92 нм) на платиновой пластине получены путем термического испарения в вакууме 2∙10-3 Па. 4) Образцы Bi2O3 получены путем нанесения суспензии оксида висмута на подложку платины. 5) Образцы Bi2O3 получены путем нанесения пленок висмута толщиной (d = 84 нм) и их последующего полного окисления в атмосферных условиях при Т = 673 К. * После предварительной тепловой обработки при Т = 673 К в течение 90 мин. Предварительный прогрев пленок висмута в вакууме при 673 К приводит к значительному уменьшению значений КРП [70 – 73, 85, 86]. Причем, значения КРП для пленок висмута, подвергнутых предварительной тепловой обработке в вакууме, практически совпадают со значениями КРП измеренными для пленок Bi2O3. При термической обработке пленок Bi2O3 в 74 вакууме КРП незначительно уменьшается [71, 72, 85, 86]. На рис. 3.16 и 3.17 представлены значения термоэлектронных работ выхода (T) висмута и оксида висмута (III) до и после тепловой обработки при Т = 673 К в течение 90 мин [71, 72, 85]. Рис. 3.16 Термоэлектронные работы выхода пленок висмута (T(Bi)) до и после тепловой обработки при 673 K (T(Pt) = 5,3 эВ). Формирование на поверхности пленок висмута оксидного слоя Bi2O3,а также десорбция адсорбированных на поверхности пленок Bi2O3 донорных газов приводят к изменению значений КРП [87, 88]. Известно [88], наличие поверхностных уровней приводит к заряжению поверхности полупроводника. От заряда поверхности зависит работа выхода полупроводника, из этого следует, что работа выхода зависит и от степени заполнения поверхности адсорбированными частицами и от их природы. 75 Рис. 3.17 – Термоэлектронные работы выхода пленок оксида висмута (III) (T(Bi2O3)) до и после тепловой обработки при 673 K (T(Pt) = 5,3 эВ). На рис. 3.18 приведена диаграмма энергетических зон полупроводника оксида висмута (III). Рис. 3.18 Диаграмма энергетических зон полупроводника Bi2O3. 76 Термоэлектронную работу выхода (φT) и фотоэлектрическую работу выхода (φP) согласно [88] можно представить: T = s + + VD, (3.13) P = u + + VD, (3.14) s = v – Vs. (3.15) где Величины u и (где u – ширина запрещенной зоны, – энергия сродства свободного электрона к решетке) фиксируются природой полупроводника; они нечувствительны к адсорбции. Величина v также нечувствительна к адсорбции; она определяется природой и состоянием полупроводника (его «биографией», температурой, природой и концентрацией содержащейся в нем примеси). В то же время величины Vs и VD зависят от природы и количества адсорбированных частиц. Величина Vs характеризует загиб, обусловленный заряжением поверхности. Величина VD представляет собой дипольную составляющую работы выхода; она обусловлена падением потенциала в двойном электрическом слое, создаваемом адсорбированными молекулами. Если обозначить через (T) изменения термоэлектронной и (P) фотоэлектрической работы выхода, вызываемые адсорбцией, а через Vs и VD – соответствующие изменения поверхностного потенциала и дипольной составляющей работы выхода, то [88]: T = Vs + VD, (3.16) P = VD. (3.17) Согласно экспериментальным данным [88], в большинстве случаев: Vs >> VD. (3.18) Следовательно, по изменению термоэлектронной работы выхода можно судить об изменении поверхностного потенциала [88, 89]: T = Vs. (3.19) При адсорбции акцепторных молекул Vs > 0, при адсорбции донорных 77 молекул Vs < 0. Исходя из этого, по знаку T можно судить об акцепторной или донорной природе адсорбируемых молекул. Для образцов оксида висмута (III), для которых при адсорбции T < 0, характерна сорбция донорных молекул. Термоэлектронная работа выхода висмута меньше термоэлектронной работы выхода оксида висмута (III) (табл. 3.5) [71 – 73, 85], поэтому в процессе установления термодинамического равновесия электроны будут переходить из висмута в оксид висмута. Уровни Ферми при этом выравниваются, а валентная зона и зона проводимости у оксида висмута (III) у контакта с висмутом изгибаются вниз [71 – 73, 85]. На основании приведенных в табл. 3.5 данных была рассчитана напряженность электростатического поля в системе Bi – Bi2O3 по формуле [71 – 73, 85]: E = VКРП / d, (3.20) где VКРП – контактная разность потенциалов; d – толщина слоя Bi2O3. Таким образом, расчетная напряженность электростатического поля на границе контакта Bi – Bi2O3 (для пленок висмута различной толщины) составляет ~ 106 В/см и уменьшается к наружной поверхности оксидного слоя по мере увеличения толщины (d) оксидной пленки [69 – 72, 76, 85]. На рис. 3.19 приведена диаграмма энергетических зон контакта Bi – Bi2O3, при построении которой использованы результаты измерений КРП, UФ, спектров поглощения и отражения пленок Bi2O3 и Bi. 78 Рис. 3.19 Диаграмма энергетических зон системы Bi–Bi2O3. EV – уровень потолка валентной зоны, EC – уровень дна зоны проводимости, EF – уровень Ферми, EO – уровень вакуума, T+п – уровни поверхностных электронных состояний. Полярность UФ (рис. 3.19, переходы 1, 2) соответствует положительному знаку со стороны оксида висмута (III) [68 – 75, 85, 86]. Генерация UФ прямо свидетельствует о формировании в процессе термообработки пленок висмута гетеросистем Bi – Bi2O3, переходы носителей заряда на границе раздела которых обеспечивают наблюдаемые изменения спектров поглощения (рис. 3.3 – 3.6) и отражения, а также кинетических кривых степени превращения (рис. 3.7 – 3.15) [66, 68 – 73, 80]. 79 3.1.4 Модельные представления процесса окисления наноразмерных пленок висмута Согласно существующим представлениям [29, 63, 64, 88, 90] рост тонких пленок в результате взаимодействия твердого тела с газовой средой связан с процессами химической адсорбции газов (или их ингредиентов) на поверхности и в приповерхностной области твердого тела, формирования на поверхности или в приповерхностной области сначала «зародышей», а после образования нескольких периодов решетки и переноса ионов разного знака и электронов в сформированном слое – нового вещества (или веществ). В процессе химической адсорбции [88, 90] частицы, находящиеся в адсорбированном состоянии, отличаются по своей природе от соответствующих молекул в газовой фазе, представляя собой не сами молекулы, а отдельные части этих молекул, которые ведут на поверхности самостоятельное существование. Мы предполагаем, что при химической адсорбции О2 свободный электрон (в решетке Bi2O3, построенной из ионов Bi3+ и О2-, свободному электрону соответствует состояние Bi2+, а свободной дырке состояние О-, блуждающие по регулярным ионам Bi3+ и О2-) решетки оксида висмута (III) (по мере приближения молекулы кислорода к поверхности Bi2O3) все в большей степени локализуется около той точки на поверхности (S), к которой приближается молекула кислорода [69 – 72, 81]. При этом между атомами кислорода и поверхностью Bi2O3 возникают связи, обеспечиваемые локализующимися электронами (e S e) и упрочняющиеся по мере приближения молекулы кислорода. Связь между атомами кислорода постепенно ослабевает. В итоге атомы кислорода оказываются связанными прочными связями с поверхностью Bi2O3 [69 – 72] О + e S e = S О2-. Необходимые для ионизации хемосорбированных атомов кислорода электроны могут туннелировать из металла через слой оксида висмута (III) [29, 63, 64, 88, 90] 80 Bi → Bi3+ + 3е, и образоваться в результате дополнительного окисления части ионов висмута (Bi3+) оксида висмута (III) с образованием дырок (О-) [69 – 72] Bi3+ + е → Bi2+, О2- → О- + е. В начальный период окисления скорость роста пленки оксида висмута (III) постоянна и определяется стадией поверхностной реакции, а экспериментальные данные удовлетворительно описываются в рамках линейного закона (рис. 3.12 кривая 1) [71 – 73, 75]. Образующийся в процессе получения и термической обработки пленок висмута слой оксида висмута (III) будет препятствовать перемещению катионных вакансий от поверхности Bi2O3 к границе контакта Bi – Bi2O3 и, таким образом, тормозить взаимодействие висмута с кислородом [69 – 72, 81]. Одним из условий [29, 61 – 64, 90], характеризующих способность оксида висмута (III) тормозить процесс взаимодействия, является сплошность оксидной пленки. Согласно критерию Пиллинга и Бедвортса, который для висмута составляет 1,23, следует ожидать образования сплошной оксидной пленки [71 – 73, 81]. В том случае, когда толщина пленки Bi2O3 менее 5 нм электрическое поле на контакте Bi – Bi2O3 (напряженность электрического поля составляет ~ 106 В/см) способно вырывать ионы из металла и перемещать их через слой оксида [29, 61 – 64, 90]. При этом скорость роста пленки определяется скоростью вырывания ионов из металла, а экспериментальные данные удовлетворительно описываются в рамках обратного логарифмического закона (рис. 3.12 кривая 2; рис. 3.13-3.14 кривая 1) [67, 69 – 72, 80]. Из-за достаточно высокой подвижности электронов и низкой скорости движения катионных вакансий в системе Bi – Bi2O3 возникает потенциал. Этот потенциал создает электрическое поле в слое Bi2O3, которое стимулирует движение катионных вакансий к поверхности Bi2O3. При этом, согласно [29, 61 – 64, 90] для оксидов, у которых основными носителями 81 заряда являются логарифмический дырки, законы может роста быть тонких реализован пленок кубический (оксид висмута и – полупроводник p-типа) [69 – 72, 81]. По видимому, скорость роста оксида висмута (III) на поверхности пленок висмута толщиной d = 3 – 120 нм (термическая обработка при Т = 373 К и 473 К), d = 35 – 120 нм (Т = 573 К), толщиной d = 80 – 120 нм (Т = 673 К) при малых временах термической обработки (рис. 3.7 – 3.15) будет определяться скоростью вырывания ионов Bi3+ из висмута, а при увеличении времени и температуры термообработки, а также толщины пленок Bi скоростью диффузии катионных вакансий от поверхности Bi2O3 (лимитирующей стадией процесса является диффузия катионных вакансий через слои оксида висмута к границе контакта Bi – Bi2O3), дальнейший рост которой будет замедляться по мере увеличения толщины пленки Bi2O3 [67, 69 – 72, 81]. Скорость диффузии катионных вакансий, в свою очередь, будет пропорциональна напряженности электрического поля [29, 61 – 64, 90]. Ионы кислорода (О2-) в приповерхностной области оксида висмута (III) создают новые узлы. Вследствие этого в приповерхностной области Bi2O3 появляется недостаток занятых катионами узлов кристаллической решетки, т. е. формируются катионные вакансии (Vк3-), наличие которых облегчает перемещение катионов Bi3+ от металла к внешней поверхности формируемой системы Bi – Bi2O3 [69 – 73, 81]. 82 3.2 Фотохимическое окисление наноразмерных пленок висмута Исследование закономерностей процессов, протекающих при воздействии света различной интенсивности в наноразмерных слоях висмута, стимулируется стремлением к разработке методов и средств использования излучения солнечного диапазона и более рациональной утилизации подведенной энергии [8, 56, 58, 91 – 94]. В данном параграфе приведены результаты исследований фотостимулированных превращений в наноразмерных пленках висмута различной толщины (d = 3 – 55 нм) при облучении их светом = 360 нм различной интенсивности I = 1,8·1015 – 7,0·1015 квант·см-2·с-1. 3.2.1 Спектры оптического поглощения и зеркального отражения наноразмерных пленок висмута до и после облучения светом Исследование оптического поглощения в твердом теле позволяет выяснить энергетику и природу электронных переходов, происходящих под действием света, в частности, получить информацию о создании в изучаемых объектах неравновесных электронов и дырок. При исследовании оптических свойств наноразмерных пленок висмута, нанесенных на стеклянные подложки, до, в процессе и после воздействия света = 360 нм различной интенсивности (I = 1,8·1015 – 7,0·1015 квант·см-2·с1 ) в атмосферных условиях при Т = 293 К, прежде всего, было установлено, что спектры поглощения и отражения, масса наноразмерных пленок висмута разной толщины (d = 3 – 55 нм) значительно изменяются [95 – 104]. Причем, наблюдаемые изменения в значительной степени зависят от первоначальной толщины пленок висмута длины волны и интенсивности падающего света, времени облучения [95 – 104]. На рис. 3.20 приведены спектры поглощения пленок висмута толщиной d = 26 нм до и после облучения светом = 360 нм. Видно, что наблюдаемые изменения не аддитивны в рассматриваемом спектральном диапазоне длин волн. 83 Рис. 3.20 Спектры поглощения пленки висмута толщиной 26 нм до (1) и после облучения светом = 360 нм и интенсивности I = 7∙1015 квант∙см-2∙с-1 в течение: 2 – 9 мин, 3 – 15 мин, 4 – 30 мин, 5 – 40 мин, 6 – 60 мин, 7 – 85 мин, 8 – 100 мин, 9 – 160 мин, 10 – 185 мин, 11 – 230 мин, 12 – 370 мин. При = 320 нм наблюдается изобестическая точка. Наряду с уменьшением в интервале = 320 – 1100 нм и увеличением в интервале 320 нм оптической плотности образца формируется спектр поглощения нового вещества [95 – 104]. Оцененная по длинноволновому порогу поглощения, который находится при ≈ 387 нм, оптическая ширина запрещенной зоны, образующегося вещества составляет, Е ≈ 3,2 эВ. Полученное значение ширины запрещенной зоны вещества удовлетворительно совпадает с шириной запрещенной зоны оксида висмута (III) [71 – 73, 84, 95 – 104]. Поэтому, было сделано предположение, что при облучении пленок висмута основным продуктом взаимодействия их с кислородом окружающей среды является оксид висмута (III). При увеличении интенсивности толщины падающего пленок света) висмута наблюдается (при постоянной последовательное уменьшение эффектов изменения оптической плотности образцов во всем 84 исследованном спектральном диапазоне [95, 97, 98, 102, 104]. По мере увеличения интенсивности падающего света (при одинаковой исходной толщине пленок висмута) наблюдается возрастание эффектов изменения оптической плотности [95, 97, 98, 102, 104]. 3.2.2 Кинетические закономерности стимулированных светом превращений наноразмерных пленок висмута Для выяснения закономерностей взаимодействия пленок висмута с кислородом окружающей среды (используя результаты гравиметрических исследований, измерений спектров поглощения и отражения пленок висмута разной толщины до и после облучения образцов светом = 360 нм и разной интенсивности) были рассчитаны и построены кинетические зависимости степени превращения α = (). Установлено [97 – 99], что спектры поглощения пленок висмута, измеренные при различных временах облучения светом = 360 нм, пересекаются в одной (изобестической) точке, в которой оптическая плотность не зависит от времени облучения, а интенсивность поглощения слоями висмута и оксида висмута (III) одинакова. Слева и справа от изобестической точки оптическая плотность (А) пленки висмута зависит от времени облучения, а при определенном времени облучения будет складываться из оптической плотности, связанной с наличием слоя висмута (АBi) и оксида висмута (III) (АBi2O3) [97 – 99]: А = АBi + АBi2O3. (3.21) Для построения кинетических кривых α = () был выбран диапазон длин волн = 400…900 нм, в котором пленки висмута имеют значительное поглощение, а поглощением Bi2O3 можно пренебречь [71, 72, 97 – 99]. 85 Рис. 3.21 Зависимость степени превращения от толщины пленок висмута: 1 – 3 нм; 2 – 16 нм; 3 – 26 нм; 4 – 40 нм; 5 – 55 нм ( = 360 нм, интенсивность I = 7∙1015 квант∙см-2∙с-1). Если обозначить через α степень фотохимического превращения пленок висмута в оксид висмута (III), при длине волны ( = 800 нм), то на основании приведенных выше уравнений (3.5), (3.6) получаем выражение для степени фотохимического превращения пленки висмута в оксид висмута (III) [97 – 99, 103, 104]: Аобр.= А1Bi (1 – α) + А1Bi2O3·α, (3.22) α = (А1Bi – А обр.) / (А1Bi – А1Bi2O3). (3.23) Учитывая [46, 77], что падающая по нормали на поверхность какой-либо системы световая волна от источника излучения, претерпевает зеркальное отражение, рассеяние, поглощение и пропускание для расчета истинного вызванного поглощением света в веществе значение оптической плотности после преобразований (3.10), (3.11), примет вид: Aобр. = A + lg(1 – R). (3.24) Степень фотохимического превращения пленок висмута зависит от первоначальной толщины, времени облучения и интенсивности падающего света (рис. 3.21, 3.22) [95 – 104]. Видно, что с уменьшением толщины пленок 86 висмута (при постоянной интенсивности падающего света) наблюдается увеличение степени превращения (рис. 3.21) [96, 97, 98, 100, 103]. По мере увеличения интенсивности падающего света в диапазоне I = 1,8·1015 – 7,0·1015 квант·см-2·с-1 (при постоянной толщине пленки висмута) степень превращения возрастает (рис. 3.22) [96, 97, 98, 100, 103]. При сопоставлении кривых α = () установлено, что степень превращения пленок висмута облучаемых светом = 360 нм значительно больше, чем при термической обработке при Т = 293 К (рис. 3.22) [95, 97 – 104]. Рис. 3.22 Зависимость степени превращения пленок висмута толщиной 26 нм от интенсивности падающего света = 360 нм: 1) 7∙1015, 2) 5,2∙1015, 3) 3,9∙1015, 4) 2,7∙1015, 5) 1,8∙1015 квант·см-2∙с-1, термообработки при Т = 293 К (6). Кинетические кривые степени превращения пленок висмута разной толщины условно можно разбить на несколько участков (табл. 3.6) [95 – 104]: линейный (α = Kτ + A) (кривая 1), обратный логарифмический (K / α = B – lgτ) (кривая 2), кубический (α3 = Kτ + B) (кривая 3) и логарифмический (α = K lg(Bτ + 1)) (кривая 4), где К – константа скорости формирования оксида висмута (III), A и B – постоянные интегрирования, τ – время взаимодействия. 87 Таблица 3.6. Зависимость продолжительности линейного, обратного логарифмического, кубического, логарифмического участков от толщины пленок висмута в процессе облучения светом = 360 нм интенсивностью I = 7∙1015 квант∙см-2∙с-1. d,нм α= Kτ + A K/α = B – lgτ 3 16 26 40 55 0 – 3мин 0 – 20мин 0 – 40мин 0 – 80мин 0 – 140мин 3 – 5мин 20 – 30мин 40 – 80мин 80 – 100мин 140 – 160мин 5 – 8мин 30 – 50мин 80 – 100мин 100 – 140мин 160 – 240мин 8 – 15мин 50 – 70мин 100–120мин 140 – 180мин 240 – 500мин α3 = Kτ + B 15 – 40мин 70 – 100мин 120–240мин 180 – 370мин 500 – 1400мин 40 – 55мин 100–140мин 240–300мин 370 – 600мин 1400–1600мин α= K lg(Bτ+1) 55 – 80мин 140–180мин 300–370мин 600 – 960мин 1600–1800мин На рисунках 3.23, 3.24 в качестве примера представлены кинетические кривые степени превращения пленок висмута толщиной d = 16, 26 нм соответственно, при облучении светом = 360 нм, I = 7·1015 квант·см-2·с-1. 88 Рис. 3.23 Кинетические кривые степени превращения пленок висмута толщиной d = 16 нм: 1 – линейный; 2 – обратный логарифмический закон; 3 – кубический закон; 4 – логарифмический закон ( = 360 нм, I = 7∙1015 квант∙см-2∙с-1). Наличие соответствующих участков, а также их продолжительность определяется толщиной пленок висмута и интенсивностью падающего света. По мере увеличения толщины пленок висмута и уменьшения интенсивности падающего света наблюдается увеличение продолжительности участков кинетических кривых степени превращения [95 – 104]. 89 Рис. 3.24 Кинетические кривые степени превращения пленок висмута толщиной d = 26 нм: 1 – линейный; 2 – обратный логарифмический закон; 3 – кубический закон; 4 – логарифмический закон ( = 360 нм, I = 7∙1015 квант∙см-2∙с-1). 3.2.3 Фотоэлектрические свойства систем Bi - Bi2O3 Для выяснения причин, вызывающих наблюдаемые изменения спектров поглощения и отражения, а также кинетических кривых степени превращения пленок висмута при воздействии света = 360 нм были измерены UФ систем Bi – Bi2O3 и КРП. Из анализа результатов измерений КРП (табл.3.7) и UФ было установлено, что в области контакта Bi – Bi2O3 (изза несоответствия между работами выхода из контактирующих партнеров) возникает двойной электрический слой [95 – 99, 102, 104]. 90 Таблица 3.7 Контактная разность потенциалов между пленками висмута, оксида висмута (III) и относительным электродом из платины при 293 К. КРП, В Образец Давление, Па 1∙105 1∙10-5 Bi 1) +1,11 +1,11 Bi2O3 2) +0,22 +0,21 1) Пленка висмута толщиной 55 нм. Образец Bi2O3 получен путем полного окисления пленки висмута толщиной 55 нм при облучении светом = 360 нм и интенсивности I = 7,0∙1015 квант∙см-2∙с-1. 2) Согласно уравнению (3.20) напряженность электрического поля на границе контакта Bi – Bi2O3 для пленок висмута различной толщины может составить ~ 106 – 107 В/см [95 – 99, 102, 104]. Знак UФ со стороны оксида висмута – положительный [95 – 99, 102, 104]. Генерация UФ прямо свидетельствует о перераспределении неравновесных носителей заряда и формировании в процессе облучения пленок висмута гетеросистем Bi – Bi2O3, фотоэлектрические процессы на границе раздела которых обеспечивают наблюдаемые изменения спектров поглощения (рис. 3.20), а также кинетических кривых степени превращения (рис. 3.21 – 3.24) [95, 97, 98, 102 – 104]. Фотохимические проявления фотоэлектрических процессов в таких системах могут быть вызваны перераспределением под действием контактного поля генерированных светом носителей заряда [63 – 65, 71, 72, 95 – 98, 102, 104 – 108]. Эти процессы приведут к существенным изменениям условий протекания реакции окисления пленок висмута в системе Bi – Bi2O3 по сравнению с окислением в атмосферных условиях. На рис. 3.25 приведена диаграмма энергетических зон контакта Bi – Bi2O3, при построении которой использованы результаты измерений КРП (табл. 3.7), UФ, спектров поглощения и отражения пленок Bi и Bi2O3. 91 Рис. 3.25 Диаграмма энергетических зон системы Bi–Bi2O3. EV – уровень потолка валентной зоны, EC – уровень дна зоны проводимости, EF – уровень Ферми, E0 – уровень вакуума, TП+ – уровни ПЭСК, R+ – центр рекомбинации. 3.2.4 Модельные представления стимулированного светом процесса окисления наноразмерных пленок висмута При воздействии света на систему Bi – Bi2O3 из области собственного поглощения оксида висмута имеет место интенсивная генерация электрондырочных пар в оксиде висмута (рис. 3.25, переход 1) А2- p + е и фотоэмиссия дырок из металла в валентную зону оксида висмута (рис. 3.25, переход 2) [97 – 99]. Генерированные в области пространственного заряда оксида висмута неравновесные носители заряда рекомбинируют (рис. 3.25, переходы 3) [97] 92 R+ + е R0 + р R+, где R+ – центр рекомбинации, перераспределяются в контактном поле, сформированном из-за несоответствия между термоэлектронными работами выхода оксида висмута, висмута и наличия структурных дефектов (Tп+) на границе Bi – Bi2O3, с переходом неравновесных электронов из зоны проводимости оксида висмута на уровни Tп+ или непосредственно в металл (Ме+) (рис. 3.25, переходы 4, 5) [97 – 99] Тп+ + е → Тп0, Ме+ + е → Ме0 и захватываются поверхностными центрами (S) на наружной поверхности Bi2O3 S + e → S e + e → e S e. Одновременно с отмеченными переходами, в результате которых формируется Uф и происходит смещение энергетических уровней у контактирующих партнеров, имеют место потоки равновесных носителей заряда [97 – 99]. Эти процессы, во-первых, приводят к возрастанию концентрации носителей заряда в Bi2O3; во-вторых, могут стимулировать диффузию катионных вакансий (VК3-) от наружной поверхности Bi2O3 к Bi. Как уже было отмечено ранее (раздел 3.1.4), и согласно [29, 61 – 64, 88, 90] рост тонких пленок в результате взаимодействия твердого тела с газовой средой связан с процессами химической адсорбции газов на поверхности твердого тела, формирования на поверхности или в приповерхностной области сначала «зародышей», а после образования нескольких периодов решетки и переноса ионов разного знака и электронов в сформированном слое – нового вещества (или веществ). В начальный период окисления скорость роста пленки оксида висмута (III) постоянна и определяется стадией поверхностной реакции, а экспериментальные данные удовлетворительно описываются в рамках линейного закона (рис. 3.23, 3.24 кривая 1) [95, 98, 99, 102 – 104]. 93 Образующийся в процессе облучения пленок висмута слой оксида висмута (III) будет препятствовать перемещению катионных вакансий от поверхности Bi2O3 к границе контакта Bi – Bi2O3 и, таким образом, тормозить процесс взаимодействия висмута с кислородом [97 – 99]. Сплошность оксидной пленки является одним из условий [29, 61 – 64, 90], характеризующих способность оксида висмута (III) тормозить процесс взаимодействия (критерий Пиллинга и Бедвортса равен 1,23) [97 – 99]. Мы полагаем, что при химической адсорбции О2 свободный электрон оксида висмута (III) (по мере приближения молекулы кислорода к поверхности Bi2O3) все в большей степени локализуется около той точки на поверхности (S), к которой приближается молекула кислорода [97 – 99]. При этом между атомами кислорода и поверхностью Bi2O3 возникают связи, обеспечиваемые локализующимися электронами (e S e) и упрочняющиеся по мере приближения молекулы кислорода. Связь между атомами кислорода постепенно ослабевает[97 – 99]. В итоге атомы кислорода оказываются связанными прочными связями с поверхностью Bi2O3 О + e S e = S О2-. Необходимые для ионизации хемосорбированных атомов кислорода электроны могут туннелировать из металла через слой оксида висмута (III) [29, 61 – 64, 88, 90] Bi → Bi3+ + 3е. В том случае, когда толщина пленки Bi2O3 менее 5 нм электрическое поле на контакте Bi – Bi2O3 (напряженность электрического поля составляет ~ 106 В/см) способно вырывать ионы из металла и перемещать их через слой оксида [29, 61 – 64, 90, 97 – 99]. При этом скорость роста пленки определяется скоростью вырывания ионов из металла, а экспериментальные данные удовлетворительно описываются в рамках обратного логарифмического закона (рис. 3.23, 3.24 кривая 2) [95, 97, 98, 102 – 104]. По мере увеличения толщины оксидной пленки (за пределами области пространственного заряда оксида висмута (III)) процесс взаимодействия 94 висмута с кислородом будет тормозиться диффузией катионных вакансий от поверхности Bi2O3 (лимитирующей стадией процесса является диффузия катионных вакансий через слои оксида висмута к границе контакта Bi – Bi2O3), дальнейший рост которой будет замедляться по мере увеличения толщины пленки Bi2O3 [97 – 99]. При этом, согласно [29, 61 – 64, 90] для оксидов, у которых основными носителями заряда являются дырки, может быть реализован кубический и логарифмический законы роста тонких пленок (оксид висмута – полупроводник p-типа) (рис. 3.23, 3.24 кривая 3, 4) [95, 97, 98, 102 – 104]. Скорость диффузии катионных вакансий, в свою очередь, будет пропорциональна напряженности электрического поля [29, 61 – 64, 90]. Ионы кислорода (S О2-) в приповерхностной области оксида висмута (III) создают новые узлы. Вследствие этого в приповерхностной области Bi2O3 появляется недостаток занятых катионами узлов кристаллической решетки, т. е. формируются катионные вакансии (Vк3-), наличие которых облегчает перемещение катионов Bi3+ от металла к внешней поверхности формируемой системы Bi – Bi2O3 [97 – 99]. 95 3.3 Модификация наноразмерных пленок висмута в атмосфере аммиака Отсутствие к настоящему времени в отечественной и зарубежной литературе систематических исследований о влиянии размерных эффектов на газостимулированные процессы в наноразмерных пленках висмута, ставят правомерной и своевременной задачу комплексного исследования свойств наноразмерных пленок висмута, в частности, выяснения степени общности процессов, протекающих на границе между металлом и окружающей атмосферой[63, 64, 109 - 112], так и в связи с необходимостью разработки принципиально новых материалов, стабильных в условиях коррозионного воздействия окружающей среды. В данном параграфе представлены результаты исследований, направленные на выяснение природы и закономерностей процессов, протекающих в наноразмерных пленках висмута различной толщины (d = 1 – 56 нм) при Т = 293 К в зависимости от времени воздействия газообразного аммиака ( = 1 мин – 5400 час). 3.3.1 Влияние газообразного аммиака на оптические свойства наноразмерных пленок висмута Спектры поглощения и отражения пленок висмута разной толщины (нанесенных на стеклянные подложки) до, в процессе и после воздействия газообразного аммиака при Т = 298 К существенно зависят от толщины указанных образцов [71, 72, 97, 98, 113 – 122]. На рис. 3.26 представлены спектры поглощения пленок висмута толщиной d = 1…56 нм. 96 Рис. 3.26 Спектры поглощения пленок висмута толщиной: 1 – 56 нм, 2 – 49 нм, 3 – 43 нм, 4 – 39 нм, 5 – 33 нм, 6 – 28 нм, 7 – 25 нм, 8 – 20 нм, 9 – 16 нм, 10 – 11 нм, 11 – 4нм, 12 – 1нм. Видно, что в исследуемом диапазоне длин волн на спектрах поглощения образцов толщиной более 28 нм можно выделить характерные для висмута полосы поглощения (в частности – максимумы поглощения при λ 410 нм и 735 нм) [71, 72, 74, 97, 98, 119 – 122]. По мере уменьшения толщины пленок висмута на спектрах поглощения и отражения постепенно перестают проявляться характерные для висмута полосы поглощения и отражения. Для пленок висмута толщиной d 28 нм наблюдается бесструктурное поглощение и отражение в диапазоне = 190…1100 нм [71, 72, 97, 98, 119 – 122]. В результате взаимодействия пленок висмута разной толщины с газообразным аммиаком при температуре Т = 298 К спектры поглощения, отражения и масса образцов претерпевают существенные изменения. Причем, наблюдаемые изменения массы, спектров поглощения и отражения после воздействия газообразного аммиака на пленки висмута в значительной степени зависят от первоначальной толщины пленок висмута и времени их взаимодействия с аммиаком [113 – 122]. 97 На рисунках 3.27, 3.28 в качестве примера приведены спектры поглощения пленок висмута толщиной d = 22 нм, 31 нм соответственно, до и после взаимодействия с газообразным аммиаком при 293 К. Рис. 3.27 Спектры поглощения пленки висмута толщиной (d = 22 нм) до (1) и после взаимодействия с газообразным аммиаком при Т = 293 К в течение: 2 – 17 мин, 3 – 21 мин, 4 – 16 ч, 5 – 37 ч, 6 – 61 ч, 7 – 73 ч, 8 – 85 ч, 9 – 133 ч, 10 – 226 ч, 11 – 576 ч. Рис. 3.28 Спектры поглощения пленки висмута толщиной (d = 31 нм) до (1) и после взаимодействия с газообразным аммиаком при Т = 293 К в течение: 2 – 3 мин, 3 – 12 мин, 4 – 32 мин, 5 – 8 ч, 6 – 16 ч, 7 – 40 ч, 8 – 64 ч, 9 – 85 ч, 10 – 133 ч, 11 – 203 ч, 12 – 227 ч, 13 – 395 ч, 14 – 720 ч, 15 – 2880 ч. 98 Видно, что воздействие газообразного аммиака приводит к существенным изменениям вида спектров поглощения образцов. Установлено [113 – 122], что наблюдаемые изменения не аддитивны в рассматриваемом спектральном диапазоне длин волн. Наряду с уменьшением в интервале = 315…1100 нм и увеличением в диапазоне = 300…315 нм значений оптической плотности образца формируется спектр поглощения нового вещества. Оцененная по длинноволновому порогу поглощения, который находится при ≈ 336 нм, оптическая ширина запрещенной зоны образующегося вещества, согласно (3.2) и (3.3), составляет Е ≈ 3,68 – 3,70 эВ [113 – 122]. Было сделано предположение, что основным продуктом взаимодействия пленок висмута с газообразным аммиаком является нитрид висмута. Последующее продолжительное (в течение 2х лет) хранение нитрида висмута в атмосфере газообразного аммиака, а также в атмосферных условиях не приводит к заметному изменению спектров поглощения и отражения [113 – 122]. Закономерности изменения спектров поглощения при увеличении или уменьшении толщины пленок висмута сохраняются (в коротковолновой области спектра – слева от изобестической точки наблюдается увеличение оптической плотности, а в длинноволновой области спектра – справа от изобестической точки наблюдается уменьшение оптической плотности образцов) [115, 117 – 122]. 3.3.2 Кинетические закономерности взаимодействия наноразмерных пленок висмута с газообразным аммиаком Используя результаты гравиметрических исследований и измерений спектров поглощения и отражения пленок висмута разной толщины, для выяснения закономерностей протекания процесса взаимодействия пленок висмута с газообразным аммиаком при температуре 293 К, были рассчитаны и построены кинетические зависимости степени превращения (α = ()). Для построения кинетических кривых в координатах α = () был 99 применен подход следующий подход [119 – 122]. Спектры поглощения пленок висмута, измеренные при различных временах взаимодействия с газообразным аммиаком, пересекаются в одной (изобестической) точке, в которой оптическая плотность не зависит от времени взаимодействия, а интенсивность поглощения слоями висмута и нитрида висмута одинакова. Слева и справа от изобестической точки оптическая плотность (Аобр.) пленки висмута зависит от времени взаимодействия с газообразным аммиаком, а при определенном времени взаимодействия будет складываться из оптической плотности, связанной с наличием слоя висмута (АBi) и нитрида висмута (АBiN) [119 – 122]: Аобр. = АBi + АBiN. (3.25) На основании анализа спектров поглощения и отражения пленок висмута и BiN для построения кинетических кривых α = () был выбран диапазон длин волн = 400…900 нм, в котором пленки висмута имеют значительное поглощение, а поглощением BiN можно пренебречь [119 – 122]. Обозначим через α степень превращения пленок висмута в нитрид висмута, при длине волны (например, = 800 нм – рис. 3.27, 3.28), соответствующей спектральной области, в пределах которой висмут поглощает, а нитрид висмута практически не поглощает свет, текущие оптические плотности пленок висмута (АBi) и нитрида висмута (АBiN) можно представить в следующем виде АBi = А1Bi (1 – α), (3.26) АBiN = А1BiN·α, (3.27) где А1Bi, А1BiN – предельные значения оптической плотности пленок висмута и нитрида висмута при = 800 нм. В итоге получаем следующее выражение для степени превращения пленки висмута в нитрид висмута [117 – 122]: Аобр. = А1Bi (1 – α) + А1BiN·α, (3.28) α = (А1Bi – А обр.) / (А1Bi – А1BiN). (3.29) Установлено [113 – 122], что степень превращения зависит от 100 первоначальной толщины пленок висмута и времени воздействия газообразного аммиака. В качестве примера на рис. 3.29 приведены кинетические кривые степени превращения пленок висмута разной толщины при температуре 293 К в процессе воздействия газообразного аммиака. Рис. 3.29 Зависимость степени превращения от толщины пленок висмута при Т = 293 К при взаимодействии с газообразным аммиаком: 1 – 13 нм, 2 – 22 нм , 3 – 31 нм, 4 – 56 нм. C уменьшением толщины пленок висмута наблюдается увеличение скорости процесса взаимодействия Bi c газообразным аммиаком. Кинетические кривые степени превращения пленок висмута разной толщины условно можно разбить на несколько участков (табл. 3.8) [113 – 122]: линейный (α = Kτ + A), обратный логарифмический (K / α = B – lgτ), параболический (α2 = Kτ + B) и логарифмический (α = K lg(Bτ + 1)), где К – константа скорости формирования нитрида висмута, A и B – постоянные интегрирования, τ – время взаимодействия. 101 Таблица 3.8. Зависимость продолжительности линейного, обратного логарифмического, параболического, логарифмического участков от толщины пленок висмута в процессе воздействия аммиака. d,нм 13 22 31 56 α = Kτ + A 0 – 19мин 0 – 22мин 0 – 32мин 0 – 48мин K/α = B – lgτ 19мин – 61ч 22мин – 85ч 61ч – 133ч 85ч – 157ч 109ч – 298ч 133 – 370ч 133ч – 271ч 157ч – 364ч 298ч – 576ч 370 – 672ч 271ч – 364ч 364ч – 480ч 576ч – 720ч 672 – 3024ч 364ч – 1200ч 480ч – 1800ч 720ч – 2880ч 3024 – 5400ч α2 = Kτ + B α = K lg(Bτ+1) 32мин – 109ч 48мин – 133ч На рисунках 3.30, 3.31 в качестве примера приведены кинетические кривые степени превращения пленок висмута толщиной d = 13 нм, 31 нм соответственно при Т = 293 К в процессе взаимодействия с аммиаком. 102 Рис. 3.30 Кинетические кривые степени превращения пленок висмута толщиной d = 13 нм при Т = 293 К в процессе взаимодействия с аммиаком: 1 – линейный; 2 – обратный логарифмический закон; 3 – параболический закон; 4 – логарифмический закон. Продолжительность соответствующих участков определяется толщиной пленок висмута. По мере увеличения толщины пленок висмута наблюдается увеличение продолжительности участков кинетических кривых степени превращения [113 – 122]. 103 Рис. 3.31 Кинетические кривые степени превращения пленок висмута толщиной d = 31 нм при Т = 293 К в процессе взаимодействия с аммиаком: 1 – линейный; 2 – обратный логарифмический закон; 3 – параболический закон; 4 – логарифмический закон. 3.3.3 Контактная разность потенциалов для пленок Bi и BiN Для выяснения направления носителей заряда на границе раздела гетеросистем Bi – BiN, а также причин, вызывающих наблюдаемые изменения спектров поглощения и отражения, а также кинетических кривых степени превращения пленок висмута в процессе воздействия газообразного аммиака были измерены UФ систем Bi – BiN и КРП для пленок Bi, BiN. Из анализа результатов измерений КРП (табл. 3.9) было установлено, что в области контакта Bi – BiN (из-за несоответствия между работами выхода из контактирующих партнеров) возникает запорный электрический слой [115, 104 117 – 121]. Напряженность электрического поля на границе контакта Bi – BiN (для пленок висмута различной толщины) составляет ~ 106 – 107 В/см. Таблица 3.9 Контактная разность потенциалов между пленками висмута, нитрида висмута и относительным электродом из платины. Образец КРП, В Р = 1·105 Па Р = 1·10-5 Па Пленка Bi 1) +1,11 +1,11 Пленка BiN 2) +1,61 +1,60 1) Пленки висмута толщиной (d = 92 нм) получены путем термического испарения в вакууме 2∙10-3 Па. 2) Образцы BiN получены путем нанесения пленок висмута толщиной (d = 40 нм) и подвергнутых взаимодействию с газообразным аммиаком (Т = 293 К) при 100 % превращении в конечный продукт – BiN. Полярность UФ (рис. 3.32, переходы 1, 2) соответствует отрицательному знаку со стороны нитрида висмута [115, 117 – 121]. Генерация UФ прямо свидетельствует о формировании в процессе воздействия газообразного аммиака на пленки висмута гетеросистем Bi – BiN, переходы носителей заряда на границе раздела которых обеспечивают наблюдаемые изменения спектров поглощения (рис. 3.27, 3.28) и отражения, а также кинетических кривых степени превращения (рис. 3.29 – 3.31) [115, 117 – 122]. На рис. 3.32 приведена диаграмма энергетических зон контакта Bi – BiN, при построении которой использованы результаты измерений КРП, UФ, спектров поглощения и отражения пленок BiN и Bi. 105 Рис. 3.32 Диаграмма энергетических зон системы Bi – BiN. EV – уровень потолка валентной зоны, EF – уровень Ферми, EC – уровень дна зоны проводимости, E0 – уровень вакуума, T п – уровни поверхностных электронных состояний контакта. 3.3.4 Модельные представления процесса превращения наноразмерных пленок висмута в атмосфере газообразного аммиака при Т = 293 К В процессе химической адсорбции [88, 90] частицы, находящиеся в адсорбированном состоянии, отличаются по своей природе от соответствующих молекул в газовой фазе, представляя собой не сами молекулы, а отдельные части этих молекул, которые ведут на поверхности самостоятельное существование. Мы предполагаем, что при химической адсорбции NH3 свободные электроны решетки нитрида висмута (по мере приближения молекулы аммиака к поверхности BiN) все в большей степени локализуется около той точки на поверхности (S), к которой приближается молекула NH3. При этом 106 между атомами азота и поверхностью BiN возникают связи, обеспечиваемые локализующимися электронами (S 3e) и упрочняющиеся по мере приближения молекулы аммиака. Связь между атомом азота и тремя атомами водорода постепенно ослабевает. В итоге атомы азота оказываются связанными прочными связями с поверхностью BiN [115, 117, 119 – 122] N + S 3e = S N3-. Необходимые для ионизации хемосорбированных атомов азота электроны могут туннелировать из металла через слой нитрида висмута [63, 64 , 71, 72, 88, 90, 115, 117, 119 – 122] Bi → Bi3+ + 3е Скорость роста пленки нитрида висмута на этапе линейного участка кинетической кривой степени превращения (α = Kτ + A), соответствующая начальному периоду взаимодействия пленки висмута с газообразным аммиаком, постоянна и определяется стадией поверхностной реакции [115, 117, 119 – 122]. Сформированный слой нитрида висмута будет препятствовать перемещению катионов от Bi к внешней поверхности BiN и, таким образом, тормозить процесс взаимодействия висмута с газообразным аммиаком [115, 117, 119 – 122]. Из-за достаточно высокой подвижности электронов и низкой скорости движения катионов в системе Bi - BiN возникает потенциал. Этот потенциал создает электрическое поле в слое BiN, которое стимулирует движение катионов висмута к наружной поверхности нитрида висмута. В том случае, когда толщина пленки BiN менее 5 нм электрическое поле на контакте Bi - BiN (напряженность электрического поля составляет ~ 106 – 107 В/см) способно вырывать ионы из металла и перемещать их через слой нитрида [115, 117, 119 – 122]. При этом скорость роста пленки нитрида висмута определяется скоростью вырывания ионов висмута из металла, а экспериментальные данные удовлетворительно описываются в рамках обратного логарифмического закона (K / α = B – lgτ) [115, 117, 119 – 122]. При дальнейшем увеличении времени взаимодействия пленок висмута 107 толщиной d = 1 – 56 нм с газообразным аммиаком (рис. 3.29 – 3.31) скорость роста нитрида висмута будет определяться скоростью диффузии катионов Bi3+ к внешней поверхности BiN, дальнейший рост которой будет замедляться по мере увеличения толщины пленки нитрида висмута, а кинетические кривые удовлетворительно описываться в рамках параболического (α2 = Kτ + B) и логарифмического законов (α = K lg(Bτ + 1)) [115, 117, 119 – 122]. Ионы азота (N3-) в приповерхностной области нитрида висмута создают новые узлы. Вследствие этого в приповерхностной области BiN появляется недостаток занятых катионами узлов кристаллической решетки, т. е. формируются катионные вакансии (Vк3-), наличие которых облегчает перемещение катионов Bi3+ от металла к внешней поверхности формируемой системы Bi – BiN [115, 117, 119 – 122]. 108 3.4 Исследование термо- фото- и газостимулированных превращений наноразмерных пленок висмута методом кварцевого микровзвешивания Исследования изменения массы наноразмерных пленок висмута в процессе воздействия газообразного аммиака, окисления при различных температурах (Т = 373 – 673 К), а также облучения светом ( = 360 нм, I = (1,8 – 7,0)·1015 квант·см-2·с-1), осуществляли методом кварцевого микровзвешивания, основанном на определении приращения массы (∆m) на единицу поверхности кварцевого резонатора (толщиной h = 0,1 мм) после нанесения на нее пленки висмута. Разрешающая способность при термостабилизации резонатора на уровне ± 0,1 К составляет ∆m = 1∙10-8 – 1∙10-9 г/см2 [34 – 36]. Согласно уравнению Зауэрбрея (2.7), приращение массы (т) может регистрироваться с тем же разрешением, что и изменение частоты (f) резонатора. Тогда степень превращения [71 – 73, 97 – 99, 104, 117, 119 – 121]: α = f1 / f2, (3.30) f1 = fИ – fТ, f2 = fИ – fК, (3.31) где fИ – частота резонатора с нанесенной пленкой висмута, fТ – текущая частота резонатора с нанесенной пленкой висмута в процессе термической обработки, облучения светом = 360 нм или взаимодействия с газообразным аммиаком, fК – частота резонатора с нанесенной пленкой висмута, подвергнутой 100 % превращению в конечный продукт – Bi2O3 или BiN. При сопоставлении масс оксида висмута (III), определенных методом кварцевого микровзвешивания при разных температурах термической обработки [71 – 73], облучении светом = 360 нм [97 – 99, 104], при условии полного окисления пленок висмута различной толщины, а рассчитанных по уравнению реакции окисления: 2Bi + 1,5O2 = Bi2O3 установлено их удовлетворительное совпадение (табл. 3.10, 3.11). также 109 На основании вышеизложенного в процессе термической обработки [68 – 72, 80], а также облучения светом ( = 360 нм, I = (1,8 – 7,0)·1015 квант·см-2·с1 ) [97, 98, 102 – 104] образуется слой оксида висмута (III). Таблица 3.10 Сопоставление теоретических (fТ) и экспериментальных (fЭ) значений приращения частот резонатора с пленками висмута различной толщины (d), подвергнутых термообработке при различных температурах (Т) при 100 % превращении в конечный продукт – Bi2O3. Т, К d, нм fЭ, Гц fТ, Гц 373 51 169 168,9 373 25 78 78,4 373 14 45 45,4 473 41 132 132,2 473 21 68 67,7 473 59 190 190,5 573 16 51 51,3 573 50 161 161,2 573 30 97 96,7 673 51 169 168,9 673 95 294 294,1 110 Таблица 3.11 Сопоставление теоретических (fТ) и экспериментальных (fЭ) значений приращения частот резонатора с пленками висмута различной толщины (d), подвергнутых облучению светом = 360 нм при интенсивности I = 7,0∙1015 квант∙см-2∙с-1 при 100 % превращении в конечный продукт – Bi2O3. d, нм fЭ, Гц fТ, Гц 3 10 9,7 26 84 83,9 40 129 128,9 16 51 51,3 55 175 175,3 На рисунках 3.33 и 3.34 представлены, в качестве примера, зависимости степени термо- и фотохимического превращения наноразмерных пленок висмута от толщины, рассчитанные по результатам гравиметрических исследований методом кварцевого микровзвешивания и измерений спектров поглощения и отражения пленок висмута в процессе термообработки и облучении светом = 360 нм, соответственно. Рис. 3.33 Зависимость степени превращения от толщины пленок висмута при Т = 573 К: 1 – 5 нм, 2 – 13 нм, 3 – 30 нм, 4 – 56 нм, 5 – 92 нм. – метод кварцевого микровзвешивания, – спектрофотометрический метод 111 Рис 3.34 Зависимость степени фотохимического превращения от толщины пленок висмута: 1 – 3 нм, 2 – 16 нм, 3 – 20 нм, 4 – 34 нм ( = 360 нм, интенсивность I = 7·1015 квант·см-2·с-1). – метод кварцевого микровзвешивания, – спектрофотометрический метод В результате сопоставления масс нитрида висмута, определенных методом кварцевого микровзвешивания при условии полного превращения пленок висмута различной толщины в нитрид висмута, а также рассчитанных по уравнению реакции 2Bi + 2NH3 = 2BiN + 3H2 установлено их удовлетворительное совпадение (табл. 3.12) [119 – 121]. Таблица 3.12 Сопоставление теоретических (fТ) и экспериментальных (fЭ) значений приращения частот резонатора с пленками висмута различной толщины (d), подвергнутых взаимодействию с газообразным аммиаком (Т = 293 К) при 100 % превращении в конечный продукт – BiN. d, нм fЭ, Гц fТ, Гц 56 173 172,8 40 123 123,3 37 114 114,1 30 93 92,8 22 66 66,2 13 40 40,4 112 На рисунке 3.35 приведена зависимость степени превращения от толщины пленок висмута, рассчитанная по результатам гравиметрических исследований методом кварцевого микровзвешивания и измерений спектров поглощения и отражения пленок висмута в процессе воздействия газообразного аммиака [117 – 122]. Рис. 3.35 Зависимость степени превращения от толщины пленок висмута при Т = 293 К при взаимодействии с газообразным аммиаком: 1 – 13 нм, 2 – 22 нм , 3 – 31 нм, 4 – 56 нм. – метод кварцевого микровзвешивания, – спектрофотометрический метод Видно, что кинетические кривые степени превращения наноразмерных пленок висмута в процессе воздействия газообразного аммиака, рассчитанные по результатам гравиметрических исследований, методом кварцевого микровзвешивания и измерений спектров поглощения и отражения образцов, спектрофотометрическим методом – совпадают. Представленные результаты исследований являются дополнительным свидетельством того, что в процессе взаимодействия пленок висмута с газообразным аммиаком образуется слой нитрида висмута [117 – 122]. 113 3.5 Термостимулированные превращения в наноразмерной системе Bi – MoO3 Выяснения степени общности процессов, протекающих на границе между металлом, оксидом и окружающей атмосферой, а также необходимость разработки принципиально новых материалов, стабильных в условиях коррозионного воздействия окружающей среды, ставят правомерной задачу комплексного исследования свойств индивидуальных и двухслойных наноразмерных слоев оксида молибдена (VI) и висмута. В данном разделе приведены результаты исследований термостимулированных превращений в наноразмерной системе Bi - MoO3 в зависимости от толщины индивидуальных пленок Bi (d = 3 - 92 нм) и MoO3 (d = 5 - 40 нм), температуры (Т= 373 – 673 К) и времени термической обработки ( = 0,02 - 2560 мин). 3.5.1 Изменение оптической плотности и отражательной способности наноразмерной системы Bi – MoO3 под действием температуры В результате исследований оптических свойств наноразмерных пленок MoO3, Bi и двухслойных систем Bi-MoO3 было установлено, что спектры поглощения образцов до термической обработки в значительной степени зависят от толщины каждого из слоев MoO3 и Bi [71, 72, 78, 123 – 129]. При этом на спектрах поглощения систем Bi-MoO3 проявляются полосы поглощения индивидуальных пленок MoO3 и Bi. На рис. 3.36 в качестве примера приведены представительные спектры поглощения системы Bi-MoO3 с различной толщиной подслоев до термической обработки. 114 Рис. 3.36 Спектры поглощения системы Bi - MoO3 толщиной: 1) d(Bi) = 63 нм, d(MoO3) = 28 нм, 2) d(Bi) = 62 нм, d(MoO3) = 26 нм, 3) d(Bi) = 60 нм, d(MoO3) = 24 нм, 4) d(Bi) = 44 нм, d(MoO3) = 29 нм, 5) d(Bi) = 27 нм, d(MoO3) = 33 нм, 6) d(Bi) = 28 нм, d(MoO3) = 23 нм, 7) d(Bi) = 17 нм, d(MoO3) = 20 нм, 8) d(Bi) = 14 нм, d(MoO3) = 12 нм, 9) d(Bi) = 10 нм, d(MoO3) = 9 нм, 10) d(Bi) = 6 нм, d(MoO3) = 5 нм. Спектры поглощения системы Bi-MoO3 в коротковолновой области спектра (λ = 300–500 нм) по мере уменьшения толщины пленок висмута в значительной степени определяются поглощением пленок MoO3 (рис. 3.36, 3.38) [124, 127 - 129]. В длинноволновой области спектра λ = 500–1100 нм (рис. 3.36, 3.38) в большей степени проявляются полосы поглощения пленок висмута [124, 127 - 129]. По мере увеличения толщины пленок висмута наблюдается увеличение оптической плотности системы Bi-MoO3 в диапазоне λ = 300-1100 нм [124, 127 - 129]. При увеличении толщины пленок MoO3 оптическая плотность системы Bi-MoO3 также возрастает, однако, при этом в большей степени проявляется полоса поглощения в коротковолновой области спектра (λ = 300-600 нм) [124, 127 - 129]. При анализе спектров зеркального отражения было установлено, что отражательная способность системы Bi-MoO3 также зависит от толщины пленок висмута и оксида молибдена (VI) [123 – 131]. 115 На рис. 3.37 представлены спектры отражения системы Bi-MoO3 с различной толщиной подслоев до термической обработки. Рис. 3.37 Спектры отражения чистого стекла (подложки) (8) и систем Bi – MoO3 толщиной: 1) d(Bi ) = 126 нм, d(MoO3) = 24 нм; 2) d(Bi ) = 92 нм, d(MoO3) = 8 нм; 3) d(Bi) = 78 нм, d(MoO3) = 14 нм; 4) d(Bi ) = 63 нм, d(MoO3) = 18 нм; 5) d(Bi) = 59 нм, d(MoO3) = 28 нм; 6) d(Bi) = 45 нм, d(MoO3) = 33 нм; 7) d(Bi) =25 нм, d(MoO3) = 11 нм. По мере увеличения толщины пленок висмута наблюдается увеличение отражательной способности системы Bi-MoO3 в диапазоне λ = 600-1100 нм [127 – 131]. При увеличении толщины пленок MoO3 (в диапазоне d=5-33 нм) и уменьшении толщины пленок Bi (в диапазоне d=126-3 нм) отражательная способность системы Bi-MoO3 в диапазоне λ = 270-450 нм возрастает [127 – 131]. При последовательном уменьшении толщины пленок MoO3 (в диапазоне d=33-8 нм) и увеличении толщины пленок Bi (в диапазоне d=45-92 нм) обнаружен «эффект просветления» - отражательная способность стеклянной подложки уменьшается практически до нулевого значения при определенных длинах волн в диапазоне λ = 610-410 нм [126 – 132]. Для выяснения возможного взаимодействия между пленками висмута и оксида молибдена (VI) в процессе приготовления системы Bi-MoO3 были 116 сопоставлены экспериментальные спектры поглощения систем с рассчитанными спектрами поглощения, полученными суммированием при каждой длине волны значений оптической плотности индивидуальных пленок MoO3 и Bi аналогичной толщины. Рассчитанные и экспериментальные спектры поглощения всех исследованных системы Bi-MoO3 не совпадают [125 – 128, 130, 131, 133]. На рис. 3.38 приведены экспериментальные спектры поглощения пленок MoO3, Bi, системы Bi-MoO3 и рассчитанный спектр поглощения системы Bi-MoO3. Рис. 3.38 Экспериментальные (1,3,5) и рассчитанные (2, 4) спектры поглощения: Bi–MoO3 (1, 2), Bi (3), MoO3 (4, 5) (d(Bi) = 17 нм, d(MoO3) = 20 нм). На экспериментальных кривых в длинноволновой области спектра в диапазоне λ = 450-1100 нм проявляется широкая полоса поглощения с максимумом при 870 нм, а в коротковолновой области спектра в диапазоне λ = 300 - 450 нм наблюдается уменьшение оптической плотности и смещение края полосы поглощения системы Bi-MoO3 в коротковолновую область спектра [126 – 133]. 117 В процессе термической обработки оптическая плотность наноразмерных пленок MoO3 (толщиной d=8-130 нм) в коротковолновой области спектра ( = 300-450 нм с максимумом = 350 нм) уменьшается и, как следствие, наблюдается смещение края полосы собственного поглощения в коротковолновую область спектра, а в длинноволновой области спектра ( = 450-1100 нм с максимумом = 870 нм) возрастает [23, 134 – 137]. При термической обработке пленок Bi (толщиной d=3-120 нм) в длинноволновой области спектра (=330–1100 нм) имеет место уменьшение, а в коротковолновой ( = 300-330 нм) увеличение оптической плотности [69 – 73, 82]. В результате термической обработки системы Bi-MoO3 (приготовленных из пленок висмута и оксида молибдена (VI) разной толщины) в интервале температур Т = 373-673 К в атмосферных условиях спектры поглощения и отражения образцов претерпевают существенные изменения [51, 123, 124, 127, 130]. Причем, наблюдаемые изменения спектров поглощения и отражения, а также предельные значения оптической плотности после термической обработки образцов зависят от первоначальной толщины пленок Bi и MoO3, температуры и времени термообработки [51, 123, 124, 127 – 129]. На рисунках 3.39, 3.40 представлены спектры поглощения системы Bi - MoO3 до и после термической обработки при Т = 473 К, 573 К соответственно. Видно, что слева от изобестической точки ( = 350нм) наблюдается увеличение оптической плотности, в длинноволновой области спектра – справа от изобестической точки наблюдается уменьшение оптической плотности образцов [51, 124, 127 – 129]. По мере увеличения температуры, уменьшения толщины пленок оксида молибдена (VI) и висмута при термообработке системы Bi-MoO3 в диапазоне Т = 373-673 К наблюдается увеличение эффектов изменения оптической плотности и уменьшение времени достижения ее предельного значения [51, 124, 127 – 129]. 118 Рис. 3.39 Спектры поглощения системы Bi - MoO3 (d(Bi) = 21 нм, d(MoO3) = 6 нм) до (1) и после термической обработки при Т = 473 К: 2 – 20с, 3 – 1,5мин, 4 - 3мин, 5 – 5мин, 6 – 7мин, 7 – 10мин, 8 – 15мин, 9 – 20мин, 10 – 30мин, 11 – 60мин, 12 – 240мин. Рис. 3.40 Спектры поглощения системы Bi - MoO3 (d(Bi) = 92 нм, d(MoO3) = 14 нм) до (1) и после термической обработки при Т = 573 К: 2 – 2с, 3 – 20с, 4 - 2мин, 5 – 3,5мин, 6 – 5мин, 7 – 6мин, 8 – 7мин, 9 – 10мин, 10 – 12мин, 11 – 60мин, 12 – 120мин. 119 3.5.2 Кинетические закономерности степени термического превращения наноразмерных пленок MoO3 в процессе термической обработки системы Bi – MoO3 Для выяснения характера влияния пленок висмута на термические превращения в пленках оксида молибдена (VI) в процессе термической обработки системы Bi-MoO3 были рассчитаны, построены и сопоставлены кинетические зависимости степени превращения α = () (где – время термической обработки) пленок MoO3 и висмута разной толщины, нанесенных на стеклянные подложки, при различных температурах термообработки. Для расчета значений оптической плотности пленок MoO3 из экспериментальных спектров поглощения системы Bi-MoO3 вычитали спектры поглощения индивидуальных пленок висмута, до, и в процессе термообработки систем Bi-MoO3 и пленок висмута при разных температурах. При построении кинетических кривых степени превращения воспользовались подходом предложенным в [23, 71, 72, 105, 127 – 129, 138]. Спектры поглощения пленок MoO3 пересекаются в одной (изобестической) точке, в которой оптическая плотность не зависит от времени термической обработки [22, 23]. Слева и справа от изобестической точки поглощение (Аобр) зависит от времени термической обработки, а наблюдаемая оптическая плотность при определенном времени термической обработки будет складываться из поглощения, связанного с наличием центра Т1 (АТ1) и центра Т2 (АТ2) [22, 23, 127 – 129]: Аобр = АТ1 + АТ2 (3.32) Учитывая [83], что падающая по нормали на поверхность какой-либо системы световая волна от источника излучения, претерпевает зеркальное отражение, рассеяние, поглощение и пропускание для расчета истинного вызванного поглощением света в веществе значения оптической плотности воспользовались уравнением [23, 127 – 129]: Aобр = A + lg(1 - R) (3.33) где A – измеряемое в реальных условиях на спектрофотометре полное 120 значение оптической плотности, включающее несколько составляющих A = Aобр + Aотр + Aрас, (3.34) где Aобр – значение оптической плотности образца; A отр – значение оптической плотности, обусловленное потерями на зеркальное отражение света поверхностью образца; Aрас – значение оптической плотности, обусловленное потерями на диффузное рассеяние света поверхностью образца. Итоговое выражение для определения степени термического превращения центра Т1 в центр Т2 [22, 23 127, 128, 130, 131]: α = (Аобр – АТ11) / (АТ21 – АТ11), (3.35) где АТ21, АТ11 – предельные оптические плотности при максимальной и минимальной концентрации центров Т2 при = 870 нм. Было установлено, что степень превращения центра Т2 пленок MoO3 в системе Bi-MoO3 зависит от первоначальной толщины пленок Bi и MoO3, температуры и времени термической обработки [125, 127 – 130]. По мере увеличения времени термообработки степень превращения пленок MoO3 в системе Bi-MoO3 (рассчитанная по изменению оптической плотности в полосе поглощения центра Т2) возрастает (рис. 3.41, 3.42) [127 – 131]. На рисунках 3.41 и 3.42 представлены зависимости степени превращения центра 2 пленки MoO3 в системе Bi–MoO3 и в пленках MoO3 в интервале температур T = 373 – 673 К, и при постоянной толщине пленок висмута и различной толщине слоев MoO при Т = 573 К, соответственно. Увеличение температуры термообработки системы Bi-MoO3 (при постоянной толщине пленок MoO3 и Bi) приводит к возрастанию скорости термического превращения центра Т2 пленок MoO3 (рис. 3.41) [125, 127 – 131]. При увеличении толщины пленок MoO3 в системе Bi-MoO3 при постоянной температуре термообработки степень превращения во всем исследованном интервале температур уменьшается (рис. 3.42) [127 - 129]. Видно (рис. 3.41, 3.42), что скорость превращения центра Т2 пленок MoO3 в системе Bi-MoO3 больше, чем в индивидуальных пленках MoO3. 121 Рис. 3.41 Температурная зависимость степени превращения центра 2 пленки MoO3 в системе Bi–MoO3 (1, 3) и в пленках MoO3 (2, 4) (d(Bi) = 30 нм, d(MoO3) = 17 нм) 1, 2 - 5 мин; 3, 4 - 1 мин. Рис. 3.42 Зависимости степени превращения центра 2 пленки MoO3 в системе Bi–MoO3 (1, 3) и в пленках MoO3 (2, 4) при постоянной толщине пленок висмута (d = 30 нм) и различной толщине пленок MoO3 1, 2 – 5 мин; 3, 4 – 1 мин, при 573 К. 122 3.5.3 Контактная разность потенциалов для наноразмерных пленок Bi и MoO3 Полученные в диссертационной работе результаты исследований свидетельствуют о контактной природе эффектов изменения висмутом скорости термического превращения пленок MoO3 [64, 65, 71, 72, 105, 107, 124, 127, 128, 138 – 142]. Для выяснения причин, вызывающих наблюдаемые изменения металлом оптических свойств MoO3 в разных спектральных областях были измерены величина и знак Uф для системы Bi-MoO3, КРП между MoO3, Bi и электродом сравнения из платины в условиях атмосферы (Р = 1·105 Па) и высокого вакуума (Р = 1·10-5 Па) [50, 51, 124, 127, 128, 131]. Таблица 3.13 Контактная разность потенциалов между пленками висмута, оксида молибдена (VI) и электродом сравнения из платины при Т = 293 К Образец КРП, В Р = 1·105 Па Р = 1·10-5 Па Пленка MoO3 (d=90 нм) +0,72 +0,71 Пленка Bi (d=92 нм) +1,11 +1,11 Из таблицы видно, что значения КРП между оксидом молибдена (VI) и электродом сравнения из платины при понижении давления в измерительной ячейке уменьшаются. Наблюдаемое отличие в значениях работ выхода Bi и MoO3 (табл. 3.13) свидетельствует о возможности при формировании плотного контакта и установлении в системе Bi-MoO3 состояния термодинамического равновесия результирующего потока электронов из висмута в оксид молибдена (VI) [127 – 129]. В результате измерений Uф для системы Bi-MoO3 в диапазоне = 300-1100 нм было установлено, что в процессе облучения светом формируется Uф положительного потенциала со стороны слоя MoO3 [124, 125, 127 – 130, 132]. Формирование Uф для системы Bi-MoO3 прямо свидетельствует о разделении неравновесных носителей заряда на границе раздела. Из анализа результатов измерений Uф и КРП 123 (табл. 3.13) было установлено, что при создании контакта оксида молибдена (VI) с висмутом в результате электронных переходов со стороны MoO3 образуется обогащенный электронами антизапорный слой [50, 51, 124, 126 – 132]. Диаграмма энергетических зон системы Bi-MoO3, при построении которой использованы результаты измерений спектров поглощения и отражения, спектрального распределения UФ, КРП (табл.3.13) представлена на рис. 3.43. Рис. 3.43 Диаграмма энергетических зон системы Bi–MoO3: EV – уровень потолка валентной зоны, EС – уровень дна зоны проводимости, EF – уровень Ферми, E0 – уровень вакуума, T1, Т2, – уровни центров захвата, ТП+ – уровни ПЭСК, R+ – центр рекомбинации. 3.5.4 Модельные представления термостимулированных превращений наноразмерных пленок оксида молибдена (VI) в системе Bi – MoO3 Авторами [19, 22, 23] было установлено, что полоса поглощения в диапазоне = 300-400 нм с максимумом при = 350 нм (центр Т1) в области края собственного поглощения для монокристаллов MoO3 связана со стехиометрическим недостатком кислорода и обусловлена вакансиями кислорода с одним захваченным электроном [(Vа)++ е] (аналог F-центра) [19]. Этот центр, формируется в процессе приготовления микрокристаллов, 124 кристаллов и пленок MoO3 различной толщины на стеклянных положках [22]. Глубина залегания [(Vа)++ е]-центра составляет EТ1 = 3.54 эВ [22, 23]. Наблюдаемые изменения на экспериментальных спектрах поглощения системы Bi-MoO3 по сравнению с рассчитанными, видимо, связаны с формированием перехода Bi-MoO3. Мы полагаем, что уменьшение оптической плотности в диапазоне = 300-610 нм и, как следствие, смещение края поглощения в коротковолновую область, а также увеличение оптической плотности в диапазоне =610-1100 нм с максимумом при =870 нм в процессе приготовления системы Bi-MoO3 взаимосвязанные процессы [127 – 129, 131]. В процессе установления термодинамического равновесия из-за несоответствия работ выхода MoO3 и Bi электроны из висмута переходят в оксид молибдена (VI) на уровни некоторых [(Vа)++ е] – центров (T1) с формированием [е (Vа)++ е] – центров (T2) [23, 124, 127, 128, 131, 133] е + [(Vа)++ е] [е (Vа)++ е] (3.36) где (Vа)++ – анионная вакансия. Для того чтобы при термическом возбуждении электронной подсистемы твердого тела осуществить переход электрона с нижнего заполненного уровня на верхний незаполненный [83] и обеспечить достаточную скорость этого процесса необходимо, чтобы средняя энергия фонона (kT) соответствовала величине преодолеваемого энергетического барьера [22, 23, 27]. Преобразование [(Vа)++ е]-центра можно осуществить путем перевода электрона с уровня залегания центра на дно зоны проводимости (для обеспечения этого процесса потребуется энергия EТ1 (формульное выражение 1.4)) [22, 23] либо путем перевода электрона с потолка валентной зоны с образованием дырок (р) на уровень центра (для обеспечения этого процесса потребуется энергия E = Eшзз – EТ1, где Eшзз – термическая ширина запрещенной зоны MoO3 (формульное выражение 1.5)) [22, 23]. Оценим возможность осуществления указанных процессов в реальных условия эксперимента. Фононы не моноэнергетичны. Их распределение по 125 энергиям подчиняется уравнению Больцмана [83]. Согласно уравнению Больцмана всегда есть вероятность того, что при температурах Т = 373 - 673 К будет существовать фонон с энергией равной EТ1 = 3,28 эВ или E = 0,26 эВ. (Для обеспечения термически активируемых переходов затраты энергии будут составлять 0,2 – 0,3 эВ от оптических [19]). Уравнение для скорости процесса термического возбуждения электрона с уровней центра Т1 на дно зоны проводимости или термического возбуждения электрона с потолка валентной зоны на уровни центра Т1 можно представить в следующем виде [22, 23 127 – 129]: W = Nexp (- ∆Е / k0 T), (3.37) где – частотный фактор (для фононов по порядку величины составляет 1013-1014), N – концентрация [(Vа)++ е]-центров, ∆Е – величина преодолеваемого барьера (EТ1 = 3.28 эВ, E = 0.26 эВ), k0 – постоянная Больцмана (8,5710-5 эВ/Т), Т – температура (575 К). Методами электронной микроскопии и рентгеновской дифракции продуктов последовательного восстановления оксида молибдена (VI) МоО3-γ, установлено, что область нестехиометрии, в которой сохраняется неизменная структура МоО3, очень мала и соответствует значению γ < 0.001 [10]. Если оценить концентрацию [(Vа)++ е]-центров 1018 см-3 (и считать, что все анионные вакансии в MoO3 заняты по одному электрону в каждой), то в идеальном случае (когда все электроны достигнут предназначенного для них места и не примут участия в других процессах) скорости процессов термического возбуждения электрона с уровней [(Vа)++ е]-центра на дно зоны проводимости и термического возбуждения электрона с потолка валентной зоны на уровни [(Vа)++ е]-центра составят W1 1102 см-3с-1 и W2 51031 см-3с-1 соответственно [22, 23, 127 - 129]. Отсюда следует, что при термическом возбуждении электронов с уровней [(Vа)++ е]-центра в зону проводимости в см3 МоО3 за одну секунду переходит 102 электронов – малое количество. Скорость процесса термического возбуждения электронов с потолка валентной зоны на уровни [(Vа)++ е]-центра достаточно велика, 126 чтобы обеспечить дальнейшие превращения слоя МоО3 [22, 23, 127 – 129]. По-видимому, широкая полоса поглощения с максимумом при = 870 нм, связана с формированием [е (Vа)++ е]-центров. Дырки (p) могут захватываться собственными (Vк6-) и примесными (Т-) дефектами с выделением кислорода и освобождением анионных вакансий[22, 23, 124, 127 – 129, 131]: р + Vк6- [Vк6- р] + р [р Vк6- р] О2 + 2е + 2Va++ + Vк6-, р + Т- Т0 + р Т+ О2 + 2е + 2Va++ + Т- (3.38) (3.39) где Vк6- и Va++ – катионная и анионная вакансии. Анионные вакансии будут взаимодействовать с электронами переходящими из висмута с формированием центров Т1 и Т2 [22, 23, 124, 127 – 129, 131]: е + (Vа)++ е + [(Vа)++ е] [е (Vа)++ е] (3.40) Увеличение концентрации электронов со стороны MoO3 в состоянии термодинамического равновесия системы Bi-MoO3 во-первых обеспечивает превращение части центров Т1 в центры Т2 в пленке MoO3 и во вторых должно привести к увеличению скорости термостимулированного процесса превращения оставшихся центров [е (Vа)++] в центры [е (Vа)++ е] [124, 127, 131]. Наблюдаемые закономерности изменения висмутом оптических свойств MoO3 соответствуют изложенной модели процессов [124, 127 – 129, 131]. 127 3.6 Образование центров окраски в наноразмерных пленках оксида молибдена (VI) под действием света Оксид молибдена (VI) и системы на его основе привлекают внимание исследователей различного профиля [10, 11, 23, 65, 140, 143 – 153]. MoO3 применяется для получения молибдена (его сплавов и соединений), как составная часть керамических глин, глазурей, эмалей, красителей. Его используют в качестве катализатора в органическом синтезе, при переработке нефти (крекинг, гидроочистка, риформинг), он добавляется в качестве присадки к моторным маслам. Оксид молибдена (VI), нанесенный на различные носители (диоксид титана, кремнезем), вызывает фотостимулированную конверсию метана и метансодержащих газовых смесей (в различных газовых композициях) с достаточно высоким выходом метанола, формальдегида, СО, СО2 [151, 154], а также может быть рекомендован к использованию в качестве чувствительного элемента электрохромных и фотохромных дисплеев, электрохромных зеркал или светоперераспределяющих фильтров, сенсоров для контроля содержания газов в атмосфере [147, 148, 150, 152, 153]. Изучение фотохимических процессов в наноразмерных пленках оксида молибдена (VI), который сочетает достоинства модельных соединений (имеет относительно несложный состав и структуру, обладает достаточной фотохимической чувствительностью и внутренним фотоэффектом [140]), актуально как в научном, так и практическом отношении. В данном разделе представлены результаты исследований закономерностей процессов, протекающих в условиях атмосферы в наноразмерных слоях MoO3 различной толщины (d = 5-54 нм) при облучении их светом из области собственного поглощения MoO3 в зависимости от интенсивности падающего света (I = (1,5 – 7)·1015 квант см-2с-1 , = 320 нм) и времени облучения. 128 3.6.1 Влияние облучения на оптические свойства наноразмерных пленок оксида молибдена(VI) Из анализа спектров поглощения и отражения наноразмерных пленок оксида молибдена (VI) разной толщины (d = 5-54 нм) было установлено, что оптические свойства MoO3 при облучении образцов в атмосферных условиях и Т = 293 К светом из области собственного поглощения существенно изменяются [155 – 161]. Наблюдаемые изменения спектров поглощения и отражения, а также предельные значения оптической плотности в максимумах и минимумах полос поглощения (реализуемых после воздействия на образцы света) зависят от толщины пленок MoO3, интенсивности падающего света и времени облучения [155, 156, 158 – 160]. На рис. 3.44 в качестве примера приведены спектры поглощения пленки MoO3 толщиной d = 24 нм до и после облучения светом из области собственного поглощения MoO3 ( = 320 нм). Рис. 3.44 Спектры поглощения слоя оксида молибдена (VI) толщиной 24 нм до (1) и после облучения светом = 320 нм I = 7 1015 квантсм-2с-1 при 293 К в течение 2 – 1 мин, 3 – 5 мин, 4 – 20 мин, 5 – 60 мин, 6 – 120 мин. На спектрах поглощения образца можно выделить характерные для 129 пленок, поли- и монокристаллов MoO3 – коротковолновую λ 400 нм и длинноволновую λ 400 нм области поглощения [22, 23, 65, 140, 143, 145 – 148, 152 – 153]. В процессе облучения край полосы поглощения пленки MoO3 смещается в коротковолновую область спектра [155, 156, 158 – 160, 161]. Как уже отмечалось ранее (раздел 3.5.4), область нестехиометрии, в которой сохраняется неизменная структура оксида молибдена (VI), очень мала и соответствует значению γ < 0,001 [10]. Концентрация анионных вакансий (Vа)++ при этом составит 1018 см-3. Было установлено [23, 145], что полоса поглощения в диапазоне = 310-400 нм с максимумом при = 350 нм (центр Т1) в области края собственного поглощения монокристаллов и пленок MoO3 связана со стехиометрическим недостатком кислорода и обусловлена вакансиями кислорода с одним захваченным электроном [(Vа)++ е] [22, 23]. Этот центр формируется в процессе приготовления пленок MoO3 различной толщины, а при воздействии света = 320 нм претерпевает фотохимическое превращение - значения оптической плотности уменьшаются и, как следствие, край полосы поглощения пленки MoO3 смещается в коротковолновую область спектра [22, 23, 155, 156, 158 – 160, 162]. Определение истинного края полосы поглощения пленок MoO3 в значительной степени осложнено из-за наличия полосы поглощения в интервале = 310-400 нм с максимумом при λ 350 нм, но после предварительного облучения образцов светом = 320 нм полоса поглощения с максимумом λ 350 нм практически полностью исчезала [155, 158 – 160]. Оптическую ширину запрещенной зоны пленок MoO3 оценивали по формулам [22, 23, 83], используя спектры поглощения образцов нанесенных на кварцевые подложки и подвергнутых предварительному облучению. Установлено, что край полосы поглощения пленок MoO3 находится при λ 328 нм. Это значение удовлетворительно совпадает с краем полосы поглощения и оптической шириной запрещенной зоны (3,76 эВ) [22, 23], определенным по спектрам диффузного отражения мелкокристаллических 130 порошков и по результатам измерений спектров поглощения и пропускания тонких нанесенных на кварцевую подложку пленок MoO3 подвергнутых предварительной тепловой обработке [23]. В длинноволновой области спектра наблюдается увеличение значений оптической плотности в интервале = 400-1100 нм с максимумом = 870 нм (формируется центр Т2). Необходимо отметить, что при облучении светом одинаковой интенсивности по мере увеличения толщины образцов (в диапазоне 5-54 нм) наблюдается последовательное возрастание эффектов изменения оптической плотности в диапазоне = 400-1100 нм [157 – 160]. Кроме того с увеличением интенсивности падающего света в диапазоне (I = 1,5 – 7 1015 квант см-2с-1) при одинаковой толщине пленок MoO3 наблюдается возрастание эффектов изменения оптической плотности. При воздействии на предварительно облученные при λ = 320 нм пленки MoO3 светом = 870 нм наблюдается уменьшение значений оптической плотности в диапазоне = 400-1100 нм с максимумом = 870 нм [155, 157 – 162]. Одновременно длинноволновую наблюдается область смещение спектра, края которое полосы связано поглощения с в увеличением оптической плотности в коротковолновой области спектра = 310-400 нм с максимумом = 350 нм [157 – 162]. 3.6.2 Кинетические зависимости степени фотохимического превращения центров окраски наноразмерных пленок MoO3 Для выяснения закономерностей протекания стимулированного светом = 320 нм и I = 1,5 – 7 1015 квант см-2с-1 процесса в пленках оксида молибдена (VI) различной толщины были рассчитаны и построены кинетические кривые степени превращения α = () (где – время облучения) при = 870 нм в зависимости от толщины пленок MoO3 и интенсивности падающего света. При построении кинетических кривых степени превращения был применен подход, предложенный в [23, 71, 72, 107, 138, 155, 158]. 131 Спектры поглощения пленок MoO3 предварительно облученных светом из области собственного поглощения при различных интенсивностях и временах облучения пересекаются в изобестической точке, в которой оптическая плотность не зависит от времени воздействия света [155, 158, 161]. Слева и справа от изобестической точки поглощение (Аобр) зависит от времени фотохимической обработки, а наблюдаемая оптическая плотность его при определенном времени облучения будет складываться из поглощения, связанного с наличием центра Т1 (АЦ1) и центра Т2 (АЦ2): Аобр = АЦ1 + АЦ2 (3.41) Для расчета истинного вызванного поглощением света в веществе значения оптической плотности воспользовались уравнением 3.33 [22, 23]: Итоговое выражение для определения степени фотохимического α = (А обр – АЦ11) / (АЦ21 – АЦ11), (3.42) превращения центра Т1 в центр Т2 [23]: где АЦ11, АЦ21 – предельная оптическая плотность центра Т1 и центра Т2 при = 870 нм. Установлено [158 – 160], что степень фотохимического превращения центра Т1 в центр Т2 зависит от первоначальной толщины пленок MoO3, времени облучения и интенсивности падающего света [155 – 160]. Независимо от толщины пленок MoO3 и интенсивности падающего света при увеличении времени облучения степень превращения возрастает. На рис. 3.45 в качестве примера приведены кинетические кривые степени превращения в зависимости от толщины образцов. Видно, что при облучении образцов светом из области собственного поглощения по мере увеличения толщины пленок MoO3 степень превращения уменьшается [156, 158 – 160]. Увеличение интенсивности падающего света (при постоянной толщине пленок MoO3) приводит к возрастанию скорости фотохимического превращения (рис. 3.46) [155, 158 – 162]. При облучении пленок MoO3 светом = 870 нм наблюдается уменьшение оптической плотности в диапазоне 132 = 400-1100 нм с максимумом = 870 нм и смещение края полосы поглощения MoО3 в длинноволновую область спектра [156 – 162]. Рис. 3.45 Зависимость степени превращения центра 2 от толщины пленок оксида молибдена (VI) при облучении светом = 320 нм и интенсивности I = 2,41015 квантсм-2с-1: 1 – 5 нм; 2 – 18 нм; 3 – 29 нм; 4 – 38 нм; 5 – 54 нм. Рис. .46 Зависимость степени превращения центра 2 пленок оксида молибдена (VI) толщиной 29 нм от интенсивности падающего света (I = квантсм-2с-1): 1) 7•1015, 2) 3,8•1015, 3) 2,4•1015, 4) 2,0•1015, 5) 1,5•1015. Уменьшение максимума поглощения при = 350 нм, а также формирование максимума поглощения при = 870 нм в процессе облучения 133 пленок MoO3 по-видимому взаимосвязанные процессы и являются результатом стимулированного светом из области собственного поглощения оксида молибдена (VI) преобразования центра Т1 ([(Vа)++ е]) [155 – 162]. 3.6.3 Модельные представления фотостимулированных процессов в наноразмерных пленках оксида молибдена (VI) Диаграммы энергетических зон дают ценную информацию при изучении механизма стимулированных светом превращений пленок MoO3 и позволяют на основе общих физических представлений объяснить химические процессы, протекающие в наноразмерных пленках MoO3 [22, 35]. На рис. 3.47 приведена диаграмма энергетических зон МоО3, при построении которой использованы результаты измерений спектров поглощения и отражения образцов разной толщины до и после воздействия света из различных спектральных областей, а также результаты [23]. Рис. 3.47 Диаграмма энергетических зон оксида молибдена (VI), EV - уровень потолка валентной зоны, EC - уровень дна зоны проводимости, EF - уровень Ферми, EO - уровень вакуума, R - центр рекомбинации, Т1 - центр [(Vа)++ е], Т2 – центр [е (Vа)++ е]. При облучении оксида молибдена (VI) светом из области собственного поглощения имеет место интенсивная генерация электрон-дырочных пар в МоО3 (рис. 3.47, переход 1) [155, 158 – 160] 134 А2- p + е (3.43) Часть неравновесных носителей заряда рекомбинирует (рис. 3.47, переходы 2,3) [155, 158 – 160] R+ + е R0 + р R+, (3.44) где R+ – центр рекомбинации. Другая часть неравновесных электронов может восстанавливать Мо6+ е+ Мо6+ е+ Мо5+ е+ Мо4+ … Мо0 + Vк6-, (3.45) а также переходить из зоны проводимости на уровни центра Т 1 (рис. 3.47, переход 4) участвуя в образовании центра Т2 [155, 158 – 160] е + [(Vа)++ е] [е (Vа)++ е]. (3.46) Дырки могут захватываться собственными (Vк6-) и примесными (Т-) дефектами с выделением кислорода и освобождением анионных вакансий (формульные выражения 3.38, 3.39) [22, 23, 155, 158 – 160]:. При облучении пленок МоО3 светом из длинноволновой области спектра ( = 870 нм) имеет место стимулированный светом переход электронов с уровней центра Т2 в зону проводимости МоО3 (рис. 3.47, переход 6) [155, 158 – 160] [е (Vа)++ е] е + [(Vа)++ е] е + (Vа)++. Уменьшение концентрации соответствующему уменьшению [е (Vа)++ е] - оптической центров (3.47) приведет плотности в и к диапазоне = 400-1100 нм с максимумом = 870 нм. Неравновесные электроны могут принимать участие в процессе восстановления Мо6+, а также взаимодействовать с анионными вакансиями с образованием центров Т1 е + (Vа)++ [(Vа)++ е]. (3.48) Формирование центров Т1 приведет к увеличению оптической плотности в диапазоне = 310-400 нм с максимумом при = 350 нм и, как следствие, к смещению края полосы поглощения МоО3 в длинноволновую область спектра [155, 156, 158 – 160]. 135 ЗАКЛЮЧЕНИЕ Исследованы оптические свойства наноразмерных пленок висмута, в зависимости от толщины (d = 3 – 120 нм), температуры (Т = 373 – 673 К) и времени термообработки ( = 0,05 – 2500 мин), интенсивности падающего света ( = 360 нм, I = (1,8 – 7,0)·1015 квант·см-2·с-1) и времени облучения, а также времени воздействия газообразного аммиака ( = 1 мин – 5400 час) при Т = 293 К. В результате исследований установлено, что: - увеличение толщины пленок висмута приводит к увеличению оптической плотности и отражательной способности образца. Для образцов толщиной более 28 нм можно выделить характерные для висмута полосы поглощения (в частности – максимумы поглощения при λ 410 нм и 735 нм); - на спектрах поглощения, фото- и термостимулированных пленок висмута, наряду с уменьшением в интервале = 320, 330 – 1100 нм и увеличением в интервале 320, 330 нм оптической плотности образца формируется спектр поглощения нового вещества – оксид висмута (III); - воздействие газообразного аммиака приводит к последовательному уменьшению в интервале = 315…1100 нм и возрастанию в диапазоне = 300…315 нм оптической плотности наноразмерных пленок висмута и формированию спектра поглощения нового вещества – BiN. Методом оптической спектроскопии установлено, что стимулированное светом увеличение оптической плотности пленок MoO3 в интервале = 400-1100 нм с максимумом = 870 нм и уменьшение в диапазоне = 310-400 нм с максимумом = 350 нм связано с расходованием центра [е (Vа)++] и формированием центра [е (Vа)++ е] пленок MoO3. Выявлены кинетические закономерности протекания процессов взаимодействия наноразмерных пленок висмута различной толщины с газообразным аммиаком при Т = 293 К, с кислородом окружающей, а также в процессе облучения светом. Установлено, что степень превращения пленок 136 висмута возрастает по мере уменьшения их толщины, увеличения времени контакта с газообразным аммиаком, температуры обработки, интенсивности падающего света. Кинетические кривые степени термо- и фотостимулированного окисления пленок висмута описываются в рамках линейного, обратного логарифмического, кубического и логарифмического законов, а кинетические кривые степени превращения пленок висмута в атмосфере газообразного аммиака – линейного, обратного логарифмического, параболического и логарифмического законов. В результате анализа кинетических кривых степени термического превращения наноразмерных пленок MoO3 (d = 5 – 40 нм) в процессе термической обработки систем Bi – MoO3 в интервале температур (Т = 373 – 673 К) установлено, что наноразмерный висмутовый подслой увеличивает скорость превращения центров [е (Vа)++] в центры [е (Vа)++ е] пленок MoO3 Методом контактной разности потенциалов определены значения термоэлектронных работ выхода до и после воздействия газообразного аммиака при Т = 293 К, термической обработки (Т = 673 К) и облучения светом наноразмерных пленок висмута, оксида висмута (III), нитрида висмута, оксида молибдена (VI) при различных внешних условиях (Р = 1·105, 1·10-5 Па; Т = 293 К). Установлены изменения КРП пленок висмута подвергнутых термической обработки, облучению светом, а также взаимодействию с газообразным аммиаком, связанные с формированием конечного продукта (оксида висмута (III), нитрида висмута). Методами оптической спектроскопии и кварцевого микровзвешивания обнаружено, что в процессе облучения светом ( = 360 нм, I = (1,8 – 7,0)·1015 квант·см-2·с-1) и термической обработки (Т = 373 – 673 К) наноразмерных пленок висмута образуется оксид висмут (III), а в результате воздействия газообразного аммиака при Т = 293 К – образуется нитрид висмута. Предложены модели термо- и фотостимулированных превращений наноразмерных пленок висмута в оксид висмута (III), а также модель 137 процесса превращения наноразмерных пленок висмута в атмосфере газообразного аммиака при Т = 293 К с формированием нитрида висмута. Таким образом в работе проведены систематические исследования закономерностей процессов термо-, фото- и газостимулированных превращений наноразмерных пленок висмута, оксида молибдена (VI) и системы на их основе, а экспериментальные данные, полученные разными методами, находятся в согласии между собой, повторяют и дополняют друг друга, что свидетельствует о достоверности результатов, полученных в работе. В практическом плане можно сделать следующие рекомендации: Обнаруженная зависимость степени окисления пленок висмута от их толщины позволят получать при определенной толщине коррозионностойкие покрытия на металлическом висмуте. На основании полученных зависимостей степени превращения центра Т1 в Т2 MoO3 от толщины, температуры, интенсивности облучения и наличие висмутового подслоя можно определить условия получения наноразмерных пленок оксида молибдена (VI), для создания более эффективных и чувствительных газовых сенсоров. Обнаруженный «эффект просветления» (уменьшение отражательной способности практически до нулевого значения) на наноразмерной системе Bi - MoO3 может быть рекомендован для создания специальных покрытий, обеспечивающих невидимость объектов в области видимого света (λ = 410 610 нм). 138 ВЫВОДЫ 1. Установлено влияние размерного эффекта на оптическую плотность и отражательную способность наноразмерных пленок висмута. Для образцов толщиной более 28 нм можно выделить характерные для висмута полосы поглощения (в частности, максимумы поглощения при λ 410 нм и 735 нм); 2. Установлен качественный и количественный состав продуктов превращения наноразмерных пленок висмута при контакте с кислородом и газообразным аммиаком методом оптической спектроскопии и кварцевого микровзвешивания. При взаимодействии с кислородом в интервале температур (Т = 373 – 673 К) и облучении светом ( = 360 нм) интенсивностью (I = (1,8 – 7,0)·1015 квант·см-2·с-1) при Т= 293К образуется оксид висмута (III), при взаимодействии с газообразным аммиаком при Т=293К – нитрид висмута. 3. Показано: на степень превращения наноразмерных пленок висмута в оксид висмута (III), висмута в нитрид висмута влияют размерные эффекты, температура, интенсивность облучения и время контакта с газообразным аммиаком. При температуры уменьшении обработки и толщины пленок висмута, интенсивности падающего увеличении света степень превращения увеличивается. 4. Обнаружено увеличении степени превращения центра Т1 в Т2 наноразмерных пленок оксида молибдена (VI) при повышении температуры, интенсивности облучения и наличии висмутового подслоя. 5. Впервые определена контактная разность потенциалов (работа выхода электрона) до и после воздействия газообразного аммиака, термической обработки и облучения светом наноразмерных пленок висмута, оксида висмута (III), нитрида висмута, оксида молибдена (VI). Установлено: работа выхода электрона в ряду Bi2O3 → BiN → MoO3 → Bi увеличивается. 6. Предложены модели термо- и фотостимулированных превращений наноразмерных пленок висмута, включающие стадии адсорбции кислорода, 139 генерации, рекомбинации и перераспределения неравновесных носителей заряда в контактном поле системы Bi – Bi2O3, диффузии катионных вакансий к внешней поверхности Bi2O3 и формирования оксида висмута (III). 7. Предложена модель процесса превращения наноразмерных пленок висмута в атмосфере газообразного аммиака при Т = 293 К, включающая стадии адсорбции аммиака, перераспределения носителей заряда в контактном поле Bi – BiN, диффузии катионных вакансий к внешней поверхности BiN, и формирования нитрида висмута. 140 СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ 1. Балобанов В.И. Нанотехнологии. Наука будущего. – М.: Эксмо, 2009. – 256 с. 2. Рудской А.И. Нанотехнологии в металлургии. – СПб.: Наука, 2007. – 186 с. 3. Коршунов А.В. Размерные и структурные эффекты в процессах окисления металлов: монография. – Томск, 2013. – 360 с. 4. Таланов В.М., Ерейская Г.П., Юзюк Ю.И. Введение в химию и физику наноструктур и наноструктурированных материалов. – М.: Академия Естествознания, 2008. – 389 с. 5. Лусис, А.Р. Электрохромные зеркала – твердотельные ионные устройства / А.Р. Лусис, Я.Я. Клеперис // Электрохимия. – 1992. – Т. 28. – Вып. 10. – С. 1450-1455. 6. Юхин Ю. М., Михайлов Ю. И. Химия висмутовых соединений и материалов. — Новосибирск: Издательство СО РАН, 2001. — 360 с. 7. Денисов В.М., Белоусова Н.В., Моисеев Г.К. и др. Висмутсодержащие материалы: строение и физико-химические свойства. Екатеринбург: Уро РАН, 2000. 527 с. 8. Рыжонков Д.И., Левина В.В., Дзидзигури Э.Л. Наноматериалы. М.: БИНОМ, 2008. 365 с. 9. Полывянный И.Р., Абланов А.Д., Батырбекова С.А. Висмут. Алма-Ата: Наука, 1989. 316 с. 10. Порай-Кошиц, М.А. Кристаллохимия и стереохимия координационных соединений молибдена / М.А. Порай-Кошиц, Л.О. Атовмян. – М.: Изд-во «Наука», 1974. – 232 c. 11. Гуревич, Ю.Я. Твердые электролиты. – М.: Изд-во «Наука», 1986. – 176 с. 12. Химическая энциклопедия: в 5 томах: том 1. / Под ред. И.Л. Кнунянц. – М.: Советская энциклопедия. 1988. - 623с. 13. Информационный портал: i-Think.ru [Электронный ресурс]. – Москва. Режим доступа: http://www.i-think.ru/wikimet/?type=metall&section_id=393/. 141 14. – Свойства элементов: Справочник. / Под ред. Дрица М. Е. – М.: Металлургия, 1985. - 672 с. 15. Справочник по редким металлам. / Под ред. В.Е. Плющева. – М.: МИР, 1965. – 946с. 16. Юхин Ю. М., Михайлов Ю. И. Висмутовые соединения и материалы // Журн. неорганической химии, 1993, Т. 38, №. 3. С. 559—560. 17. Асидзава Т., Исихара М., Симода С. Способ получения сверхпроводника, содержащего висмут// Кокай токке кохо, 1991. — Сер. 3(1), № 53. С. 273— 278. 18. Service R. Superconductivity turns 10 // Science. 1996. V. 271, № 5257. — P. 1804— 1806. 19. Школьник А.Л. Оптические свойства МоО3 // Известия АН СССР. Серия «Физика». – 1967. – Т. 31. – № 12. – С. 2050-2051. 20. Yang Y.A. Microstructures of electrochromic MoO3 thin films colored by injection of different cations / Y.A. Yang, Y.W. Cao, B.N. Loo, J.N. Yao // J. Phys. Chem. – 1998. – V. 102. – P. 9392-9396. 21. Махаев Е.Т. Химия соединений молибдена (VI) и вольфрама (VI). – Новосибирск: Изд-во «Наука», 1979. – 324 с. 22. Борисова Н.В. Термопревращения в наноразмерных слоях алюминия, оксида молибдена (VI) и системах на их основе: Автореф. дис. канд. хим. наук. – Кемерово: КемГУ, 2007. – 25 с. 23. Суровой Э.П., Борисова Н.В. Термопревращения в наноразмерных слоях MoO3 // Журнал физической химии, 2008, Т. 82, С. 2120 – 2125. 24. Лусис А.Р. Электрохимические процессы в твердотельных электрохромных системах / А.Р. Лусис, Я.К. Клявинь, Я.Я. Клеперис // Электрохимия. – 1982. – Т. 18. – № 11. – С. 1538-1541. 25. Tubbs, M.R. Optical Properties, Photographic and Holographic Applications of Photochromic and Electrochromic Layers // Brit. J. Appl. Phys. – 1964. – V. 15. – P. 181-198. 142 26. Arnoldussen, Thomas C. Electrochromism and photochromism in MoO3 films // J. Electrochem. Sol.: Solid-State Science and Technology. – 1976. – V. 123. – P. 527-531. 27. Бродский А.М., Гуревич Ю.Я. Теория електронной эмиссии из металлов. – М.: Наука, 1973. - 256 с. 28. Семенова И.В., Флорианович Г.М., Хорошилов А.С. Коррозия и защита от коррозии / Под ред. И.В. Семеновой. – М.:ФИЗМАТЛИТ, 2002. – 336 с. 29. Томашов, Н.Д. Теория коррозии и защиты металлов. – М.: Изд-во АН СССР, 1960. – 592 с. 30. Ermakova L.V. Structural transformations in a Bi2O3 crystal and in Bi2O3based solid solutions in the temperature interval 25-750°C / L.V. Ermakova, V.N. Strekalovskii, E.G. Vovkotrub et al. // J. Appl. Spectroscopy. . 2002. . V. 69, № 1. . P. 152-154. 31. Антонова Л.Т., Денисов В.М., Талашманова Ю.С. Получение оксидных соединений окислением жидких сплавов металлов и полупроводников // Современные наукоемкие технологии. – 2005. – № 8 – С. 33-34. 32. Белоусова Н.В. Взаимодействие жидких металлов и сплавов с кислородом / Н.В. Белоусова, В.М. Денисов, С.А. Истомин и др. . Екатеринбург: УрО РАН, 2004. . 285 с. 33. Антонова Л.Т., Денисов В.М., Талашманова Ю.С. Взаимодействие расплавов системы висмут – серебро с кислородом воздуха // Вестник красноярского государственного университета. Естественные науки, 2006 г. №2, – С. 69 – 82. 34. Технология тонких пленок / Под ред. Л. Майссела, Р. Гленга. Т. 1. – М.: Советское радио, 1977. – 664 с. 35. Титов, И.В. Исследование процесса окисления наноразмерных слоев меди: дисс… канд. хим. наук: 02.00.04 / Титов Илья Вячеславович. – Кемерово, 2006. – 118 с. 36. Методы исследования неорганических материалов: учеб. пособие / Н.В. Борисова, Л.Н. Бугерко, С.М. Сирик, Э.П. Суровой, И.В. Титов; ГОУ ВПО 143 «Кемеровский государственный университет». – Кемерово: Кузбассвузиздат, 2008. – 182 с. 37. Методы исследования неорганических материалов. Часть 2. Оптическая спектроскопия: учеб. пособие / Н.В. Борисова, Л.Н. Бугерко, С.М. Сирик, Э.П. Суровой, Л.И. Шурыгина; ГОУ ВПО «Кемеровский государственный университет». – Томск: Издательство ТГПУ, 2008. – 136 с. 38. Полифункциональные неорганические материалы на основе природных и искусственных соединений / В.И. Верещагин, В.В. Козик, В.И. Сырямкин и др. – Томск: Изд-во Том. ун-та, 2002. – 359 с. 39. Суровой, Э.П. Направленное регулирование процесса фотолиза азидов свинца, серебра, таллия металлами и неорганическими полупроводниками: дисс… доктора хим. наук: 02.00.04 / Суровой Эдуард Павлович. – Кемерово, 2000. – 310 с. 40. Милнс А., Фойхт Д. Гетеропереходы и переходы металл-полупроводник. – М.: Мир, 1975. – 432 с. 41. Электронные явления на поверхности полупроводников: Сб. статей / Под ред. В. И. Ляшенко. – Киев: Наукова думка, 1968. – 364 с. 42. Матосов, М.В. Физика контактной разности потенциалов / М.В. Матосов; Ред. совет фак-та №4 Московского авиационного института. – Москва, 1987. – 188 с. – Библиограф. 186. – Деп. в ВИНИТИ 14.05.87., № 4470-В87. 43. Фоменко, В.С. Эмиссионные и адсорбционные свойства веществ и материалов. Справочник / В.С. Фоменко, И.А. Подчерняева. – М.: Атомиздат, 1975. – 320 с. 44. Гаркуша, Ж.М. Основы физики полупроводников. – М.: Высш. школа, 1982. – 245 с. 45. Стриха В. И., Бузанева Е. В., Радзиевский И. А. Полупроводниковые приборы с барьером Шоттки. – М.: Советское радио, 1974. – 248 с. 46. Гуревич, М.М. Фотометрия. Л.: Энергоатомиздат, 1983. – 272 с. 47. Суровая В.Э. Релаксация тока в наноразмерных пленках оксида молибдена (VI) / В.Э. Суровая, С.В. Бин, А.М. Аникушина // «Студент и 144 научно-технический прогресс»: материалы XLVIII Международной научной студенческой конференции. – Новосибирск, 2010. – С. 196. 48. Суровая В.Э. Оптические и электрофизические свойства наноразмерных систем оксида молибдена (VI) – висмут / В.Э. Суровая, С.В. Бин, Г.О. Еремеева, А.М. Аникушина // «Ломоносов-2010»: материалы XVII Международной конференции студентов, аспирантов и молодых ученых. – Москва, 2010. 49. Суровая В.Э. Исследование темнового и фототока в наноразмерных системах Bi – MoO3 – Bi / В.Э. Суровая, С.В. Бин // «Научное творчество молодежи. Часть 1»: материалы XV Всероссийской научно-практической конференции. - Анжеро-Судженск, 2011. – С. 264-267. 50. Суровая В.Э. Исследование электрофизических свойств наноразмерных систем Bi – MoO3 – Bi / В.Э. Суровая, Л.Н. Бугерко, С.В. Бин // Ползуновский вестник. – Барнаул, 2011. № 4. – С. 105 – 109. 51. Суровая В.Э. Исследование влияния MoO3 на термические превращения наноразмерной пленки Bi / В.Э. Суровая, Л.Н. Бугерко, Г.О. Еремеева // «Научное творчество молодежи. Часть 2»: материалы XVI Всероссийской научно-практической конференции. - Анжеро-Судженск, 2012. – С. 241-244. 52. Методы получения, применение и исследование тонкослойных неорганических материалов: учебное пособие / Н.В. Борисова, Л.Н. Бугерко, Э.П. Суровой и др. – Кемерово: Кузбассвузиздат, 2006. – 140 с. 53. Уэйн, Р. Основы и применения фотохимии. – М.: Мир, 1991. – 304 с. 54. Рабек, Я. Экспериментальные методы в фотохимии и фотофизике. Т. 2. – М.: Мир, 1985. – 544 с. 55. Самсонов Г. В., Кулик О. П., Полищук В. С. Получение и методы анализа нитридов. – Киев: Наукова думка, 2000. – 317 с. 56. Елисеев А. А., Лукашин А. В. Функциональные наноматериалы / Под ред. Ю.Д. Третьякова. – М.: ФИЗМАТЛИТ, 2010. – 456 с. 57. Самсонов Г. В. Тугоплавкие соединения: Справочник по свойствам и применению. – М.: Металлургиздат, 1993. – 398 с. 145 58. Гусев А. И. Наноматериалы, наноструктуры, нанотехнологии. – М.: ФИЗМАТЛИТ, 2009. – 414 с. 59. Ягодин Г.А., Синегрибова О.А., Чекмарев А.М. Технология редких металлов в атомной технике. – М.: Атомиздат, 1974. – 344 с. 60. Краткая химическая энциклопедия. Т.1. – М.: Советская энциклопедия, 1961. – 1263 с. 61. Кофстад П. Отклонение от стехиометрии, диффузия и электропроводность в простых окислах металлов. – М.: Мир, 1975. – 399 с. 62. Бенар Ж. Окисление металлов. – М.: Металлургия, 1969. - 448 с. 63. Хауффе К. Реакции в твердых телах и на их поверхности. – М.: Иностр. лит-ра, 1962. – 415 с. 64. Кубашевский О., Гопкинс Б. Окисление металлов и сплавов. – М.: Металлургия, 1965. – 429 с. 65. Лазарев В.Б., Соболев В.В., Шаплыгин И.С. Химические и физические свойства простых оксидов металлов. – М.: Наука, 1983. – 239 с. 66. Суровая В.Э. Формирование наноразмерных систем Bi – Bi2O3 в процессе термообработки пленок висмута / В.Э. Суровая, Г.О. Еремеева // «Ломоносов-2011»: материалы XVIII Международной конференции студентов, аспирантов и молодых ученых. – Москва, 2011. 67. Суровая В.Э. Кинетические особенности окисления наноразмерных пленок висмута / В.Э. Суровая, С.В. Бин // «Студент и научно-технический прогресс»: материалы XLIX Международной научной студенческой конференции. – Новосибирск, 2011. – С. 196. 68. Суровая В.Э. Поведение наноразмерных пленок висмута в процессе термообработки при Т = 373К / В.Э. Суровая, Г.О. Еремеева // «Научное творчество молодежи. Часть 1»: материалы XV Всероссийской научнопрактической конференции. - Анжеро-Судженск, 2011. – С. 303-306. 69. Суровая В.Э. Термические превращения в наноразмерных пленках висмута / В.Э. Суровая, Э.П. Суровой, Т.Г. Черкасова, Л.Н. Бугерко, С.В. Бин 146 // материалы I Международной Российско-Казахстанской конференции по химии и химической технологии. – Томск, 2011. – С. 40 – 43. 70. Бугерко Л.Н. Термостимулированные превращения в наноразмерных пленках висмута при Т = 573К / Л.Н. Бугерко, В.Э. Суровая, Т.Г. Черкасова // Ползуновский вестник. – Барнаул, 2011. № 4. – С. 101 – 105. 71. Суровой Э.П. Кинетические закономерности термических превращений в наноразмерных пленках висмута / Э.П. Суровой, В.Э. Суровая, Л.Н. Бугерко, С.В. Бин // Журнал физической химии, 2012. Т. 86. № 4. - С. 702-709. 72. Surovoy E.P. Kinetic Regularities of Thermal Transformations in Nanosize Bismuth Films / E.P. Surovoy, L.N. Bugerko, V.E. Surovaia // Journal of Physical Chemistry, 2012. V. 86. № 4. – P. 621-627. 73. Surovoy E.P. Kinetic Regularities of Thermal Transformations in Nanosize Bismuth Films / [Электронный ресурс] E.P. Surovoy, // URL: L.N. Bugerko, V.E. Surovaia http://www.springerlink.com/content/ p141r6l338q08r54/ 74. Золотарев В.М., Морозов В.Н., Смирнова Е.В. Оптические постоянные природных и технических сред. Справочник. - Л.: Химия, 1984. - 216 с. 75. Суровая В.Э. Окисление наноразмерных пленок висмута / В.Э. Суровая, С.В. Бин // «Теоретическая и экспериментальная химия жидкофазных систем»: материалы VI конференции молодых ученых. – Иваново, 2011. – С. 133-134. 76. Суровая В.Э. Закономерности формирования оксида висмута (III) на поверхности наноразмерной пленки висмута / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // материалы IV Всероссийской конференции по химии и химической технологии. – Москва, 2012. – С. 71 – 75. 77. Эпштейн М.И. Измерения оптического излучения в электронике. - Л.: Энергоатомиздат, 1990. - 256 с. 78. Афанасьев В.А. Оптические измерения. – М.: Высшая школа, 1981. – 229 с. 147 79. Самсонов Г.В. Физико-химические свойства окислов. Справочник. – М.: Металлургия, 1978. - 472 с. 80. Суровая В.Э. Закономерности формирования систем Bi - Bi2O3 / В.Э. Суровая, Л.Н. Бугерко // «Россия Молодая»: материалы VI Всероссийской, 59 научно-практической конференции молодых ученых. – Кемерово, 2014. http://science.kuzstu.ru/wp-content/Events/Conference/RM/2014/materials/pdf/ IHNT/ХТНВ/суровая/index.html. 81. Суровая В.Э. Изучение механизма термостимулированного окисления наноразмерных пленок висмута // «Физикохимия и технология неорганических материалов»: материалы XI Российской конференции молодых научных сотрудников и аспирантов. – Москва, 2014. – С. 545 - 546. 82. Суровая В.Э. Термопревращения в наноразмерных слоях висмута / В.Э. Суровая, Г.О. Еремеева, А.М. Аникушина // «Научное творчество молодежи. Часть 1»: материалы XIV Всероссийской научно-практической конференции. - Анжеро-Судженск, 2010. – С. 206-209. 83. Панков Ж. Оптические процессы в полупроводниках. – М.: Мир, 1973. – 456 с. 84. Бьюб Р. Фотопроводимость твердых тел. – М.: ИЛ, 1962. - 558 с. 85. Суровая В.Э. Определение работы выхода наноразмерных пленок висмута и оксида висмута (III) / В.Э. Суровая, Л.Н. Бугерко // «Полифункциональные химические материалы и технологии»: материалы Всероссийской c международным участием научной конференции. – Томск, 2013. – С.87-88. 86. Суровая В.Э. Применение модифицированного метода Кельвина для определения работы выхода Bi и Bi2O3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Теоретическая и экспериментальная химия жидкофазных систем»: материалы VIII Всероссийской школы-конференции молодых ученых. – Иваново, 2013. – С. 139. 148 87. Суровой Э.П. Контактная разность потенциалов для азидов свинца, серебра и таллия / Э.П. Суровой, И.В. Титов, Л.Н. Бугерко // Известия Томского политехнического университета. 2005. Т. 308. № 2. – С. 79-83. 88. Волькенштейн Ф.Ф. Физико - химия поверхности полупроводников. – М.: Наука, 1972. – 399 с. 89. Ляшенко В.И. Электронные явления на поверхности полупроводников / В.И. Ляшенко, В.Г. Литовченко, И.И. Степко. – Киев: «Наукова думка», 1968. – 400 с. 90. Барре П. Кинетика гетерогенных процессов. – М.: Мир, 1976. - 400 с. 91. Индутный И.З., Костышин М.Т., Фотостимулированные взаимодействия в Касярум О.П. и структурах металл др. – полупроводник. – Киев: Наукова думка. 1992. – 240 с. 92. Груздков Ю.А., преобразование Савинов Е.Н., солнечной Пармон В.Н. энергии. Фотокаталитическое Гетерогенные, гомогенные молекулярные структурно-организованные системы. – Новосибирск: Наука, 1991. – 138 с. 93. Стриха В.И., Бузанева Е.В. Физические основы надежности контактов металл-полупроводник в интегральной электронике. – М.: Радио и связь. 1987. – 254 с. 94. Халманн М. Энергетические ресурсы сквозь призму фотохимии и фотокатализа. – М.: Мир, 1986. – 578 с. 95. Суровая В.Э. Фотостимулированные превращения в системах Bi – Bi2O3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой, Т.Г. Черкасова // «Химия и химическая технология: достижения и перспективы»: материалы Всероссийской конференции. – Кемерово, 2012. – С. 49 – 51. 96. Суровая В.Э. Фотостимулированное формирование систем Bi – Bi2O3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Химия и технология полимерных и композиционных материалов»: материалы Всероссийской молодежной научной школы. – Москва, 2012. – С. 284. 149 97. Суровой Э.П. Кинетика фотостимулированных превращений в наноразмерных пленках висмута / Э.П. Суровой, Л.Н. Бугерко, В.Э. Суровая // Журнал физической химии, 2013. Т. 87. № 9. - С. 1565-1571. 98. Surovoi E.P. Kinetics of Photostimulated Transformations in Nanosized Bismuth Films / E.P. Surovoy, L.N. Bugerko, V.E. Surovaia // Journal of Physical Chemistry, 2013. V. 87. № 9. - P. 1556-1561. 99. Surovoi E.P. Kinetics of Photostimulated Transformations in Nanosized Bismuth Films / E.P. Surovoy, L.N. Bugerko, V.E. Surovaia [Электронный ресурс] // URL: http://www.springerlink.com/openurl.asp?genre =article&id=doi:10.1134/S0036024413090239 100. Суровая В.Э. Окисление наноразмерных пленок висмута в процессе облучения / В.Э. Суровая, Г.О. Рамазанова // «Ломоносов-2013»: материалы XX Международной конференции студентов, аспирантов и молодых ученых. – Москва, 2013. 101. Суровая В.Э. Влияние облучения на оптические свойства висмута / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Теоретическая и экспериментальная химия жидкофазных систем»: материалы VIII Всероссийской школыконференции молодых ученых. – Иваново, 2013. – С. 140. 102. Суровая В.Э. Закономерности получения наноразмерной пленки Bi2O3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Современные тенденции и инновации в науке и производстве»: материалы III Международной научнопрактической конференции. – Междуреченск. 2014. – С. 173 – 174. 103. Суровая В.Э. Создание наноразмерных систем Bi – Bi2O3 в процессе облучения / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Природные и интеллектуальные ресурсы Сибири», «СИБРЕСУРС 2014»: материалы ХV Международной научно-практической конференции. – Кемерово, 2014. http://science.kuzstu.ru/wp-content/Events/Conference/Sibresource/2014/materials /pages/Articles/himiya_i_himicheskaya_tehnologiya/surovaya.pdf. 150 104. Суровая В.Э. Модификация наноразмерных пленок висмута под действием света / В.Э. Суровая, Л.Н. Бугерко // Ползуновский вестник. – Барнаул, 2014. № 3. – С. 74 – 78. 105. Surovoi E.P. Regularities of Photostimulated Conversions in Nanometer Aluminum Layers / E.P. Surovoi, N.V. Borisova // Journal of Physical Chemistry, 2009. V. 83. № 13. – P. 2302-2307. 106. Суровой Э.П. Фотостимулированные изменения в спектрах наноразмерных пленок WO3 / Э.П. Суровой, С.В. Бин, Н. В. Борисова // Журнал физической химии, 2010. Т. 84. № 8. - С. 1539 - 1543. 107. Суровой Э.П. Термические превращения в наноразмерных системах Pb WO3 / Э.П. Суровой, С.В. Бин // Журнал физической химии, 2012. Т. 86. № 2. – С. 337-343. 108. Суровой Э.П. Термостимулированное газовыделение из систем азид серебра – металл / Э.П. Суровой, Л.Н. Бугерко // Химическая физика, 2002. Т. 21. № 7. - С. 74 – 78. 109. Тот JI. Карбиды и нитриды переходных металлов: Пер. с англ. / Под ред. П. В. Гельда. – М.: Мир, 1974. – 294с. 110. Кипарисов С. С., Левинский Ю. В. Внутреннее окисление и азотирование сплавов. – М.: Металлургия, 1979. – 200с. 111. Свойства, получение и применение тугоплавких соединений. Справочник / Под ред. Т. Я. Косолаповой. – М.: Металлургия, 1986. – 928с. 112. Самсонов Г. В. Нитриды. – Киев: Наукова думка, 1969. – 380 с. 113. Суровая В.Э. Исследование взаимодействия наноразмерных пленок Bi с аммиаком / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Функциональные наноматериалы и высокочистые вещества»: материалы III Всероссийской молодёжной конференции с элементами научной школы. – Москва, 2012. С. 548 – 549. 114. Суровая В.Э. Влияние аммиака на оптические свойства наноразмерной пленки висмута / В.Э. Суровая, Л.Н. Бугерко // «Научное творчество 151 молодежи. Часть 2»: материалы XVI Всероссийской научно-практической конференции. - Анжеро-Судженск, 2012. – С. 237-240. 115. Суровая В.Э. Индикация аммиака наноразмерными пленками висмута / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Исследования и метрология функциональных материалов»: материалы V Школы-семинара сети центров коллективного пользования. – Томск, 2012. – С. 199 – 207. 116. Суровая В.Э. Особенности взаимодействия висмута с газообразным аммиаком / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Теоретическая и экспериментальная химия жидкофазных систем»: материалы VII Всероссийской школы-конференции молодых ученых. – Иваново, 2012. – С. 157. 117. Суровая В.Э. Модификация наноразмерных пленок висмута в атмосфере аммиака / В.Э. Суровая, Л.Н. Бугерко, Т.Г. Черкасова // Ползуновский вестник. – Барнаул, 2013. № 1. – С. 114 – 118. 118. Суровая В.Э. Формирование наноразмерных систем Bi – BiN / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Высокие технологии в современной науке и технике»: материалы II Всероссийской научно – технической конференции. – Томск, 2013. – С. 250 – 254. 119. Суровой Э.П. Кинетические закономерности взаимодействия наноразмерных пленок висмута с аммиаком / Э.П. Суровой, Л.Н. Бугерко, В.Э. Суровая // Журнал физической химии, 2013. Т. 87. № 6. - С. 1020-1026. 120. Surovoi E.P. Kinetic Patterns of the Interaction between Ammonia and Nanoscale Films of Bismuth / E.P. Surovoy, L.N. Bugerko, V.E. Surovaia // Journal of Physical Chemistry A, 2013. V. 87. № 6. - P. 1009-1014. 121. Surovoi E.P. Kinetic Patterns of the Interaction between Ammonia and Nanoscale Films of Bismuth / E.P. Surovoy, L.N. Bugerko, V.E. Surovaia [Электронный ресурс] // URL: http://www.springerlink.com/openurl.asp?genre =article&id=doi:10.1134/S0036024413060265 122. Суровой Э.П. Особенности процессов формирования нитрида висмута на поверхности наноразмерных пленок висмута / Э.П. Суровой, Л.Н. Бугерко, 152 В.Э. Суровая // Наноматериалы и наноструктуры – XXI век, 2013. Т. 4. № 1. – С. 035 – 038. 123. Суровая В.Э. Термические превращения в наноразмерных системах МоО3− Bi и Bi − МоО3 / В.Э. Суровая, Г.О. Еремеева, А.М. Аникушина // «Студент и научно-технический прогресс»: материалы XLVIII Международной научной студенческой конференции. – Новосибирск, 2010. – С. 168. 124. Суровая В.Э. Термопревращения в наноразмерных системах Bi – MoO3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой, Т.Г. Черкасова, С.В. Бин // материалы II Международной Казахстанско-Российской конференции по химии и химической технологии. – Караганда, 2012. Т. 1. - С. 239 – 243. 125. Суровая В.Э. Оптические свойства наноразмерных систем Bi – MoO3 / В.Э. Суровая, Г.О. Еремеева // «Ломоносов-2012»: материалы XIX Международной конференции студентов, аспирантов и молодых ученых. – Москва, 2012. 126. Суровая В.Э. Влияние термообработки на оптические свойства гетеросистемы Bi - MoO3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Функциональные наноматериалы и высокочистые вещества»: материалы III Всероссийской молодёжной конференции с элементами научной школы. – Москва, 2012. С. 550 – 551. 127. Суровой Э.П. Термостимулированные превращения в наноразмерных системах Bi–MoO3 / Э.П. Суровой, В.Э. Суровая, Л.Н. Бугерко // Журнал физической химии, 2013. Т. 87. № 5. – С. 842-848. 128. Surovoi E.P. Thermostimulated Transformations in Nanosized Bi–MoO3 Systems / E.P. Surovoi, V.E. Surovaia, L.N. Bugerko // Journal of Physical Chemistry A, 2013. V. 87. № 5. - P. 826-831. 129. Surovoi E.P. Thermostimulated Transformations in Nanosized Bi–MoO3 / E.P. Surovoy, L.N. Bugerko, V.E. Surovaia [Электронный ресурс] // URL: http://www.springerlink.com/openurl.asp?genre=article&id=doi:10.1134/S003602 4413050257 153 130. Суровая В.Э. Влияние висмута на отражательные свойства MoO3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Химия и химическая технология: достижения и перспективы»: материалы Всероссийской конференции. – Кемерово, 2012. – С. 52 – 54. 131. Суровая В.Э. Исследование закономерностей процессов в наноразмерных системах Bi – MoO3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Современное состояние и проблемы естественных наук»: материалы Всероссийской научно-практической конференции молодых ученых, аспирантов и студентов. – Юрга, 2014. – С.184 – 189. 132. Суровая В.Э. Влияние висмута на термические превращения наноразмерной пленки MoO3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Теоретическая и экспериментальная химия жидкофазных систем»: материалы VII Всероссийской школы-конференции молодых ученых. – Иваново, 2012. – С. 158. 133. Суровая В.Э. Влияние висмута на оптические свойства наноразмерных пленок MoO3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой, Т.Г. Черкасова // «Полифункциональные химические материалы и технологии»: материалы Общероссийской с международным участием научной конференции. – Томск, 2012. С. 87-88. 134. Суровая В.Э. Оптические свойства предварительно активированных пленок триоксида молибдена / В.Э. Суровая, Н.В. Борисова, А.М. Аникушина // «ВНКСФ – 15»: материалы XV Всероссийской научной конференции студентов-физиков и молодых ученых. – Кемерово, 2009. – С. 463 – 465. 135. Суровая В.Э. Деградация центров окраски в наноразмерных пленках МоО3 / В.Э. Суровая, Н.В. Борисова, А.М. Аникушина // «Студент и научнотехнический прогресс»: материалы XLVII Международной научной студенческой конференции. – Новосибирск, 2009. – С. 150. 136. Суровая В.Э. Оптические свойства наноразмерных слоев МоО3 / В.Э. Суровая, Н.В. Борисова, А.М. Аникушина, С.П. Говорина // «Россия 154 Молодая»: материалы I Всероссийской 54 научно-практической конференции. – Кемерово, 2009. – С. 155 – 158. 137. Суровой Э.П. Оптические свойства наноразмерных слоев MoO3 / Э.П. Суровой, В.Э. Суровая, Н.В. Борисова, Л.Н. Бугерко, А.М. Аникушина, Т.Г. Черкасова // Химия – XXI век: Новые технологии, новые продукты. – Кемерово, 2009. – С. 97 – 99. 138. Суровой Э.П. Термические превращения в наноразмерных слоях меди / Э.П. Суровой, Н.В. Борисова // Журнал физической химии, 2010. Т. 84. № 2. – С. 307 – 313. 139. Суровой Э.П. Закономерности формирования продуктов фотолиза смесей азидов свинца с кадмием / Э.П. Суровой, Л.Н. Бугерко, С.В. Расматова // Журнал физической химии, 2006. Т. 80. № 7. – С. 1308 – 1313. 140. Surovoi, E.P Investigation of energy action influence on WO3 (MoO3) – metal system / E.P Surovoi, N.V. Borisova, I.V. Titov // Известия высших учебных заведений. Физика, 2006. № 10. Приложение. – С. 338-340. 141. Суровой Э.П. Закономерности формирования твёрдофазного продукта фотолиза гетеросистем азид свинца – металл / Э.П. Суровой, Л.Н. Бугерко, Ю.А. Захаров и др. // Материаловедение, 2002. № 9. – С. 27 – 33. 142. Суровой Э.П. Исследование состояния поверхности азидов свинца, серебра и таллия в процессе фотолиза методом КРП / Э.П. Суровой, И.В. Титов, Л.Н. Бугерко // Материаловедение, 2005. № 7. – С. 15 – 20. 143. Epifani M. Synthesis and Characterization of MoO 3 Thin Films and Powders from a Molybdenum Chloromethoxide / M. Epifani, P. Imperatori, L. Mirenghi, M. Schioppa, P. Siciliano // Chem. Mater. 2004. Т. 16. № 25. – С. 5495. 144. Ivanova T. Structure And Optical Properties Of CVD-Molybdenum Oxide Films For Electrochromic Application / T. Ivanova, K. Gesheva, A. Szekeres // J. Solid State Electrochem. 2002. Т. 7. № 1. – Р. 21. 145. Вертопрахов В.Н., Сальман Е.Г. Термостимулированные токи в неорганических веществах. – Новосибирск: Наука, Сибирское отд-е. 1979. – 336 с. 146. Суровой Э.П. Термостимулированные изменения оптических свойств 155 предварительно активированных слоев оксида молибдена (VI) / Э.П. Суровой, Н.В. Борисова // Материаловедение. 2009. № 7. – С. 43. 147. Prasad A. K. Synthesis and sensing properties to NH3 of hexagonal WO3 metastable nanopowders / A. K. Prasad, D. J. Kubinski, P. I. Gouma // Sens. and Actuators. B. 2003. Т. 93. № 1. – Р. 25. 148. Sian Tarsame S. Infrared spectroscopic studies on Mg intercalated crystalline MoO3 thin films / Sian Tarsame S., G.B. Reddy // Sol. Energy Mater. and Sol. Cells. 2004. 82. № 3. – Р. 375. 149. Раманс Г.М. Структура и морфология аморфных пленок триоксида вольфрама и молибдена // Электрохромизм. – Рига: ЛГУ им. П.Стучки, 1987. – С. 121-129. 150. Андреев В.Н. Исследование фотохромных кластерных систем на основе оксидов Мо методом ЭПР-спектроскопии / В.Н. Андреев, С.Е. Никитин // Физика твердого тела, 2001. Т. 43. № 4. – С. 755-758. 151. Chary Komandur V. R. Structure and Catalytic Properties of Molybdenum Oxide Catalysts Supported on Zirconia. In / V. R. Chary Komandur, Reddy Kondakindi Rajender, Kishan Gurram, Niemantsverdriet J. W., Mestl Gerhard. // J. Catal. 2004. Т. 226. №2. – Р. 283. 152. Yao, J.N. Enhancement of Photochromism and Electrochromism in MoO3/Au and MoO3/Pt Thin Films / J.N. Yao, Y.A. Yang, B.H. Loo // J. Phys. Chem. B, 1998. V. 102. – P. 1856-1860. 153. Гончаров, И.Б. Ионный циклотронный резонанс в реакциях ионных кластеров оксида молибдена с аммиаком / И.Б. Гончаров, У.Ф. Фиалко // Журнал физической химии, 2002. Т. 76. № 9. – С. 1610-1617. 154. Груздков Ю.А., преобразование Савинов Е.Н., солнечной Пармон В.Н. энергии. Фотокаталитическое Гетерогенные, гомогенные молекулярные структурно-организованные системы. – Новосибирск: Наука, 1991. – 138 с. 155. Суровая В.Э. Влияние облучения на оптические свойства MoO3 / В.Э. Суровая, Л.Н. Бугерко, Н.В. Борисова, Г.О. Еремеева // Ползуновский вестник. – Барнаул, 2013. № 1. – С. 77 – 82. 156 156. Суровая В.Э. Образование центров окраски в наноразмерных пленках оксида молибдена (VI) под действием света / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой, Н.В. Борисова // «Высокие технологии в современной науке и технике»: материалы II Всероссийской научно – технической конференции. – Томск, 2013. – С. 254 – 259. 157. Суровая В.Э. Модификация оксида молибдена под действием света // «Инновации а материаловедении»: материалы Всероссийской молодежной научной конференции. – Москва, 2013. – С. 321. 158. Суровой Э.П. Закономерности фотостимулированных превращений в наноразмерных пленках MoO3 / Э.П. Суровой, Л.Н. Бугерко, Н.В. Борисова, В.Э. Суровая, Г.О. Рамазанова // Журнал физической химии, 2013. Т. 87. № 12. – С. 2105 – 2109. 159. Surovoi E.P. Photostimulated Transformations in Nanosized MoO3 Films / E.P. Surovoy, L.N. Bugerko, N.V. Borisova, V.E. Surovaia, G.O. Ramazanova // Journal of Physical Chemistry A, 2013. V. 87. № 12. – P. 2063 – 2067. 160. Surovoi E.P. Photostimulated Transformations in Nanosized MoO3 Films / E.P. Surovoy, L.N. Bugerko, N.V. Borisova, V.E. Surovaia, G.O. Ramazanova [Электронный ресурс] // URL: http://www.springerlink.com/openurl.asp?genre =article&id=doi:10.1134/S0036024413120248 161. Суровая В.Э. Фотохимические превращения в наноразмерных пленках MoO3 / В.Э. Суровая, Л.Н. Бугерко, Э.П. Суровой // «Химия и химическая технология: достижения и перспективы»: материалы II Всероссийской конференции. – Кемерово, 2014. – http://ihnt.kuzstu.ru/conf/pages/Articles/ ximia_i_ximicheskaya_tehnologiya_neorganicheskix_veshestv_i_materialov/Suro vaya_Bugerko.pdf. 162. Суровая наноразмерных В.Э. Особенности пленок MoO3 // фотостимулированных «Ломоносов-2014»: превращений материалы XX Международной конференции студентов, аспирантов и молодых ученых. – Москва, 2737.pdf. 2014. - http://lomonosov-msu.ru/archive/Lomonosov_2014/2737/ 157 ПРИЛОЖЕНИЕ