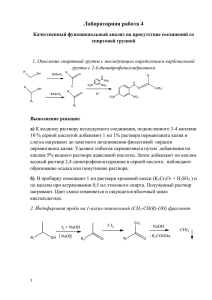
Министерство образования и науки Российской Федерации Федеральное агентство по образованию
реклама

Министерство образования и науки Российской Федерации Федеральное агентство по образованию Южно-Уральский государственный университет Кафедра органической химии 547(07) К40 Д.Г. Ким, А.В. Журавлёва, Т.В. Тюрина ОРГАНИЧЕСКАЯ ХИМИЯ Учебное пособие Челябинск Издательство ЮУрГУ 2009 УДК 547(076.5) К40 Одобрено учебно-методической комиссией химического факультета Рецензенты: Д. х. н., профессор Козьминых В.О., Оренбургский государственный университет Кафедра органической химии Пермского государственного университета, зав. кафедрой, д.х.н., профессор Шуров С.Н. Органическая химия: учебное пособие/ Д.Г. Т.В.Тюрина. - Челябинск: Изд. ЮУрГУ, 2009. - 125 с. Ким, А.В.Журавлёва, Практикум по органической химии составлен в соответствии с программой дисциплины «Химия» для студентов нехимических специальностей высших учебных заведений. Пособие включает темы: изучение состава органических соединений их очистка и определение физических констант; ознакомительный (малый) практикум; методы синтеза органических соединений; идентификация органических соединений. Раздел «Изучение состава органических соединений их очистка и определение физических констант» включает качественный и функциональный анализ, тонкослойную хроматографию, перегонку, перекристаллизацию, возгонку, определение температуры плавления и кипения, показателя преломления. Ознакомительный (малый) практикум включает получение и изучение свойств важнейших представителей основных классов органических соединений: углеводородов, галогенпроизводных углеводородов, кислород- и азотсодержащих соединений и гетероциклических соединений. В части «Методы синтеза органических соединений» студенты осваивают основные методы синтеза основных представителей органических соединений с использованием различных реакций: нитрование, сульфирование, галогенирование, алкилирование, ацилирование, окисления и восстановления. Завершает пособие часть «Идентификация органических соединений», в которой студенты идентифицируют и устанавливают структуры органических соединений с использованием физических, химических и физико-химических методов анализа (инфракрасная спектроскопия, масс-спектрометрия, тонкослойная хроматография). В приложении приведены необходимые справочные данные для выполнения лабораторных работ. 2 1. ОСНОВНЫЕ ПРАВИЛА И ОРГАНИЗАЦИЯ РАБОТЫ В ЛАБОРАТОРИИ ОРГАНИЧЕСКОЙ ХИМИИ 1.1. Техника безопасности 1. Общие положения Работа в лаборатории должна производиться только в халатах; запрещается работать в верхней одежде и головных уборах. Студенты обязаны соблюдать в лаборатории чистоту и порядок; во время работы следует поддерживать тишину. В лаборатории запрещается принимать пищу или класть продукты на рабочий стол. В случае какого бы то ни было происшествия необходимо немедленно сообщить об этом преподавателю и лаборанту. 2. Перед началом лабораторной работы Получив от преподавателя разрешение на выполнение работы, студент должен обратиться к дежурному лаборанту и получить от него инструктаж по правилам работы на используемой установке; после этого с разрешения лаборанта студент может приступать к самостоятельной работе. Без разрешения преподавателя и лаборанта работать на приборах и установках категорически запрещается. 3. При выполнении работы Категорически запрещается пробовать вещества на вкус, нюхать вещества из горлышек склянок, при растворении веществ в пробирке зажимать отверстие пальцем. Опыты проводить только с таким количеством веществ, которые обозначены в методическом руководстве по проведению каждого опыта. Наливать или насыпать вещества можно только над столом или специальным поддоном; просыпанные или случайно пролитые вещества надо немедленно убирать специальной тряпочкой. Горелку (или спиртовку) зажигать только спичкой или лучиной; спичку зажигают движением от себя (и от соседа). Нельзя ни в коем случае держать голову близко к пламени или наклоняться над ним. 3 При нагревании вещества в пробирке ее сначала необходимо целиком прогреть над пламенем, нельзя сразу направлять пламя на границу поверхности жидкости в пробирке; отверстие пробирки во время ее нагревания должно быть направлено от себя и соседа. Нельзя ставить горячие пробирки в штатив из пластмассы. Необходимо соблюдать осторожность при работе со всякого рода электроприборами и электрооборудованием; при обнаружении неисправности следует прекратить работу, отключить прибор от сети и обратиться к преподавателю или лаборанту. 4. По окончании работы По окончании работы студенты обязаны: выключить электроприборы, вымыть использованную посуду, закрыть водопроводные краны, убрать свое рабочее место, сдать лаборанту используемую посуду и реактивы и получить разрешение преподавателя покинуть лабораторию. 5. Оказание первой доврачебной помощи В случае пореза стеклом необходимо сначала убедиться, что в ранке нет осколков, и затем ватой, смоченной спиртом или раствором перманганата калия, удалить кровь, после чего края раны смазать иодом и забинтовать. При небольших термических ожогах следует смазать обожженное место глицерином или спиртом; при более значительных ожогах обожженное место следует смазать мазью от ожогов или концентрированным раствором перманганата калия и наложить повязку. Нельзя смазывать рану вазелином или жиром. При попадании на кожу кислоты необходимо немедленно смыть кислоту большим количеством воды, затем промыть пострадавшее место 3%-ным раствором соды и смазать мазью от ожогов. При ожогах щелочью обожженное место следует промыть водой и затем раствором борной кислоты. При попадании кислоты или щелочи в глаз необходимо промыть его водой, затем раствором соды или борной кислоты соответственно и внести в глаз каплю касторового масла. В более тяжелых случаях следует вызвать скорую помощь. 4 Источники опасности при работе с органическими растворителями Органические растворители занимают особое место среди применяемых в лабораториях огнеопасных веществ. Они легко воспламеняются, быстро горят и с трудом тушатся. Пары многих органических растворителей даже при комнатной температуре способны образовывать с воздухом пожаровзрывоопасные смеси. Опасность применения и хранения органических растворителей зависит от ряда условий — количества и горючести жидкости, температуры, герметичности аппаратуры или тары, наличия источников воспламенения и т. д. Классификация ЛВЖ по степени опасности В зависимости от температуры вспышки ЛВЖ принято условно относить к одному из трех разрядов, которые представлены в таблице 1.1.: Таблица 1.1. Температура вспышки, °С: Разряд Характеристика жидкости в открытом тигле в закрытом тигле I Особо опасные до -18 до -13 II Постоянно опасные от -18 до 23 от -13 до 27 III Опасные при повышенной температуре от 23 до 61 от 27 до 66 К I разряду относятся: акролеин, аллилхлорид, ацеталь, ацетальдегид, ацетон, бензины, гексан, диметилдихлорсилан, дипропиловый эфир, диэтиламин, диэтиловый эфир, изопропиламин, изопропилхлорид, метилметакрилат, метилформиат, пропиленоксид, петролейный эфир, пропилхлорид, сероуглерод, тетрагидрофуран, фуран, циклогексан, циклогексен, этиламин, этилформиат и др. Ко II разряду относятся: акрилонитрил, аллилбромид. аллиламин, амилхлорид, ацетонитрил, бензол, бутиламин, 2-метил-2-пропанол, бутилхлорид, винилацетат, гексаметилдисилоксан, гептан, дибутиловый эфир, диоксан, дихлорэтан, диэтилхлорсилан, диэтилкетон, изопропилацетат, 2-пропанол, изопропилформиат, изопропилкарбонат, лигроин, метилацетат, метанол, метилтрихлорсилан, метилхлорформиат, метилэтилкетон, пиперидин, пиридин, толуол, триэтиламин, циклогексиламин, этилакрилат, этилацетат, этилбензол, этиленамин, этанол и др. К III разряду относятся: амилацетат, анизол, ацетилацетон, бензилхлорид, бромбензол, бутанол, гексилхлорид, декан, диамиловый эфир, дикетен, диметиламиноэтанол, диметилсульфат, N,N-диэтиламиноэтанол, диэтилкарбонат изоамилацетат, керосины, ксилол, метилакрилат, морфолин, муравьиная кислота, 5 октиламин, пентанол, пропилбензол, пропанол, скипидар, стирол, уксусная кислота, уксусный ангидрид, хлорбензол, циклогексанон и др. Жидкости, имеющие температуру вспышки выше 61 °С в закрытом тигле или выше 66 °С в открытом тигле и способные гореть после удаления источника зажигания, относятся к ГЖ. Работа с легковоспламеняющимися жидкостями. При работе с ЛВЖ следует придерживаться трех основных принципов: 1. работать только в вытяжном шкафу 2. не допускать попадания горючих паров в атмосферу (предотвращать образование пожаровзрывоопасных смесей); 3. исключать возможность воспламенения при случайном образовании пожаровзрывоопасной концентрации паров (исключать возникновение источников зажигания, работать вдали от открытого пламени и электронагревательных приборов); 4. При перегонке легковоспламеняющихся жидкостей необходимо использовать водяной холодильник и тщательно собирать прибор во избежание утечек жидкости через соединения; во время перегонки следует надевать защитные очки. 5. Запрещается выливать отходы ЛВЖ в канализацию. Выливать органические растворители следует только в склянки, предназначенные для их слива. 6. Хранят ЛВЖ только под тягой в герметично закрытой толстостенной таре. 7. заранее принимать все возможные меры, чтобы последствия аварии, если она все же произойдет, были минимальными. Поэтому любые работы с ЛВЖ, при которых в окружающее пространство могут выделяться горючие пары, следует проводить при выключенных горелках, электрических приборах и потенциальных источниках зажигания. Нагревание ЛВЖ можно производить только в приборах и установках, обеспечивающих полную конденсацию образующихся паров. В качестве нагревательных приборов нельзя использовать электроплитки с открытой спиралью, а также открытое пламя. При необходимости нагревания ЛВЖ, относящихся к I разряду, особенно диэтилового эфира и сероуглерода, следует либо использовать предварительно нагретые в другом месте жидкостные бани, либо применять бани с нагревательным элементом. 6 Источники опасности при работе со щелочными металлами Натрий (плотность 0,97 г/см3, температура плавления 97,7 °С). Вследствие высокой химической активности натрия работа с ним в лаборатории представляет серьезную опасность. Натрий взаимодействует с кислородом воздуха при комнатной температуре с образованием оксида. Реагируя с влагой воздуха, оксид переходит в гидроксид. Куски натрия, оставленные на воздухе, быстро обрастают расплывающейся коркой гидроксида. Работа с металлическим натрием: 1. Нельзя допускать соприкосновения щелочных метало в водой и галогенсодержащими соединениями; 2. Хранят металлический натрий и калий в толстостенной посуде из темного стекла под слоем керосина или трансформаторного масла в вытяжном шкафу; 3. Для работы натрий нужно извлечь пинцетом из-под защитного слоя, остатки керосина промокнуть сухой фильтровальной бумагой, отрезать ножом необходимое количество металла и сразу же использовать его по назначению; 4. Большие обрезки натрия следует собрать под слоем керосина, а небольшие кусочки натрия, посуду и бумагу с остатками металла залить этиловым спиртом. При мытье посуды водой в таких случаях иногда происходят взрывы, которые особенно опасны из-за их неожиданности. Во избежание несчастных случаев следует уничтожать остатки натрия сразу после их образования. 5. Категорически запрещается выбрасывать остатки металлического натрия и калия (а так же необработанную этиловым спиртом бумагу) в мусорное ведро и раковину или оставлять их в пустых пробирках или колбах; 6. При нагревании реакционных смесей, содержащих щелочные металлы, можно использовать только воздушные или песчаные бани. Правила работы с бромом. Первая помощь при отравлениях бромом Бром - жидкость темно-бурого цвета с плотностью d=3,102, растворимая в эфире, спирте, тетрахлориде углерода и др., выделяет оранжево-красные пары, имеющие резкий запах. При попадании на кожу бром быстро впитывается и вызывает долго не заживающие ожоги и язвы. Особенно тяжело поражаются не огрубевшие участки кожи (например, между пальцами и под ногтями). Пары брома, соприкасаясь со слизистыми оболочками, вызывают жжение в области носоглотки. 7 1. Отмеривание необходимого количества брома и выполнение синтезов с его участием обязательно проводятся в вытяжном шкафу с полуопущенными створками, а применением резиновых перчаток и защитных очков или маски. 2. Сосуд для хранения брома должен быть достаточно прочным, с притертой пробкой и не превышать одного литра по объему. Хранить сосуды с бромом следует на песчаной бане. При переливании брома в другие емкости вынимать сосуд из бани с песком не разрешается. 3. Бром, предназначенный для синтеза, следует наливать в плоскодонную колбу через коническую воронку, закрепленную над колбой в кольце штатива. Носик воронки должен находиться внутри колбы и быть значительно уже ее горла (чтобы не затруднять выход воздуха при заполнении колбы бромом). В противном случае возможно разбрызгивание брома, находящегося в воронке. Колбу-приемник с бромом следует немедленно закрыть каучуковой или полиэтиленовой пробкой и отнести к рабочему месту, расположенному в вытяжном шкафу. 4. Колбу, после использования брома, надо сразу же вымыть в раковине, находящейся в вытяжном шкафу. Для этого ее ставят в раковину так, чтобы струя воды поступала в сосуд, закрывают створку вытяжного шкафа и полностью вытесняют водой оставшиеся в колбе пары брома. После окончания синтеза все детали прибора, соприкасавшиеся с бромом, моются аналогичным образом. 5. Колбу, после использования брома, надо сразу же вымыть в раковине, находящейся в вытяжном шкафу. Для этого ее ставят в раковину так, чтобы струя воды поступала в сосуд, закрывают створку вытяжного шкафа и полностью вытесняют водой оставшиеся в колбе пары брома. После окончания синтеза все детали прибора, соприкасавшиеся с бромом, моются аналогичным образом. При отсутствии раковины в вытяжном шкафу посуду после брома оставляют под тягой до полного его испарения, а затем моют водой. Тяжесть травм, наносимых бромом, в значительной степени зависит от того, насколько своевременно и правильно будет оказана первая помощь. Во избежание тяжелых травм, который может нанести жидкий или парообразный бром при несвоевременном удалении его с пораженного места, необходимо одновременно с реактивами необходимыми для выполнения синтеза, получить флакон с 5-10 мл этилового спирта и ватный тампон. В случае попадания жидкого брома на кожу, его необходимо немедленно снять с пораженного места сухим ватным тампоном, несколько раз обработать ватным тампоном, смоченным спиртом, и приложить спиртовой компресс. Если болевые ощущения не проходят, а кожа на пораженном месте приобретает розовый цвет, то следует обратиться к врачу. При попадании паров брома в дыхательные пути следует, смочив вату спиртом, в течение некоторого времени вдыхать через нос его пары, а затем выйти на свежий воздух. При этом противопоказаны резкие движения, приводящие к учащенному дыханию. Если 8 вышеописанные действия не снимают болевых ощущений в носоглотке, следует обратиться к врачу. Средства первой помощи. При легком отравлении - вдыхание паров аммиака (3-5 % в воздухе). Промывание глаз, рта и носа 5%-ным раствором бикарбоната натрия или разбавленным этиловым спиртом. При сильном отравлении - свежий воздух (в лежачем положении), покой, вдыхание кислорода. При попадании брома на плоскость стола или пола необходимо, предварительно приложив ко рту и носу ватный тампон или марлевую повязку, смоченные этиловым спиртом, надев резиновые перчатки, дегазировать загрязненную бромом площадь сухим карбонатом натрия или калия. Правила работы с едкими веществами (кислоты, щелочи) 1. Едкие (агрессивные, вызывающие химические ожоги) вещества (концентрированные кислоты - соляная, азотная, серная, фтористоводородная и хромовый ангидрид, сухие щелочи - едкий натр, едкое кали и их концентрированные растворы, а также растворы аммиака), попадая на кожу, вызывают ожоги. Особая опасность заключается в возможности поражения глаз. 2. Хранить едкие вещества только в толстостенной стеклянной посуде емкостью не более 2-х литров в вытяжном шкафу. 3. Переливать кислоты только при включенной тяге в вытяжном шкафу. Дверцы шкафа должны быть по возможности, прикрыты. 4. Разбавление кислот производить приливанием кислоты в воду и только в жаростойких и фарфоровых стаканах, т.к. при этом происходит значительное выделение тепла. Склянки с концентрированными кислотами, а также бромом переносить только в ведре, а при переливании склянку нельзя держать за горлышко. Растворять едкие щелочи следует путем медленного прибавления к воде небольших кусочков; куски щелочи брать щипцами или шпателем. 5. При работе с дымящей азотной кислотой и олеумом одевать очки. 6. Запрещается применять серную кислоту в вакуум - эксикаторах в качестве водопоглощающего средства. 7. Работа с плавиковой кислотой требует особой осторожности. Обязательно надевать резиновые перчатки, защитные очки и все работы проводить только под тягой. 8. Разлитые кислоты и щелочи следует немедленно нейтрализовать и лишь после этого проводить уборку. 9. Запрещается набирать растворы кислот и щелочей, всасывая их ртом в пипетку. 10. использованные реактивы следует выливать в специально отведенные склянки 9 Правила безопасности работы со стеклянной химической посудой и ампулами 1. При закрывании толстостенного сосуда пробкой следует держать его за верхнюю часть горла ближе к пробке; руки должны быть защищены полотенцем. 2. Нагретый сосуд нельзя закрывать притертой пробкой до тех пор, пока он не охладится. 3. При переливании жидкостей следует пользоваться воронкой. 4. При нагревании жидкости в пробирке отверстие последней должно быть направлено в сторону от себя и соседей. 5. При переносе сосуда с горячей жидкостью нужно пользовался полотенцем, держа сосуд за дно и горловину. 6. Работы, при которых возможно образование давления в сосуде, перегрев его, поломка и разбрызгивание горячих жидкостей, необходимо вести в вытяжном шкафу и по фронту работ устанавливать предохранительные щитки из оргстекла. 7. Смешение и разбавление веществ, сопровождающееся выделением тепла, следует проводить в термостойкой стеклянной или фарфоровой посуде. Правила безопасной работы с электрооборудованием и электроприборами 1. При работе с электрооборудованием и электроприборами возможны поражения электрическим током, причинами которых могут быть: а) работа при неисправном электрооборудовании (рубильники, двигатели и т.п.); б) прикосновение к случайно оказавшимися под током металлическим конструкциям, корпусам электрооборудования и к другим незаземленным металлическим предметам; в) контакт с находящимися под током неизолированными проводами или проводами с поврежденной изоляцией; г) нарушение правил применения индивидуальных средств защиты. Электрический ток может вызвать пожары и взрывы, источником которых могут быть искры, нагретые (накаленные) токопроводящие части электроустановки, короткое замыкание. 2. Работы в лаборатории должны проводиться при наличии исправного электрооборудования. При обнаружении дефектов в изоляции проводов, неисправности рубильников, штепселей, розеток, вилок, а также заземления, следует сообщить об этом ответственному за ТБ данной лаборатории для принятия им соответствующих мер. Все неисправности электроприборов, электросети и электрооборудования должны устраняться только электромонтером. 3. Запрещается переносить включенные приборы и ремонтировать оборудование, находящееся под током. 4. Шкафы, в которых установлены щитки электрорубильников, должны быть закрыты. 10 В случае загорания проводов или электроприборов, находящихся под током, необходимо немедленно выключить ток и тушить огонь сухим углекислотным огнетушителем, сухим песком, покрывалом из асбеста и другого негорючего. 11 1.2. Правила оформления, ведения рабочего (лабораторного) журнала и составление отчета Перед тем, как приступить к лабораторной работе, каждый студент должен получить допуск. При допуске студенту необходимо знать основные принципы используемых методов и основные свойства исследуемых соединений. А так же студент должен иметь представление о ходе проведения каждого опыта и знать технику безопасности. При выполнении лабораторной работы студент обязан вести рабочий (лабораторный) журнал, который предназначен для всех наблюдений за ходом эксперимента и полученных результатов. Делая записи в журнале, необходимо четко излагать суть проведенного опыта или синтеза. Такие записи проводят или в процессе выполнения работы, или сразу же после ее окончания. Необходимо указывать продолжительность некоторых операций, а так же все изменения или возможные отступления от методики, описанной в руководстве к лабораторному практикуму. Отчеты ко всем проделанным лабораторным работам оформляются вместе (в одной тетради) последовательно. Первый лист тетради - титульный лист оформляется, как показано в Приложении 1. Каждая лабораторная работа начинается с ее номера и названия и оформляется с нового листа. В отчете по каждой лабораторной работе обязательно пишется: 1. Краткое теоретическое введение (описывается объект исследования и применяемые методы); 2. Практическая часть (номер и название опыта; методика опыта со всеми ее изменениями, если таковые имеются; полученные результаты или наблюдения, уравнение реакции, если проводилось какое-либо взаимодействие). В уравнении реакций необходимо называть исходные и конечные соединения. В некоторых случаях описывается механизм. При необходимости в практической части зарисовывают схему установки или прибора данного опыта; 3. Выводы о проделанной работе (анализируются полученные результаты). При защите отчета студенту необходимо объяснить полученные результаты, сделать выводы, ответить на вопросы, представленные в конце каждой работы в лабораторном практикуме, а так же ответить на дополнительные вопросы. 12 2. ЛАБОРАТОРНАЯ ХИМИЧЕСКАЯ ПОСУДА 2.1. Перечень и краткое описание лабораторной посуды В химических лабораториях обычно используют стеклянную посуду. Она изготавливается, как правило, из специального стекла, которое устойчиво к кислотам, щелочам и большинству химических реагентов, и обладает сравнительно небольшим коэффициентом линейного расширения (что очень важно при переменных температурах химического эксперимента). Посуда из стекла очень удобна – она прозрачна, хорошо моется и сушится и легко поддается термической обработке. Основным ее недостатком является хрупкость. Стаканы обычно изготавливаются из термического стекла и бывают различной вместимости (от 50 до 1000 мл). Они служат для вспомогательных работ с органическими жидкостями и водными растворами. Пробирки бывают различной величины и диаметра. Обычные лабораторные пробирки изготавливают из легкоплавкого стекла, но для особых работ, например, при высоких температурах, применяют пробирки из тугоплавкого стекла или кварца. При перемешивании реактивов пробирку держат за верхнюю часть большим, указательным и средним пальцами левой руки, а указательным пальцем правой руки ударяют косым скользящим движением по ее нижней части несколько раз. Нельзя встряхивать пробирку, закрывая ее пальцем, так как при этом загрязняются перемешиваемые вещества, а при проведении опытов с едкими веществами может быть травмирована кожа руки. Если пробирку необходимо нагреть, то ее закрепляют в держателе или в лапке штатива. Колбы бывают плоскодонные, конические, круглодонные и грушевидные. Плоскодонные и конические колбы обычно используют в качестве приемников при перегонке жидкости, для приготовления растворов и кристаллизации. Их нельзя применять при нагревании веществ до высоких температур и использовать при работе при пониженном давлении (из-за опасности разрушения колб). Круглодонные колбы используют для перегонки веществ, в том числе и под вакуумом. Длина и диаметр горла круглодонных колб могут варьировать. Эти колбы бывают двух-, трехгорлыми и т.д. Круглодонные колбы с отводной трубкой называют колбами Вюрца. Они предназначены для перегонки веществ при атмосферном давлении. В отличие от колбы Вюрца колба Кляйзена имеет на горле две шейки, от одной из которых отходит отводная трубка. Колбы Кляйзена применяют для перегонки жидкостей при пониженном давлении. Колба Бунзена вместе с воронкой Бюхнера (рис.2.1.) используется для фильтрования под вакуумом. Холодильники служат для охлаждения и конденсации паров, образующихся при кипении органических жидкостей. Чтобы избежать потерь низкокипящих компонентов, колбы снабжают обратными холодильниками, где пары охлаждаются и конденсат возвращается в реакционную смесь. При перегонке вещество конденсируется в холодильнике и отводится в приемную колбу. Такие 13 холодильники называются нисходящими (они крепятся под углом к столу в сторону приемника). Рис. 2.1. Колбы: а – плоскодонная; б – круглодонные; в – коническая; г – двух- и трехгорлые; д –Вюрца для жидкостей с низкой температурой кипения; е - Вюрца для жидкостей с высокой температурой кипения; ж – Кляйзена; з – Фаворского (с дефлегматором); и – грушевидная с дефлегматором; к – Бунзена. Простейшим является воздушный холодильник (рис.2.2.), который представляет собой длинную стеклянную трубку. Он годится только для работы с высококипящими жидкостями, поскольку эффективность воздуха как охлаждающего средства невелика. Воздушный холодильник можно использовать и как нисходящий, но при не слишком большой скорости перегонки, для жидкостей с температурой кипения больше 1500С. В холодильнике Либиха (рис.2.2.) для охлаждения и конденсации пара используется проточная вода. Его применяют в качестве нисходящего для перегонки жидкостей с температурой кипения меньше 1600С. В качестве обратного холодильника он мало эффективен, так как имеет небольшую охлаждающую поверхность. Шариковый холодильник (рис.2.2.) используют только как обратный, поскольку его охлаждающая поверхность значительно больше, чем у холодильника Либиха. Змеевиковый холодильник (рис.2.2.) никогда не следует использовать как обратный, потому что конденсат, который недостаточно хорошо стекает по изгибам змеевика, может быть выброшен из холодильника и послужить причиной несчастного случая. Змеевиковый холодильник, установленный вертикально, является наиболее эффективным нисходящим холодильником, особенно для низкокипящих веществ. Если нельзя устанавливать наклонно, так как конденсат может скапливаться внутри холодильника и не доходить до приемника. 14 Рис. 2.2. Холодильники: а – воздушный; б – Либиха; в – шариковый; г – змеевиковый; д – Димрота. а б в г д Холодильник Димрота (рис.2.2.) – очень эффективный обратный холодильник, но иногда он может быть использован как нисходящий, хотя в этом случае будут наблюдаться большие потери дистиллята на змеевике. При работе с холодильниками, в которых охлаждающим средством является вода, необходимо помнить, что к водопроводному крану всегда присоединяется нижний отросток «рубашки» холодильника, а верхний отводят в раковину. При этом холодильник должен быть полностью заполнен водой, и ее циркуляция через «рубашку» не должна прекращаться, так как отключение холодильника может привести к пожару или взрыву. Воронки. Воронки для фильтрования выпускают различных размеров – диаметром от 35 до 300 мм. Обычные воронки имеют ровную внутреннюю поверхность, но для ускоренного фильтрования иногда применяют воронки с ребристой внутренней поверхностью. Кроме того, некоторые воронки имеют удлиненный конец, внутренний диаметр которого в верхней части меньше, чем в нижней (рис.2.3.). Такая конструкция также ускоряет фильтрование. Воронки Бюхнера отличаются от обычных воронок тем, что они сделаны из фарфора и имеют перегородку с отверстиями, на которую помещают фильтр (рис. 2.4.). Воронку вставляют в колбу Бунзена, из которой затем откачивают воздух. 15 Рис. 2.3. Воронка. Рис. 2.4. Воронка Бюхнера: 1 – резиновая пробка под колбу Бунзена; 2 – перегородка с отверстиями; 3 – бумажный фильтр. Делительные воронки применяют для разделения несмешивающихся жидкостей и экстракции. Они бывают цилиндрической, шаровидной или грушевидной формы, с пробкой в верхней части и с притертым стеклянным краном в верхней части отводной трубки (рис.2.5). Рис. 2.5. Делительные воронки: а –цилиндрическая; б – шарообразная; в, г – грушевидные. д– капельная воронка. а б в г д Капельные воронки (рис.2.5) предназначены для медленного прибавления жидкости в реакционную смесь во время проведения синтеза вещества. Они похожи на делительные, но у них более тонкие стенки и более длинные отводные трубки. Капельные воронки составляют часть прибора и крепятся к горлу колбы на шлифе или при помощи резиновой пробки. Перед работой с капельной или делительной воронкой шлиф стеклянного крана нужно смазать вазелином или специальной смазкой. Дефлегматоры применяют для более тщательной фракционной перегонки веществ. В верхнее отверстие дефлегматора вставляют термометр, а отводную трубку соединяют с нисходящим холодильником (рис. 2.6.). 16 Рис. 2.6. Дефлегматоры: а, б – шариковые; б – елочный; в – с насадкой. а б в г Хлоркальциевые трубки применяют для защиты реакционной смеси от попадания в нее нежелательных примесей из воздуха (паров воды, оксида углерода IV), а так же от попадания в окружающую среду вредных веществ, образующихся в ходе химической реакции. Мерная посуда служит для измерения объема жидкости. Мерные цилиндры и мензурки (рис. 2.7.) служат для измерения больших объемов – от 5 до 2000 мл. Бюретки – приборы для измерения точных объемов жидкости, применяемые преимущественно при титровании. Пипетками отмеряют наиболее точные объемы – от 0.005 мл (для микропипеток) до 10-25 мл (для градуированных пипеток и пипеток Мора). Мерные колбы предназначены для приготовления растворов точных концентраций. Они имеют длинную шейку, на которой нанесена метка, шлиф и притертую пробку. При приготовлении раствора уровень жидкости доводят до метки. Кристаллизаторы – это низкобортные сосуды, предназначенные для охлаждения веществ при их получении или кристаллизации. Иногда в кристаллизаторах можно проводить выпаривание, но следует помнить, что нагревать их можно только на водяной бане. Эксикаторы – это емкости из толстостенного стекла, состоящие из массивного корпуса и притертой к нему стеклянной крышки. Они предназначены для упаривания растворов и высушивания твердых веществ. Различаю простые и вакуум - эксикаторы. Рис. 2.7. Мерная посуда. 17 Из вакуум – эксикаторов (рис. 2.8.) через трубку с краном при помощи водоструйного насоса откачивают воздух. Вещество помещают в эксикатор в чашке Петри. В качестве осушителя применяют прокаленный хлорид кальция, оксид фосфора (V), силикагель, натронную известь, гидроксид натрия, сульфат магния или натрия. Рис. 2.8 . Вакуум-эксикатор: 1 – осушающее вещество; 2 – высушиваемое вещество. Фарфоровая посуда позволяет вести прямой обогрев веществ до температуры 12000с. Недостатком этой посуды является ее большая масса и непрозрачность. Чашки для выпаривания применяют для нагревания и выпаривания различных растворов. Этот процесс можно вести на открытом пламени, но равномерные выпаривание растворов обычно происходит на асбестовой сетке или водяной бане. Тигли применяют для прокаливания различных веществ и для сжигания органических соединений. Из фарфоровой посуды в химической лаборатории часто применяют стаканы, ложки, шпатели и ступки. Нагревательные бани. Прямое нагревание на пламени газовой горелки или на электрической плитке может приводить к местным перегревам. Этого можно избежать при использовании нагревательных бань. В качестве теплопроводящей среды в банях используют воду, воздух, песок и масло. Простейшую воздушную баню можно получить, если между пламенем и нагреваемой колбой поместить асбестовую сетку. Песчаные бани обладают очень большой тепловой инерцией, что затрудняет регуляцию температуры. Наиболее удобные масляные и водяные бани, так как они обеспечивают равномерное нагревание колбы и благодаря незначительной тепловой инерции позволяют точно регулировать температуру реакционной смеси. Выбор бани определяется свойствами нагреваемого вещества или смеси, а так же температурой, необходимой для их нагревания. Водяные бани применяют при нагревании веществ до 1000С, масляные – до 1500С, электрические воздушные – до 2500С, песчаные – выше 4000С. Необходимо помнить. Что водяные бани нельзя использовать при работе с металлическими натрием и калием. 18 2.2. Правила сборки установок для выполнения органических синтезов Выбор установки (прибора) для синтеза определяется, в первую очередь, задачей, стоящей перед экспериментатором, условиями проведения реакции, а также свойствами исходных и конечных продуктов. Сборка установки должна проводиться с большой тщательностью и аккуратностью, так как это является непременным условием успешной и безопасной работы. Собранные установки должны быть не только грамотными конструкционно, но и иметь привлекательный вид. Общие правила сборки приборов. Отдельные части установки необходимо соединять друг с другом осторожно, подбирая пробки, трубки и другие детали еще до закрепления прибора на штативе. Если прибор собирают на шлифах, то их следует предварительно смазать. Посуду подбирают такого размера, чтобы реагирующие вещества занимали не более половины объема (или не более 2/3 объема). Если реакционная смесь будет нагреваться, то обязательно применяют круглодонную колбу соответствующего размера. После того как собраны отдельные части установки, их закрепляют в лапках штатива. Установку всегда собирают, начиная с ее предполагаемого «верха» или с основного блока. Например, при сборке установки для простой перегонки следует вначале укрепить на штативе колбу Вюрца, затем к ней присоединить нисходящий холодильник, потом аллонж и, наконец, подвести под него приемник. Вся установка должна быть собрана в одной плоскости или по одной линии (за исключением некоторых случаев), без перекосов или напряжения стеклянных частей прибора. Это особенно важно при работе со стандартными шлифами, когда они должны присоединяться друг к другу без особых усилий со стороны экспериментатора. В то же время нужно следить, чтобы при соединении отдельных частей прибора выполнялись условия герметичности. Если стеклянные части установки достаточно тяжелые (например, колба с обратным холодильником, мешалкой, капельной воронкой, термометром и т. д.), то крепить их к штативу следует несколькими лапками. При этом дефлегматоры, мешалки, обратные холодильники крепят строго вертикально, а нисходящие холодильники — наклонно, чтобы жидкость стекала в приемник, не попадая на пробки. Если установка предназначена для работы под атмосферным давлением, то необходимо, чтобы она свободно сообщалась с атмосферой во избежание повышения давления в системе. Для защиты реагирующих веществ от действия влаги воздуха (если это нужно) используют хлоркальциевые трубки. Приступая к работе, следует еще раз внимательно осмотреть прибор и убедиться в правильности его сборки. 19 3. ПРАКТИЧЕСКАЯ ЧАСТЬ 3.1. Изучение состава органических соединений, их очистка и определение физических констант Лабораторная работа №1 «Методы очистки и выделения органических веществ» Полученные при синтезе вещества, как правило, содержат некоторое количество примесей (исходные вещества, не вступившие в реакцию, побочные продукты, растворители и др.). Чтобы избавиться от них, применяют различные методы очистки и выделения органических веществ. Эти методы довольно разнообразны и зависят, в основном, от агрегатного состояния соединения Методы очистки и выделения химических соединений разнообразны и зависят от их свойств и агрегатного состояния. Для твердых веществ наиболее важным методом очистки является кристаллизация. Различают три вида кристаллизации: • Из расплавов – зонная плавка • Из растворов – перекристаллизация • Из парообразного состояния – возгонка (сублимация) Метод перекристаллизации основан на различной растворимости вещества и сопутствующих ему примесей в данном растворителе при определенной температуре. Загрязненное вещество растворяют при нагревании в подходящем растворителе, а затем горячий раствор отфильтровывают от не растворившихся примесей и дают охладиться. Выпавший осадок отфильтровывают и сушат. Возгонка (сублимация) – это переход вещества, нагретого ниже его температуры плавления, из кристаллического состояния в парообразное, минуя жидкое состояние. При охлаждении пары вновь переходят в твердое состояние. Таким образом, возгонка состоит из двух стадий, одна из которых – испарение твердого вещества, а вторая – конденсация образовавшихся паров в твердое вещество. Для очистки органических соединений возгонка удобна в том случае, когда возгоняется лишь основной продукт, а примеси не испаряются. Также для очистки твердых веществ можно использовать экстракцию, но чаще всего экстрагированию подвергают водные растворы. Экстракция, или извлечение, основана на различной растворимости веществ в двух несмешивающихся жидкостях. Другими словами, экстракцией называется перевод вещества из одной фазы, в которой оно растворено, в другую жидкую фазу. 20 Рис. 3.1. Делительные воронки. а – цилиндрическая; б – шарообразная; в, г – грушевидные. Экстрагирование растворов проводят с помощью делительной воронки (рис.3.1.), в которую помещают две несмешивающихся жидкости, одна из которой содержит извлекаемое или разделяемые вещества (экстрагируемое вещество). Изза различной растворимости разделяемых веществ, одно вещество переходит в новую фазу (экстрагент), а другое (примеси) – нет. Для извлечения нужного продукта из смеси двух веществ иногда достаточно обработать тщательно измельченную смесь растворителем при комнатной температуре или при нагревании с обратным холодильником. Оставшийся осадок отфильтровывают и из полученного раствора извлекают искомое вещество. Для разделения жидких или летучих веществ используют метод перегонки. Перегонка — это процесс разделения смеси жидкостей на компоненты, основанный на разнице температур их кипения. Этот метод заключается в нагревании жидкости до кипения с последующей конденсацией паров в холодильнике. При разгонке смеси жидкостей парообразная фаза, образующаяся над жидкостью, содержит большее количество низкокипящих компонентов, чем жидкая фаза, поэтому при конденсации пара в приемнике получают жидкость, обогащенную низкокипящей фракцией. Смеси, при перегонке которых состав пара не отличается от состава жидкости (эти вещества кипят при одной температуре), называются нераздельнокипящими или азеотропными. Способы перегонки разделяются на две группы: простая перегонка и ректификация. По условиям проведения различают три вида перегонки: при атмосферном давлении, при уменьшенном давлении (перегонка в вакууме) и с водяным паром. При простой перегонке (рис. 3.2.) пары кипящей жидкости непосредственно из перегонной колбы поступают в холодильник. Таким образом, разделение смеси жидкостей может происходить лишь на стадии испарения. Простая перегонка применяется в том случае, когда температуры кипения веществ, входящие в состав перегоняемой смеси, значительно отличается друг от друга (не менее 800С). 21 Рис. 3.2. Прибор для простой перегонки: 1 — колба Вюрца; 2 — термометр; 3 — нисходящий холодильник Либиха; 4 — аллонж; 5 — приемная колба Перегонку в вакууме (рис.3.3.) проводят в том случае, если вещество разлагается до достижения температуры кипения, или имеет очень высокую температуру кипения. Рис. 3.3. Прибор для перегонки при пониженном давлении: 1 — колба Кляйзена или круглодонная колба с насадкой Кляйзена; 2 — капилляр, соединенный с резиновым шлангом с зажимом; 3 — термометр; 4 — холодильник; 5 — аллонж; 6 — приемная колба; 7 — предохранительная склянка; 8 — манометр Перегонку с водяным паром используют при очистке веществ, загрязненных большим количеством смолистых примесей, а также для соединений, которые кипят при высокой температуре и поэтому не исключена возможность их разложения. Эти вещества должны быть мало растворимы в воде и должны обладать значительной упругостью пара при температуре кипения воды. Таким методом можно перегнать высококипящий компонент, как жидкий, так и твердый, при атмосферном давлении и температуре около 100 °С. Перегонкой с водяным паром разделяют смеси веществ, из которых только одно способно отгоняться с паром. Прибор для перегонки с водяным паром изображен на рис.3.4. 22 Сущность перегонки с водяным паром заключается в том, что высококипящие, не смешивающиеся или мало смешивающиеся, т.е. малорастворимые в воде вещества улетучиваются при пропускании в них водяного пара; затем они вместе с паром конденсируются. Рис. 3.4. Прибор для перегонки с водяным паром: 1— парообразователь; 2 — тройник с зажимом; 3 — перегонная колба; 4 — холодильник; 5 — аллонж; 6 — приемная колба Фракционной (дробной) перегонкой разделяют смеси жидкостей, которые отличаются температурой кипения на 50—80 0С и не образуют азеотропные смеси. Перегонку осуществляют в приборе (рис. 3.5.), в состав которого входит дефлегматор. В дефлегматоре часть паров конденсируется вследствие охлаждения воздухом. Конденсирующиеся пары, обогащенные высококипящими компонентами смеси, стекают в перегонную колбу. Пары, проходящие через дефлегматор, обогащены летучими фракциями смеси, которые конденсируются в холодильнике и стекают в приемную колбу. Рис. 3.5. Прибор для фракционной перегонки: 1 — перегонная колба; 2 — дефлегматор; 3 — термометр; 4 — холодильник; 5 — аллонж; 6 — приемная колба 23 Техника перегонки при атмосферном давлении. Для такой перегонки собирают прибор как показано на рис. 10. Перегонную колбу 3 с помощью воронки заполняют не более чем на две трети перегоняемой жидкостью. Перед заполнением прибора измеряют объем или вес жидкости. Перегонный прибор собирают из сухих чистых деталей и закрепляют на штативах. Включают воду для охлаждения. В качестве нагревателя используют баню (водяная, масляная) или колбонагреватель. Контролируя температуру бани с помощью закрепленного на штативе термометра 4, устанавливают такой нагрев, который обеспечивает равномерное, медленное кипение содержимого колбы. В приемник должно падать не более двух капель чистого и прозрачного дистиллята в секунду. Только при таких условиях термометр в колбе показывает температуру, соответствующую точке равновесия между паром и жидкостью; при слишком быстрой перегонке пары легко перегреваются. Температуру перегонки записывают в журнал. Перегонку нельзя продолжать досуха! По окончании перегонки определяют объем или вес дистиллята, а также остатка в перегонной колбе. Ректификация – это способ разделения или очистки жидкостей с достаточно близкими температурами кипения путем перегонки с применением специальных колонок. Фильтрование — процесс отделения твердых компонентов смеси, находящихся в осадке, от маточных растворов (жидких компонентов) посредством пористой перегородки — фильтра. В качестве фильтра обычно используют фильтровальную бумагу, которая может быть различной пористости. Фильтрами могут служить также различные ткани, пористое стекло, асбест, обычная и стеклянная вата и др. При этом необходимо помнить, что фильтрующие материалы не должны взаимодействовать ни с растворителем, ни с отделяемым осадком. Фильтрование можно проводить различными способами. Это определяется как характером растворителя, так и свойствами отделяемого вещества при фильтровании. Обычно пользуются двумя способами фильтрования: при нормальном и пониженном давлении. Фильтрование при нормальном давлении — наиболее простой и часто применяемый в лабораторной практике способ, не требующий сложных приспособлений. Для этого необходимы стеклянная воронка и фильтр. Бумажные фильтры могут быть двух видов: простые и складчатые. Последние применяются чаще, так как имеют большую фильтрующую поверхность, и это намного ускоряет процесс фильтрования. Простой фильтр можно изготовить так: квадратный кусок фильтровальной бумаги складывают вчетверо и обрезают по окружности таким образом, чтобы готовый фильтр имел вид конуса. При этом размер конуса должен соответствовать размеру фильтровальной воронки. Складчатый фильтр имеет более сложную форму. Для его изготовления вначале поступают так же, как и для получения простого фильтра. Затем 24 четвертушки бумаги разгибают и на фильтре, сложенном вдвое, Делают сгибы «гармошкой», как показано на рис. 3.6. Рис. 3.6. Последовательность действий при изготовлении простого фильтра (а) и складчатого (б). Фильтр (простой или складчатый) вставляют в воронку, укрепленную в кольце, которое присоединено к штативу зажимом. Под воронку ставят стакан, и прибор для фильтрования холодных растворов готов (рис.3.7.) Для фильтрования горячих растворов применяют специальную воронку, обогреваемую электрической спиралью или горячей водой (рис. 3.8.). При фильтровании веществ, имеющих низкую температуру плавления, используют воронки с охлаждением Фильтрование при пониженном давлении (отсасывание) применяют для ускорения процесса фильтрования. Основным прибором служат фарфоровая воронка Бюхнера и толстостенная колба Бунзена, соединенная через предохранительную склянку с Рис. 3.7. водоструйным насосом (или лабораторным насосом Установка для Комовского) (рис.3.9.). Для предотвращения последствий фильтрования возможного разрыва колбы Бунзена в ходе фильтрования ее через необходимо обвернуть полотенцем. Чтобы фильтруемая стеклянную жидкость не попала между воронкой и фильтром, на воронку. дырчатое дно воронки Бюхнера кладут бумажный фильтр, смоченный дистиллированной водой. Рис. 3.8. Приспособления для обогрева фильтровальной воронки горячей водой (а) и электрической спиралью (б): 1 — воронка для фильтрования; 2 — специальная воронка с двойными стенками («рубашкой»), между которыми находится вода; 3 — «отросток» для обогрева воды; 4 — специальная электрическая воронка для горячего фильтрования (электрическая печь); 5 — электрическая спираль Диаметр фильтра должен быть немного меньше внутреннего диаметра воронки, но таким, чтобы фильтр полностью закрывал все отверстия. Водоструйный насос при нормальном напоре воды может создавать вакуум в 1—2 кПа (8—15 мм рт. ст.). Чтобы в колбу Бунзена случайно не попала вода от водоструйного насоса, между ним и колбой устанавливают предохранительную 25 склянку (некоторые конструкции водоструйных насосов позволяют обходиться без предохранительных склянок). Фильтруемую жидкость необходимо доливать равномерно, чтобы осадок все время находился под слоем жидкости. Нужно стараться, чтобы осадок не попал между фильтром и дном воронки Бюхнера, иначе некоторая часть осадка может попасть в колбу Бунзена. После окончания фильтрации осадок на фильтре отжимают стеклянной пробкой. Если растворитель или осадок легко окисляются воздухом, или гигроскопичны, или взаимодействуют с С02 воздуха, то процесс фильтрования следует проводить в атмосфере инертного газа. Рис. 3.10. Воронка с пуговкой для отсасывания малых количеств Рис. 3.9. Воронка Бюхнера (1); дырчатое дно воронки (вид сверху) (2); колба Бунзена (3). Для отфильтровывания очень малых количеств кристаллов (порядка 0,1 г и менее) пользуются обычной маленькой стеклянной воронкой, в которую вставляют маленькую стеклянную палочку с расплюснутым концом – «пуговку». Для приготовления такой пуговки конец тонкой стеклянной палочки нагревают до размягчения и затем прижимают ко дну ступки. Стеклянная палочка должна быть настолько тонкой и длинной, чтобы она свободно входила в трубку воронки, и конец ее выдавался немного снизу. На пуговку кладут кружок фильтровальной бумаги немного большего размера так, чтобы он плотно прилегал к стенкам воронки, для чего его можно смочить водой (рис.3.10.). Практическая часть Опыт №1. Перекристаллизация бензойной кислоты из воды Реактивы: бензойная кислота (или реактив, предложенный преподавателем). Оборудование: стакан, колба, воронка, фильтр. В стакане вместимостью 50 мл растворяют при нагревании 0,1 г бензойной кислоты в 5 мл дистиллированной воды. Горячий раствор быстро отфильтровывают в колбочку через складчатый фильтр. Фильтрат охлаждают. Выпавшие кристаллы отфильтровывают и высушивают. 26 Опыт №2. Возгонка нафталина Реактивы: нафталин (или реактив, предложенный преподавателем). Оборудование: песчаная баня, фарфоровая чашка, термометр, воронка, фильтр. Технический нафталин помещают в небольшую фарфоровую чашку, накрывают ее листом фильтровальной бумаги с мелкими проколами (20-30 отверстий) и плотно прижимают фильтровальную бумагу опрокинутой стеклянной воронкой, отверстие которой закрыто ватным тампоном (рис.3.11.). Чашку с веществом нагревают на песчаной бане, снабженной термометром, ртутный шарик которого должен находиться на уровне дна фарфоровой чашки. Нагревают чашку с веществом до температуры ниже Тпл нафталина на 10-200С. После того, как на стенках воронки появится налет вещества, прибор оставляют при данной температуре (отключить нагревательный прибор) на 1-1,5 часа. Рис 3.11. Прибор для возгонки. 1 – песчаная баня; 2 – фарфоровая чашка; 3 – воронка; 4 – термометр; 5 – возгоняемое вещество; 6 – ватный тампон. За это время, возгоняющееся вещество будет проникать через отверстие в фильтровальной бумаге, и оседать на внутренних стенках воронки в виде крупных кристаллов. Определяют температуры плавления бензойной кислоты и нафталина до и после их очистки. Взвешивают очищенные вещества и определяют выход чистого вещества в процентах. Опыт №3. Экстракция анилина. Реактивы: анилин, четыреххлористый углерод. Оборудование: делительная воронка, стаканы. В делительную воронку вместимостью 100 мл наливают 30 мл смеси анилина и воды (1:2) и добавляют 10 мл четыреххлористого углерода CCl4. Четыреххлористый углерод – один из немногих органических растворителей, имеющий большую плотность, чем вода (p=1,59 г/мл), поэтому он сразу опускается на дно воронки. Закрывают воронку стеклянной пробкой и несколько раз энергично встряхивают, чтобы слои перемешались. Затем для выравнивания давления воронку переворачивают краном вверх и приоткрыванием его выпускают образовавшиеся пары. Встряхивание с последующим приоткрыванием 27 крана повторяют несколько раз. В результате анилин переходит в слой четыреххлористого углерода, т.е. экстрагируется им. После отстаивания и четкого расслоения разделяют образовавшиеся слои. В каком слое находится анилин: верхнем или нижнем? Полученный экстракт высушивают от воды путем добавления хлористого кальция. Для выделения анилина из экстракта используют перегонку (опыт №4). Опыт №4. Разделение смеси анилина и четыреххлористого углерода простой перегонкой Опыт выполняется преподавателем! Реактивы: раствор анилина в четыреххлористом углероде Оборудование: прибор для перегонки, колба, водяная баня, электрическая плитка. Собирают прибор для перегонки (рис. 3.12.). В круглодонную колбу вместимостью 50 мл через воронку наливают раствор анилина в четыреххлористом углероде, полученный в опыте №3. Помещают в колбу несколько «кипелок» для равномерного кипения. Закрывают колбу пробкой с вставленным в нее термометром так, чтобы ртутный шарик термометра находился на 0,5 см ниже отводящей трубки. Пускают воду в обратный холодильник, под колбу подводят водяную баню и начинают нагревать так, чтобы перегонка проходила с постоянной скоростью. Рис. 3.12. Прибор для перегонки. На асбестовое сито 2, закрепленное в штативе 1, ставят колбу 3 с пробками, плотно закрепленными в горловинах. В верхней пробке делают отверстие для термометра 4, а в боковой - для трубки 6 с краном 5, которая соединяет колбу с холодильником Либиха 7. Колбу 3 выбирают такого размера, чтобы подвергаемая перегонке жидкость занимала 2/3 ее объема. Аллонж 8 опускают в колбу-приемник 10. Шланг 9 подсоединяют к крану с водой. Температура кипения CCl4 76,50С. До температуры 800С перегоняется CCl4 (фракция 1). Затем убирают водяную баню, помещают под колбу асбестовую сетку и продолжают нагрев. Во время быстрого повышения температуры собирают в другую колбу промежуточную фракцию 2, а затем в интервале 1801900С перегоняют анилин (фракция 3). Температура кипения анилина 1840С. Измеряют объемы фракций цилиндром. 28 Вопросы коллоквиума: 1. Перечислите методы очистки веществ и дайте им краткую характеристику. 2. Какие вещества можно очищать возгонкой? При какой температуре проводят возгонку? 3. На каком свойстве соединений основана перегонка? Лабораторная работа №2 «Определение физических констант органических соединений» К простейшим физическим константам веществ относятся температуры плавления и кипения, плотность вещества и показатель преломления. Температура плавления вещества – это температура, при которой вещество из твердого (кристаллического) состояния переходит в жидкое. Началом плавления считается момент размягчения вещества и переход его в жидкое состояние, а концом – образование прозрачной жидкости. Если вещество химически чистое, то его температура плавления колеблется в интервале 0,5-1,00С. Четкая температура плавления вещества является признаком его чистоты. Определение температуры плавления проводят обычно в приборе, состоящем из круглодонной (или плоскодонной) колбы с длинным горлом и широкой пробирки, вставленной в это горло через корковую пробку. В пробирку помещают термометр, на шарик которого надевают резиновое кольцо для закрепления капилляра (рис.3.13а). В капилляр помещают вещество, температуру которого определяют. Колбу заполняют глицерином и греют. Температуру плавления определяют визуально. а) б) в) г) Рис. 3.13. а) прибор Тиле для определения температуры плавления с естественной циркуляцией жидкости (часть между точками 1 и 2 обматывают тонким асбестовым картоном); б), в) упрощенные приборы для определения температуры плавления; г) Прибор для определения температуры кипения (по Сиволобову). 29 Жидкость кипит, когда давление ее паров равно атмосферному давлению, т.е. температура кипения жидкости зависит от давления. Обычно температуру кипения определяют по методу Сиволобова. В стеклянную трубочку помещают несколько капель исследуемой жидкости. Туда же погружают тонкий капилляр, запаянный с верхнего конца (рис. 3.13б). Трубку с жидкостью и капилляром прикрепляют к термометру. Когда при медленном нагревании из тонкого капилляра начинают выделяться пузырьки воздуха, температура кипения жидкости считается достигнутой. В настоящее время для определения температуры плавления веществ используют специальные аппараты: блок для определения температуры плавления (рис.3.14.,3.15.) и столик Кофлера. Рис.3.14. Блок для определения температуры плавления: 1 – вольтметр; 2 – лампа для освещения; 3 - увеличительное стекло; 4 – трубка с термометром и нагревательной спиралью. Рис.3.15. Прибор для плавления с воздушным электрообогревом: определения температуры термостатированием и 1 – подставка из изолирующего материала; 2 – кожух; 3 – рубашка с электрообогревом; 4 – капилляр с веществом; 5 – термометр. Плотность вещества – это одна из его основных физических характеристик, численно равная массе единицы объема тела: ρ = m/ V где ρ – плотность вещества, m – масса, V – объем. Плотность вещества обычно уменьшается с ростом температуры (из-за теплового расширения тел) и увеличивается с повышением давления. Единицей плотности в международной системе единиц (СИ) служит кг/м3. На практике часто применяют г/см3, г/л. В ряде случаев предпочитают пользоваться относительной плотностью d, представляющей собой отношение плотности данного вещества к плотности другого при определенных условиях. 30 Относительная плотность выражается отвлеченным числом. Обычно ее определяют по отношению к плотности дистиллированной воды при 4 0С. Относительную плотность жидкостей можно определять при помощи ареометров, пикнометров и специальных весов. Ареометры применяют для быстрого определения относительной плотности жидкости (рис.3.16.,3.17.). Рис.3.16. Определение плотности вещества при помощи ареометра: 1 – шкала плотности; 2 – связующая масса; 3 – балласт (дробь); 4 – цилиндр с исследуемой жидкостью. Рис.3.17. Ареометры: а) Постоянной массы (денсиметр) б) постоянного объема; 1 – шкала плотности; 2 – балласт (дробь); 3 – связующая масса; 4 – встроенный термометр; 5 – тарелка для гирь; 6 – метка. Шкалы ареометров градуируются в единицах плотности или при определении концентрации растворов в процентах (по объему или по массе). В нижней части ареометра имеется шарик, заполненный дробью (реже – ртутью). Для определения относительной плотности жидкость наливают в широкий стеклянный цилиндр и осторожно погружают в нее ареометр. Прибор не должен касаться стенок сосуда. Чем больше относительная плотность жидкости, тем меньше в нее погружен ареометр. Значение относительной плотности показывает деление на шкале, против которого установился уровень жидкости. Для более точного определения относительной плотности пользуются пикнометрами вместимостью 1 -2 мл (рис. 3.18.,3.19.). 31 Для этого пикнометр предварительно моют ацетоном, спиртом или эфиром, высушивают в сушильном шкафу, а затем взвешивают на аналитических весах при комнатной температуре пустым, потом с водой, а затем с исследуемой жидкостью. Находят массу равных объемов исследуемой жидкости и воды. Относительную плотность жидкости определяют по формуле: d = (m – mп) / B где m – масса пикнометра с исследуемой жидкостью; mп – масса пустого пикнометра; B – водное число пикнометра (численно равное отношению массы воды в объеме пикнометра при 200С к массе воды в объеме пикнометра при 40С). Рис. 3.18. Пикнометры для определения относительной плотности жидкостей: а, б – Рейшауэра; в – Ренье; г – с капиллярной пробкой; д – Менделеева; е – Оствальда. Рис.3.19. Определение плотности жидкости с помощью пикнометра. Показатель преломления n – представляет собой отношение синуса угла падения света на поверхность раздела двух сред к синусу угла отражения света: n = sin α/ sin β. Эта величина используется для идентификации жидких веществ и характеристики их чистоты. Показатель преломления зависит от температуры и резко меняется с изменением длины волны света, поэтому измерения проводят при постоянной температуре и монохроматическом свете. Обычно опыт ведет при 200 С и при длине волны, соответствующей длине волны желтой линии натрия D (λ = 589,3 нм). Так, символ nD20 означает, что показатель преломления был определен для линии D при 200С. Для большинства жидких органических веществ показатель преломления находится в пределах от 1,3 до 1,8. 32 Рис.3.20. Рефрактометр ИРФ-22: 1- верхнее полушарие измерительной головки; 2, 9 – маховики; 3 – нижнее полушарие измерительной головки; 4 – осветительное зеркало; 5 – зеркало для освещения шкалы; 6 – окошко; 7 – зрительная труба; 8 – окуляр; 10 – термометр; 11 – ящик; 12 – гнездо для ключа. Работа на рефрактометре проводится в следующем порядке: открывают верхнее полушарие измерительной головки (рис. 3.20.) и протирают смоченной эфиром ватой гипотенузные плоскости осветительной (А, рис.3.21.) и измерительной (Б, рис.3.21.) призм и дают эфиру испариться (сильно смачивать эфиром полушария 1 и 3 не следует). Поворотом маховика 2 надо привести измерительную головку в такое положение, чтобы плоскость нижнего полушария 3 и гипотенузная плоскость измерительной призмы (Б) приняли горизонтальное положение. Затем на плоскость измерительной призмы наносят посредством стеклянной палочки или капилляра несколько капель исследуемого вещества (палочка не должна касаться призмы) и осторожно закрывают верхнее полушарие 1 измерительной головки. Осветительное зеркало 4 устанавливают так, чтобы свет от источника поступал к осветительной призме и освещал поле зрения. Рис.3.21. Измерительная головка рефрактометра ИРФ-22 (в разрезе): А – осветительная призма; Б – измерительная призма; 1 и 3 – полушария измерительной головки; 7 – зрительная труба; 8 – окуляр. 33 Зеркало 5 для освещения шкалы ставят в такое положение, чтобы свет поступал в окошко 6, освещающее шкалу прибора. Глядя в зрительную трубу 7, фокусируют окуляр 8 так, чтобы шкала прибора была отчетливо видна (рис.3.22а). Вращая маховик 2и наблюдая в окуляр зрительной трубы 8, находят границу раздела света и тени (рис. 3.22б). Если она размыта и окрашена в желто-красный или сине-зеленый цвет, надо при помощи маховика 9, вращая его в любом направлении, добиться, по возможности, более полного обесцвечивания Рис.3.22. Снятие отсчета на этой границы (при этом она становится более рефрактометре ИРФ-22: четкой, хотя в некоторых случаях может а- шкала; б- вид перекрестия понадобиться дополнительная наводка на с границей света и тени. резкость). Показатель преломления зависит от температуры, поэтому при измерении она должна быть постоянной (200С). Показатель преломления уменьшается на 3-8 единиц в четвертом знаке после запятой при повышении температуры на 10С. Для наблюдения за постоянством температуры около измерительной головки вмонтирован термометр 9. Когда в измерительной головке установится постоянная температура, при помощи маховика 2 необходимо точно совместить границу раздела света и тени с перекрестием сетки (рис. 30б) и снять отчет по шкале показателя преломления. Показатель преломления измеряется с точностью до четвертого знака после запятой. Первые три цифры (1,45…) – это ближайшие, находящиеся ниже горизонтального штриха сетки цифры шкалы. Третий знак после запятой соответствует числу целых мелких делений, расположенных между ближайшим нижним оцифрованным делением и горизонтальным штрихом сетки. Четвертый знак после запятой получается визуально интерполяцией в пределах того деления, в котором находится горизонтальный штрих сетки. Так, например, в случае, изображенном на рис. 24а, показатель преломления равен 1,4593. Практическая часть Опыт №1. Определение температуры плавления. Реактивы: реактив по указанию преподавателя. Оборудование: прибор для определения температуры плавления вещества, капилляры. По указанию преподавателя определяют температуру плавления вещества. Для этого в капилляр помещают вещество, температуру которого нужно определить. Затем аккуратно закрепляют капилляр с веществом на шарик термометра с помощью резинки. Помещают термометр в собранный заранее прибор (рис.17а) 34 для определения температуры плавления. Колбу закрепляют в лапке штатива на расстоянии 2 см от поверхности плитки и нагревают. При переходе вещества в жидкое состояние отмечают температуру плавления вещества. Опыт №2. Определение температуры кипения. Реактивы: реактив по указанию преподавателя. Оборудование: прибор для определения температуры кипения вещества, капилляры. По указанию преподавателя определяют температуру кипения вещества. Для этого в стеклянную трубочку помещают несколько капель исследуемой жидкости. Туда же погружают тонкий капилляр, запаянный с верхнего конца (рис. 17б). Трубку с жидкостью и капилляром прикрепляют к термометру. Термометр закрепляют в лапке штатива на расстоянии 2 см от поверхности плитки и нагревают. Отмечают температуру, при которой начнут выделяться пузырьки воздуха. Опыт №3. Определение показателя преломления одной из органических жидкостей. Реактивы: реактив по указанию преподавателя. Оборудование: рефрактометр. На имеющемся в лаборатории рефрактометре определяют показатели преломления некоторых жидкостей по указанию преподавателя (например, этилового спирта, толуола, хлороформа, тетрахлорида углерода, анилина и т.д.) и сравнивают их с литературными данными. Сравнивают полученные физические константы со справочными данными и делают вывод о точности данных методов определения температур плавлении, кипения и показателя преломления. Вопросы коллоквиума: 1. Что такое температура плавления, температура кипения? 2. Достаточно ли определения температуры кипения или плавления для установления чистоты вещества? 3. Назовите методы определения плотности веществ. 4. Что такое показатель преломления? Для чего и как используется эта величина? 5. Почему приборы, предназначенные для определения показателя преломления, называются рефрактометрами? 6. От чего зависит значение показателя преломления? 35 Анализ органических соединений Принадлежность органических веществ к определенным классам устанавливается функциональным анализом, их чистота – хроматографией, строение – всеми существующими физико-химическими методами исследования с учетом способа получения, а в случае необходимости и результатов встречного синтеза. Качественный элементный анализ позволяет определить, из атомов каких элементов построены молекулы органического вещества; количественный элементный анализ устанавливает состав соединения и простейшую формулу. При выполнении элементного анализа органические вещества «минерализуют», т.е. разлагают таким образом, чтобы углерод превратился в СО2, водород – в Н2О, азот – в N2, NH3 или цианид - ионы CN- и т.п. Дальнейшее определение проводят обычными методами аналитической химии. В функциональном анализе применяются химические, физические и физикохимические методы. Для качественных проб на функциональные группы выбирают реакции, при которых происходит изменение окраски или разделение фаз (выпадение осадка, выделение газа). Реакций, характерных только для какой-нибудь одной функциональной группы, известно немного, и для того, чтобы установить, к какому классу соединений относится данное вещество, нужно проделать несколько качественных реакций. Лабораторная работа № 3 «Качественный элементный анализ» Практическая часть Опыт №1. Открытие углерода и водорода сожжением вещества с оксидом меди (П). Реактивы: порошок оксида меди (П), сахароза, безводный медный купорос, известковая вода. Оборудование: пробирки, пробка с газоотводной трубкой, вата, сухое горючее. Дня проведения эксперимента в пробирку «а» (рис. 31) насыпают черного порошка оксида меди (П) на высоту около 10 мм. Добавляют одну лопаточку сахарозы, тщательно перемешивают, энергично встряхивают пробирку. В верхнюю часть пробирки «а» вводят в виде пробки небольшой комочек ваты (рис. 3.23.). Насыпают на вату тонкий слой белого порошка - безводного медного купороса. Закрывают пробирку «а» пробкой с газоотводной трубкой. При этом конец трубки должен почти упираться в вату с CuSO4. Нижний конец трубки помещают в пробирку «б», предварительно наливают в нее около 1-2 мл известковой воды. Конец газоотводной трубки должен быть погружен в известковую воду. 36 Рис.3.23. Открытие углерода и водорода Нагревают пробирку «а» на пламени горелки. Если пробка плотно закрывает пробирку, то через несколько секунд из газоотводной трубки начнут выходить пузырьки газа. Как только известковая вода помутнеет, вследствие выделения белого осадка СаСОз, пробирку «б» убирают. Пробирку «а» продолжают нагревать по всей длине до ваты, пока пары воды не достигнут белого порошка -обезвоженного медного купороса, находящегося на ватном тампоне, и не вызовут посинения его вследствие образования кристаллогидрата CuSО4 · 5Н2О. Если слишком большой кусок ваты, то она поглотит выделившиеся пары и посинения может не произойти. Опыт №2. Открытие азота сплавлением вещества с металлическим натрием. Реактивы: мочевина, металлический натрий, этиловый спирт, спиртовой раствор фенолфталеина, раствор железного купороса FeS04 , 2 н раствора НСl. Оборудование: сухое горючее, пробирки. Для открытия азота 5 - 10 мг испытуемого вещества, например, несколько кристаллов мочевины, помещают в сухую пробирку. Прибавляют к мочевине небольшой кусочек металлического натрия. Нагревают осторожно смесь в пламени горелки, пробирку вносят и выносят из пламени, не нагревая ее постоянно! Когда мочевина расплавится, следят, чтобы она смешалась с натрием (для успеха опыта необходимо чтобы натрий плавился вместе с веществом, а не отдельно от него - не на стенке пробирки!). При этом иногда наблюдается небольшая вспышка. Нагревают, пока получится однородный сплав. Когда пробирка остынет, добавляют в нее 5 капель этилового спирта для устранения остатков металлического натрия, который реагирует со спиртом не так бурно, как с водой. При этом происходит образование алкоголята натрия с выделением водорода: 2С2Н5ОН +2Na →2C2H50Na + Н2 Убедившись, что остаток натрия прореагировал со спиртом (прекращается шипение от выделения пузырьков газа), добавляют в пробирку 5 капель воды и нагревают ее на пламени горенки, чтобы все растворилось. При этом цианид натрия переходит в раствор, а алкоголят натрия с водой образует едкую щелочь: C2H5ОNa + НОН → С2Н5ОН + NaОH Добавляют в пробирку 1 каплю спиртового раствора фенолфталеина. Появление малиново-красного окрашивания показывает, что в растворе образовалась щелочь. После этого внести в пробирку 1 каплю раствора железного купороса FeS04 обычно содержащего примесь соли оксида железа (III) Fe2(S04)3. В присутствии щелочи 37 немедленно образуется грязно - зеленый осадок гидроксида железа (II) в смеси с желтым осадком гидроксида железа (III). При наличии в растворе избытка цианида натрия гидроксид железа (II) образует комплексную желтую кровавую соль: Fe(OH)2 + 2NaCN → Fe(CN)2 + 2 NaOH Fe(CN)2 + 4NaCN → Na4[Fe(CN)6] Пипеткой наносят в центр фильтровальной бумажки каплю жидкости из пробирки. Как только капля впитается, на нее наносят 1 каплю 2 н раствора НСl. После подкисления грязно-зеленый или желтоватый осадок гидроксидов железа (II) и (Ш) растворяется и при наличии азота немедленно появляется синее пятно образовавшейся берлинской лазури: Fe(OH)3 + 3 НСl → FeСl3 + 3 Н2О 3 Na4[Fe(CN)6]+ 4FeСl3 → Fe4[Fe(CN)6]3 + 12 NaСl Опыт № 3. Открытие серы сплавлением органического вещества с металлическим натрием. Реактивы: тиомочевина или сульфаниловая кислота, металлический натрий, этиловый спирт, раствора ацетата свинца Рb(СН3СОО)2. Оборудование: сухое горючее, пробирки. Для открытия серы испытуемое вещество, например, тиомочевину или сульфаниловую кислоту, помещают в сухую пробирку. Достаточно взять всего несколько кристаллов вещества.(5 – 10мг). Добавляют к веществу кусочек металлического натрия (столбик длиной около I мм). Пробирку нагревают, следя за тем, чтобы натрий плавился не отдельно, а вместе с веществом, иначе, опыт не удастся. Наблюдаемая небольшая вспышка натрия не опасна (см. предыдущий опыт). При этом органическое вещество (тиомочевина) переходит в неорганическое соединение - сульфид натрия. Когда пробирка остынет, прибавляют в нее 5 капель этилового спирта для устранения остатков металлического натрия, который со спиртом образует, алкоголят натрия C2H5ОNa. После окончания реакции (прекращение выделения пузырьков газа водорода) добавляют для растворения сплава 5 капель воды и кипятят, чтобы ускорить растворение. Сульфид натрия при этом перейдет в раствор вместе с гидроксидом натрия, который, однако, не мешает дальнейшей реакции. Для открытия серы добавляют несколько капель раствора ацетата свинца Рb(СН3СОО)2. При этом выпадает темно-коричневый осадок сульфида свинца: Рb(СН3СОО)2.+ Na2S → PbS ↓ + 2 CH3COONa Это качественная реакция на ион двухвалентной серы S-2. Опыт № 4. Открытие хлора при действии водорода на органическое вещество. Реактивы: хлороформ CHСl3, этиловый концентрированная азотная кислота HNO3. Оборудование: сухое горючее, пробирки. 38 спирт, металлический натрий, Помещают в пробирку I каплю хлороформа CHСl3. Добавляют 5 капель этилового спирта и кусочек металлического натрия (столбик длиной 1 мм). При этом происходит следующая реакция: С2Н5ОН + Na → C2H5ОNa + Н2 Обращают внимание на выделяющийся водород. Его можно зажечь у отверстия пробирки, если предварительно закрыть это отверстие пальцем, чтобы, накопить водород, а потом поднести отверстие к пламени горелки. Водород в момент выделения отщепляет хлор от хлороформа и образует хлористый водород, реагирующий затем с образовавшимся алкоголятом натрия. CHCl3 + 3H2 → CH4 + 3HCl C2H5ONa + HCl → C2H5OH + NaCl После того, как прекращается выделение водорода, для растворения образующегося белого осадка, нерастворимого в этиловом спирте, добавляют 2-3 капли воды. При этом избыток алкоголята натрия реагирует с водой, образуя щелочь: C2H5ОNa + НОН → С2Н5ОН + NaOH В присутствии щелочи нельзя отрывать ион хлора, так как добавление раствора нитрата серебра немедленно дает коричневый осадок оксида серебра, маскирующего осадок хлорида серебра: AgNO3 + 2 NaOH → Ag20 + H20 + 2 NaN03 Поэтому добавляют к раствору сначала 2 - 3 капли концентрированной азотной кислоты HNO3 (в вытяжном шкафу) для нейтрализации щелочи, а затем уже 2 капли 0,1 н раствора AgN03. При наличии хлора немедленно выпадает белый творожистый осадок хлорида серебра, нерастворимый в HNO3: NaCl + AgNO3 → AgCl ↓+ NaNO3 Ни в коем случае не следует брать для реакции больше 1 капли хлороформа, так как это только вредит чувствительности реакции. Остаток не вступившего в реакцию хлороформа еще до прибавления нитрата серебра дает с водой прочную эмульсию в виде беловатой мутной жидкости, которая будет маскировать появление белой мути от хлорида серебра. Опыт № 5. Открытие хлора по зеленой окраске пламени (проба Бейльштейна). Реактивы: хлороформ CHСl3. Оборудование: сухое горючее, медная проволока. Берут медную проволоку длиной около 10 см, загнутую на конце петлей и вставленную другим концом в небольшую корковую пробку. Держа за пробку, прокаливают петлю в пламени горелки до исчезновения посторонней окраски пламени (признак загрязнения медной петли). 2Cu + O2 → 2 CuO Остывшую петлю, покрывшуюся черным налетом оксида меди (II), опускают в пробирку, на дно которой помещают испытуемое вещество, например хлороформ. Смоченную веществом петлю вновь вносят в пламя горелки. Немедленно появляется характерная ярко-зеленая окраска пламени вследствие образования летучего 39 соединения меди с хлором. Подобную же окраску пламени дают, помимо хлористых и другие галогенсодержащие органические соединения. 2CHCl3 + 5CuO → CuCl2 +4 CuCl + 2CO2 + H2O Для очистки проволоку можно смочить соляной кислотой и прокалить. В отчете пишут уравнения соответствующих реакций и делают вывод о наличии анализируемых элементов в веществах. Вопросы коллоквиума: 1. В какие неорганические соединения переводят углерод-, водород-, азот-, серо- и хлорсодержащие органические соединения для качественного определения соответствующих элементов? Почему именно в эти неорганические соединения? 2. Для чего при открытии таких элементов, как азот, сера, хлор, добавляют этиловый спирт и воду? 3. В чем смысл пробы Бейльштейна? Лабораторная работа № 4 «Функциональный анализ» Для того чтобы отличить ароматические углеводороды от алифатических, можно использовать некоторые цветные реакции, например реакцию ароматических углеводородов с хлороформом в присутствии хлорида алюминия. Эта реакция сопровождается образованием окрашенных продуктов. Так, при взаимодействии бензола с хлороформом в присутствии AlCl3 кроме основного продукта реакции – бесцветного трифенилметана, образуется также окрашенная соль трифенилкарбения: Окрашено Эту реакцию можно также использовать для обнаружения ароматических галогенпроизводных. Опыт. К 1-2 мл хлороформа прибавляют 2-3 капли бензола, тщательно перемешивают и пробирку слегка наклоняют, чтобы смочить стенки. Добавляют 0,5-0,6 г безводного хлорида алюминия таким образом, чтобы часть порошка попала на стенки пробирки. Обращают внимание на окраску порошка на стенке и на цвет раствора. В реакции с бензолом возникает красно-оранжевая окраска, с дифенилом – пурпурная, с нафталином – синяя, с антраценом – зеленая. 40 Для того чтобы различить первичные, вторичные и третичные спирты, используется различная подвижность оксогруппы в реакции спиртов с раствором хлорида цинка в концентрированной соляной кислоте: Третичные спирты взаимодействуют с этим реактивом с большей скоростью, давая нерастворимые галогеналкилы; первичные спирты реагируют только при продолжительном нагревании или стоянии, вторичные занимают промежуточное положение. Опыт. В три пробирки наливают свежеприготовленный раствор хлорида цинка в соляной кислоте и охлаждают. В каждую пробирку добавляют по 3-4 капли соответственно первичного, вторичного или третичного спиртов, энергично встряхивают и оставляют в стакане с водой при 25-300С. О начале реакции судят по помутнению раствора вследствие образования нерастворимого галогеналкила. Отмечают время помутнения раствора в каждой пробирке. Качественные реакции карбонильных соединений многочисленны и разнообразны, что объясняется склонностью карбонильных соединений вступать в различные реакции замещения и присоединения. Альдегиды жирного ряда восстанавливают двухвалентную медь в одновалентную. В качестве реактива, содержащего ионы Cu2+, применяется реактив Фелинга. Реактив Фелинга готовят перед употреблением, смешивая свежеприготовленный гидроксид меди (II), образующийся при взаимодействии гидроксида натрия с сульфатом меди(II), и раствор сегнетовой соли. При сливании растворов образуется гидроксид меди(II), который с сегнетовой солью дает комплексное соединение типа гликолята меди: Ароматические альдегиды эту реакцию не дают. 41 Опыт. Приготавливают в пробирке реактив Фелинга, сливая по 1 мл исходных растворов, и прибавляют 2 мл карбонильного соединения. Верхнюю часть содержимого пробирки нагревают и наблюдают появление желтого или красного осадки оксида меди (I). Практическая часть Студентам выдается набор, состоящий из 6 бесцветных и прозрачных жидкостей, среди которых находятся по одному представителю алканов, ароматических углеводородов, спиртов (первичных, вторичных и третичных) и альдегидов. Названия представителей указываются преподавателем. Задача студента, предварительно ознакомившись с основами функционального анализа, представленного во введении, составить план анализа, чтобы по его завершению можно было сделать вывод о нахождении того или иного соединения в пронумерованной пробирке. В отчете пишут наблюдаемые явления, протекающие реакции и ход мышления. Делают вывод о принадлежности жидкостей к тому или иному классу и обосновывают его. Лабораторная работа №5 «Тонкослойная хроматография» Хроматография. Одним из наиболее простых и эффективных методов изучения состава смеси органических соединений, а также установление степени чистоты является тонкослойная хроматография (ТСХ). Наиболее широко применяется адсорбционный вариант ТСХ. Процесс хроматографического разделения в этом варианте основан на различии в относительном сродстве компонентов анализируемой смеси к неподвижной фазе (сорбенту) и осуществляется в результате перемещения подвижной фазы (элюента) под действием капиллярных сил по слою сорбента, нанесенного на стеклянную или алюминиевую пластинку. Хроматографирование проводится следующим образом. На пластинке отмечается стартовая и финишная линия (1-1,5 см от края пластинки). На стартовую линию в виде небольших пятен с помощью капилляра (не более 2-3 мм в диаметре) наносят раствор анализируемой смеси. Затем пластинку помещают в закрытую камеру с элюентом. Элюент представляет собой растворитель или смесь растворителей в различном соотношении. В качестве хроматографических 42 камер используют как специальные камеры, так и различную химическую посуду: эксикаторы, стаканы, чашки Петри (рис.3.24.). б) a) Рис. 3.24. а) Эксикатор, оборудованный для тонкослойной хроматографии; б) использование стакана и чашки Петри для тонкослойной хроматографии. При погружении нижней части пластинки в элюент линия старта должна находиться выше уровня растворителя. Поднимаясь по пластинке снизу вверх, растворитель разделяет нанесенные исследуемые вещества, перемещая их в слое сорбента с различной скоростью в зависимости от природы и свойств вещества. В результате компоненты смеси остаются на различном расстоянии от стартовой линии. Хроматографирование заканчивают, когда граница движущегося элюента достигнет линии финиша. Затем пластинку достают из хроматографической камеры и высушивают на воздухе. Бесцветные соединения обнаруживают оптическим (ультрафиолет) или химическими методами. Последний метод заключается в обработке хроматограммы реагентами, которые взаимодействуют с анализируемыми веществами с образованием окрашенных пятен. Наиболее доступным и универсальным методом обнаружения является обработка парами иода. Для этого хроматограмму помещают на несколько минут в эксикатор, насыщенный парами иода. После проявления пятен рассчитывают коэффициент подвижности Rf , который представляет собой отношение расстояний от стартовой линии до центра пятна к расстоянию от стартовой до финишной линий (рис.3.25): Rf=Li/L Li – расстояние от линии старта до центра пятна вещества i (см), L – расстояние от линии старта до лини финиша (см). 43 Рис.3.25. Хроматограмма, полученная при разделении смеси трех компонентов методом тонкослойной хроматографии. Так же для идентификации веществ, входящие в состав анализируемой смеси, на стартовую линию дополнительно наносят растворы известных веществ – «свидетелей». После проявления пятен и вычисления Rf сравнивают характеристики «свидетеля» и анализируемого вещества. Практическая часть Опыт №1. Обнаружение аскорбиновой кислоты (витамин С) во фруктовых соках. Реактивы: сок апельсина (лимона, мандарина, рябины, граната и др.), элюент (этанол – гексан 3:1), 1%-ный раствор аскорбиновой кислоты. Оборудование: чашки Петри, стаканы, адсорбент Sorbfil, капилляры. На стартовую линию пластинки наносят пробы отфильтрованного сока апельсина (лимона, мандарина, рябины, граната и др.) и 1%-ного раствора аскорбиновой кислоты так, чтобы расстояние пятен от боковых краев и между собой было не менее 1 см. Когда пятна подсохнут, пластинку помещают в стакан, на дно которого наливают 2 мл элюента (этанол – гексан 3:1). Чтобы элюент не испарялся с поверхности пластинки, накрывают стакан чашкой Петри. После достижения элюента финишной линии, вынимают пластинку и высушивают ее на воздухе. Для обнаружения соединений помещают пластинку в эксикатор с парами иода. Отмечают проявившиеся пятна и определяют значение Rf аскорбиновой кислоты. Опыт №2. Обнаружение лимонной кислоты в лимоне. Реактивы: сок лимона, раствор лимонной кислоты, элюент (этанол – гексан 3:1). Оборудование: чашки Петри, стаканы, адсорбент Sorbfil, капилляры. Аналогично предыдущему опыту на пластинку наносят пробы сока лимона и раствора лимонной кислоты («свидетель»). Выполняют хроматографирование и обнаружение аналогично опыту №1. Определяют значение Rf лимонной кислоты. 44 Опыт №3. Обнаружение кофеина в чае и кофе. Реактивы: растворы чая, кофе и кофеина, элюент этанол. Оборудование: чашки Петри, стаканы, адсорбент Sorbfil, капилляры. На линию старта пластинки наносят капли водного раствора чая, кофе и кофеина («свидетель»). Пластинку помещают в хроматографическую систему с этанолом в качестве элюента. Детектирование кофеина проводят парами иода. Определяют величину Rf кофеина. Опыт №4. Выделение кофеина и качественная реакция на него. Реактивы: сухой чай, 30%-ный водный раствор пероксида водорода, концентрированный раствор аммиака, 10%-ный раствор соляной кислоты. Оборудование: фарфоровая чашка, воронка, вата, асбестовая сетка, сухое горючее, предметное стекло. Кофеин можно получить из листьев чая. Для этого в фарфоровую чашку насыпают около 0,5 – 1 г сухого чая, накрывают ее воронкой с заткнутым ватным тампоном отверстием и нагревают на асбестовой сетке около 10 мин. Сначала на внутренней части воронки конденсируются капельки воды, а затем начинает возгоняться кофеин, белые тонкие кристаллы которого осаждаются на холодных стенках воронки. Нагревание прекращают и после полного охлаждения фарфоровой чашки кристаллы кофеина счищают со стенок воронки и растворяют в 1 мл воды. Для проверки наличия кофеина 1 каплю полученного раствора наносят на предметное стекло, добавляют 1 каплю 30%-ного водного раствора пероксида водорода и 1 каплю 10%-ной соляной кислоты. Смесь осторожно выпаривают досуха над пламенем сухого горючего. Стекло охлаждают и добавляют 1 каплю концентрированного раствора аммиака, а затем стекло вновь нагревают до полного испарения воды. Пурпурно красный цвет пятна указывает на наличие кофеина. В отчете делают вывод об обнаружении заявленных компонентов в соках, фруктах и чае (кофе). 1. 2. 3. 4. Вопросы коллоквиума: На чем основан метод ТСХ? Что такое коэффициент подвижности? Что такое подвижная и неподвижная фаза? Назовите методы проявления бесцветных пятен. 45 Список рекомендуемой литературы к разделу «Изучение состава органических соединений, их очистка и определение физических констант» 1. Иванов В.Г., Гева О.Н., Гаверова Ю.Г. Практикум по органической химии. М.: Академия, 2000. 2. Артеменко А.И. Практикум по органической химии. - М.: Высшая школа, 2001. 3. Гинзбург О.Ф. Практикум по органической химии. Синтез и идентификация органических соединений. - М.: Высшая школа, 1989. 46 3.2. Ознакомительный (малый) практикум. Лабораторная работа №6 «Алифатические углеводороды» Углеводороды наиболее простые органические соединения, молекулы которых состоят только из атомов углерода и водорода. Углеводы, в молекулах которых углеродные атомы соединены друг с другом в открытые цепи (прямые или разветвленные), называют ациклическими (алифатическими). От лат. Aliphatic – жирный. Первыми изученными соединениями этого класса были жиры. Алициклические углеводороды – циклические соединения, молекулы которых построены из углеродных атомов, связанных между собой σ-связью. Основными представителями алициклических углеводородов являются циклоалканы (циклопарафины) и циклоалкены (циклоолефины). По характеру связи между углеродными атомами углеводороды могут быть предельными (насыщенными) и непредельными (ненасыщенными). К предельным углеводородам относятся алканы (парафины), к непредельным – алкены (олефины), алкадиены и алкины. В алканах атомы углерода связаны между собой простой (одинарной) связью, в алкенах – двойной связью, алкинах – тройной связью. Алкадиены – это непредельные соединения, в молекулах которых имеются две двойные связи. Предельные углеводороды при обычных условиях обладают большой химической инертностью. Это объясняется тем, что все σ-связи С-С и С-Н весьма прочны (энергии этих связей порядка 380 кДж/моль). К реакциям присоединения они вообще не способны вследствие ненасыщенности всех связей атомов углерода. С большинством химических реагентов алканы или вовсе не реагируют, или реагируют чрезвычайно медленно. Сильные окислители (например, перманганат калия) при комнатной температуре тоже не действуют на алканы. При сравнительно невысоких температурах протекает лишь небольшое число реакций, при которых происходит замена атомов водорода на различные атомы или группы – реакции замещения. Алкены и алкины являются более реакционно-способными из-за наличия двойной и тройной связи соответственно, которые можно считать функциональными группами. Естественно ожидать, что реакции алкенов и алкинов будут происходить по ненасыщенной связи – реакции присоединения. Важными представителями алканов является метан СН4 – главная часть природного (до 95-98%) и попутных газов. В значительных количествах он присутствует в газах переработки. Метан используют в основном в качестве дешевого топлива (в быту и промышленности). Он бесцветен и не имеет запаха. Для обнаружения его утечки в газопроводах добавляют небольшое количество сильно пахнущего вещества (одоранта). Метан является ценным сырьем для химической промышленности. Из него получают ацетилен, галогенпроизводные, метанол, формальдегид и другие вещества. Метан служит для производства синтез - газа (водяного газа). 47 Изооктан (2,2,4-триметилпентан) С8Н18 – главная составная часть высококачественного горючего (бензина) для карбюраторных двигателей внутреннего сгорания. Средние члены гомологического ряда метана С7 – С17 используют как горючее для двигателей (бензин, керосин), а также в качестве растворителей. Высшие алканы С18 – С44 – сырье для производства моющих средств, смазочных масел, пластификаторов. К высшим алканам относится озокерит (горный воск), состоящий в основном из твердых алканов с разветвленной цепью углеродных атомов, число которых превышает 25-30. Многие алкены широко используют в качестве мономеров (исходных продуктов) для получения высоко молекулярных соединений (полимеров). Ацетилен используют для сварки и резки металлов, т.к. при горении в кислороде ацетилен создает высокотемпературное пламя (31500С). Так же ацетилен – ценный продукт для химической промышленности. Из него получают синтетический каучук, уксусный альдегид и уксусную кислоту, этиловый спирт и многие другие вещества. Практическая часть Опыт №1. Получение метана и его свойства. Реактивы: ацетат натрия, натронная известь, бромная вода, раствор перманганата калия. Оборудование: пробирка с газоотводной трубкой, штатив, лапка штатива, горелка. В пробирку с газоотводной трубкой помещают смесь, состоящую из одной части обезвоженного тонкоизмельченного ацетата натрия и двух частей натронной извести (NaOH и CaO). Общий объем смеси 1-2 см (около 1/3 по высоте пробирки). Закрепляют пробирку в штативе в горизонтальном положении, нагревают ее в пламени горелки. Поджигают метан у выхода газоотводной трубки через 2 минуты после выделения газа, т.е. после того, как улетучится гремучая смесь (смесь взрывоопасна!). Обращают внимание, что метан горит светящимся пламенем. Выделяющийся метан пропускают через растворы бромной воды и KMnO4. Изменяется ли окраска растворов? Почему? Опыт №2. Бромирование гексана. Реактивы: гексан, бромная вода. Оборудование: пробирка, стаканчик со льдом, горелка, пипетка. А) Демонстрационный опыт. В две кюветы помещают 3 мл гексана и добавляют 4-5 капель раствора брома в четыреххлористом углероде и перемешивают. Одну кювету ставят под источник УФ-света, а другую накрывают бумагой и оставляют под тягой. Через 3-4 минуты сравнивают кюветы. 48 Б) В сухую пробирку помещают 1 мл гексана и несколько капель бромной воды. Содержимое пробирки перемешивают на холоде. Отмечают, исчезает ли окраска брома. Нагревают содержимое пробирки на водяной бане до исчезновения окраски. Реакция сопровождается выделением HBr. Как можно обнаружить выделение HBr? Опыт №3. Получение этилена и изучение его свойств. Реактивы: этиловый спирт, серная кислота, песок, бромная вода, раствор перманганата калия. Оборудование: коническая колба на 50 мл с газоотводной трубкой, пробирки, горелка. В коническую колбу с газоотводной трубкой помещают 4-5 мл смеси этилового спирта и серной кислоты (1:5) и добавляют немного «кипелок» для равномерного кипения. Нагревают колбу со смесью в пламени горелки. Выделяющийся газ пропускают через раствор бромной воды, не прекращая нагревания. Отмечают, исчезает ли окраска брома. Далее опускают конец газоотводной трубки в заранее приготовленную пробирку с 3-4 мл раствора KMnO4, продолжая нагревание колбы. Отмечают, исчезает ли окраска розового раствора KMnO4. После пропускания этилена через бромную воду и раствор перманганата калия этилен можно поджечь у конца газоотводной трубки. Он горит несветящимся пламенем. Опыт №4. Получение ацетилена и изучение его свойств. Реактивы: карбид кальция, бромная вода, раствор перманганата калия. Оборудование: пробирка с газоотводной трубкой, пробирки. В сухую пробирку помещают кусочек карбида кальция и приливают воду, быстро закрывают пробирку пробкой с газоотводной трубкой и выделяющийся газ пропускают последовательно в пробирки с бромной водой, раствором KMnO4. Как изменяется окраска растворов? Поджигают газ у конца отводной трубки. Ацетилен горит коптящим пламенем. В отчете пишут наблюдения, уравнения всех проделанных реакций и называют полученные вещества. Делают вывод о сходстве и различии свойств алифатических углеводородов. Вопросы коллоквиума: 1. Предложите радикальный цепной механизм бромирования гексана и ионный механизм бромирования этилена. 2. Напишите уравнения реакций получения ацетилена и уравнение реакции ацетилена с аммиачным раствором оксида серебра [Ag(NH3)2OH]. 49 3. Приведите примеры углеводородов, содержащие первичный, вторичный и третичный атом углерода. Дайте им название. 4. Дайте определение изомерии. Изобразите возможные изомеры пентана и дайте им название. 5. Нахождение в природе важнейших углеводородов и их применение. Лабораторная работа №7 «Галогеналканы» Галогенпроизводными углеводородов называются органические соединения, образующиеся при замене атомов водорода в углеводородах на атомы галогенов. Соответственно галогеналканами называют производные алканов, в молекулах которых один или несколько атомов водорода замещены на атомы галогена. В зависимости от числа атомов водорода, замещенных галогеном, различают моно-, ди-, тригалогенпроизводные и т.д. Например: СН3Сl (хлорметан, метилхлорид), СН2Сl2 (дихлорметан, метиленхлорид), CHCl3 (трихлорметан, хлороформ), CCl4 (тетрахлорметан, четыреххлористый углерод, тетрахлорид углерода). В зависимости от типа атома углерода, связанного с галогеном, галогеналканы классифицируют как первичные, вторичные и третичные. Также как и среди углеводородов, различают предельные, непредельные, циклические и ароматические галогенпроизводные углеводородов. бромэтан (этилбромид) первичный галогеналкан 2-бромпропан (изопропилбромид) вторичный галогеналкан хлорциклобутан 2-бром-2-метилпропан (трет-бутилбромид) третичный галогеналкан бромциклогексан бромбензол Низшие алкилгалогениды – газообразные вещества, средние – жидкости, высшие – твердые вещества. Галогеналкилы почти нерастворимы в воде. Низшие члены ряда обладают характерным запахом. Химические свойства галогенпроизводных определяется главным образом атомом галогена, связанного с радикалом. Галогенпроизводные вступают в реакции замещения и отщепления. Наличие кратной связи приводит к увеличению реакционной способности. 50 Реакции с нуклеофилами – наиболее распространенные превращения галогеналканов. Практическая часть Опыт№1. Получение 2-бромпропана (бромистого изопропила). Реактивы: изопропиловый спирт, концентрированная серная кислота, бромид калия. Оборудование: пробирки с газоотводной трубкой, лед, штативы, стаканчики, плитка. В пробирку с газоотводной трубкой наливают 1,5-2 мл изопропилового спирта и 2 мл концентрированной серной кислоты. Смесь охлаждают и добавляют 1-2 мл воды. Продолжая охлаждение, всыпают в пробирку 1,5 г бромида калия. Присоединив газоотводную трубку, укрепляют пробирку наклонно в лапке штатива. Конец отводной трубки погружают в другую пробирку - приемник, содержащую 1 мл воды и помещают в стаканчик с водой и льдом. Реакционную смесь осторожно нагревают до кипения до тех пор, пока в приемник не перестанут поступать маслянистые капли, опускающиеся на дно. В случае сильного вспенивания реакционной массы нагревание на короткое время прерывают. По окончании реакции при помощи делительной воронки 2бромпропан отделяют от воды, собирая его в сухую пробирку или плоскодонную колбу. Для осушения 2-бромпропана добавляют несколько кусочков хлорида кальция. Полученный продукт используют для следующего опыта. Опыт№2. Отщепление галогена от галогеналкилов при действии щелочей. Реактивы: 2-бромпропан (опыт №1), раствор гидроксида натрия, азотная кислота, 1%-ный раствор нитрата серебра. Оборудование: Делительная воронка, пробирки, лед. Полученный в опыте №1, 2-бромпропан промывают в делительной воронке дистиллированной водой. Воду сливают, а 2-бромпропан переливают в пробирку, в которую затем добавляют 1-2 мл раствора гидроксида натрия. Смесь нагревают до начала кипения, охлаждают в ледяной бане. В этих условиях происходит щелочной гидролиз галогеналкилов с образованием галогенида натрия. Далее для обнаружения иона галогена небольшую часть смеси подкисляют азотной кислотой и добавляют несколько капель 1%-ного раствора нитрата серебра. Что происходит? Опыт№3. Свойства хлороформа (трихлорметана). Реактивы: хлороформ, 10% раствор гидроксида натрия, раствор иода в иодиде калия, 1%-ный раствор нитрата серебра, 10% раствор аммиака, 20% раствор азотной кислоты; Оборудование: пробирки, обратные холодильники, стаканы на 100 мл, лед. 51 3.1. В пробирку наливают 1 мл хлороформа и 1 мл воды. Закрывают пробирку пробкой и интенсивно встряхивают. Через некоторое время образуются два слоя, так как хлороформ практически нерастворим в воде. Пояснить где находится слой органического растворителя, а где вода и почему? А так же почему хлороформ не растворяется в воде? 3.2. В пробирку наливают 1 мл хлороформа и добавляют несколько капель раствора иода в иодиде калия. Смесь интенсивно встряхивают. Через некоторое время нижний слой приобретает розовую окраску. Хлороформ хорошо растворяет иод, при встряхивании иод переходит из водного слоя в хлороформ, окрашивая его в розовый цвет. 3.3. Щелочной гидролиз хлороформа. В пробирку наливают 1 мл хлороформа и 3 мл 10% раствора гидроксида натрия. Пробирку закрывают пробкой с обратным холодильником. Смесь осторожно нагревают до начала кипения, охлаждают в ледяной бане. В этих условиях происходит щелочной гидролиз хлороформа с образованием хлорида натрия и натриевой соли муравьиной кислоты: Далее для обнаружения хлорид - иона небольшую часть смеси подкисляют азотной кислотой и добавляют несколько капель 1%-ного раствора нитрата серебра (аналогично опыту №2). Обнаружение муравьиной кислоты в растворе основано на ее способности легко окисляться благодаря наличию карбонильной группы. К оставшейся части гидролизата хлороформа приливают свежеприготовленный аммиачный раствор гидроксида серебра. Пробирку нагревают на горячей водяной бане. Через некоторое время наблюдают образование металлического серебра (реакция «серебряного зеркала»). Напишите уравнение данных реакций. В отчете пишут наблюдения, уравнения всех проделанных реакций и называют полученные вещества. Вопросы коллоквиума: 1. Напишите примеры реакций галогенирования алканов и циклоалканов, назовите полученные вещества. 2. Напишите примеры реакций галогенирования алкенов, алкинов и ароматических углеводородов, назовите полученные вещества. На одном из примеров приведите механизм прямого галогенирования. 3. Присоединения галогенводородов к непредельным углеводородам. Напишите примеры реакций и объясните ход реакций, используя правила Марковникова или Зайцева. 4. Реакции нуклеофильного замещения галогена. Приведите механизмы SN1 и SN2. 52 Лабораторная работа №8 «Ароматические углеводороды и их производные» Арены – обширнейший класс циклических соединений, обладающих рядом специфических свойств. Их называют также ароматическими, поскольку простейшие из них – соединения ряда бензола – имеют интенсивный запах. Впоследствии не запах, а особые свойства стали служить критерием при отнесении того или иного соединения к ряду ароматических (к ряду аренов). Класс аренов непрерывно расширяется, постоянно открываются и изучаются все новые системы. Простейший представитель С6Н6 – бензол был открыт Фарадеем в 1825 г. и была установлена его брутто-формула. В 1865 г. Кекуле предложил его структурную формулу как циклогексатриена-1,3,5. Низшие члены гомологического ряда бензола представляют собой бесцветные жидкости с характерным запахом. Из-за высокого содержания углерода все ароматические соединения горят сильно коптящим пламенем. Все ароматические углеводороды нерастворимы в воде и хорошо растворимы в большинстве органических растворителей: многие из них хорошо перегоняются водяным паром. Наиболее характерной особенностью химического поведения бензола, как непредельного соединения в отличие от рассмотренных ранее алкенов, алкадиенов и алкинов является его устойчивость к действию окислителей (например, перманганата калия в кислой и щелочной среде). Также бензол не вступает в обычные реакции электрофильного присоединения, характерные для непредельных алифатических углеводородов. Наиболее важными и хорошо изученными являются реакции электрофильного замещения. Практическая часть Опыт №1. Растворение толуола. Реактивы: толуол, вода, этиловый спирт, диэтиловый эфир. Оборудование: пипетки и пробирки. В три пробирки помещают по 1 мл толуола. В первую пробирку наливают 2-3 мл воды, во вторую – 2-3 мл спирта, в третью – 2-3 мл диэтилового эфира. В какой из этих жидкостей толуол растворяется и почему? Поясняют, какой слой соответствует толуолу, а какой воде, если плотность толуола 0,8669 г/мл. 53 Опыт №2. Нитрование толуола. Реактивы: азотная и серная кислоты, концентрированные; толуол. Оборудование: колба 50 мл, стаканчик со льдом, пипетки. В колбу наливают 2 мл концентрированной азотной кислоты и 2,5 мл концентрированной серной кислоты. Колбу охлаждают. К охлажденной смеси добавляют по каплям при постоянном встряхивании 1 мл толуола. Через 5 минут помещают пробирку с реакционной смесью в холодную воду со льдом и дают отстояться. Выделение o- и n-нитротолуолов происходит в виде желтоватых капель с характерным запахом. Отмечают, что наблюдается? Теоретически сравнивают способности к нитрованию бензола и толуола. Сколько продуктов образуется при нитровании бензола и толуола? Опыт №3. Бромирование толуола и нафталина. Реактивы: раствор брома в четыреххлористом углероде, толуол, бромная вода, бром, железные опилки, нафталин, кристаллический иод. Оборудование: пробирки, обратный холодильник, водяная баня, лакмусовая бумага, пипетки, часовое стекло. А) Бромирование с катализатором. В две пробирки с 2 мл толуола помещают несколько капель бромной воды. Какой цвет имеет раствор брома в толуоле? Пробирку встряхивают. Исчезает ли окраска брома и почему? В первую пробирку с реакционной смесью прибавляют немного железных опилок, во вторую – немного кристалликов иода. В каком случае реакция протекает быстрее? Объясняют влияние различных катализаторов на скорость бромирования. Б) Бромирование без катализатора. В две сухие пробирки помещают по небольшому количеству нафталина и толуола и добавляют по 1 мл раствора брома. После встряхивания делят каждую смесь на две части, отливая половину ее объема в чистую сухую пробирку. Одну часть каждой смеси оставляют стоять в штативе; другую часть нагревают до кипения на водяной бане. Признаком реакции бромирования является исчезновение оранжевой окраски бромной воды и выделение бромистого водорода, который можно обнаружить у отверстия пробирки по покраснению смоченной водой лакмусовой бумажки. Сделайте вывод о различии скорости бромирования на холоде и при нагревании. Теоретически рассматривают условия бромирования бензола, и условия бромирования толуола в боковой цепи. Объясняют различие в свойствах галогена, стоящего в ядре и в боковой цепи на примере реакции бромбензола и бромистого бензила с AgNO3. Почему бром в бензольном ядре малоподвижен? 54 Опыт №4. Сульфирование толуола. Реактивы: серная кислота концентрированная, толуол, насыщенный раствор хлорида натрия. Оборудование: колба 100 мл, стаканчик со льдом, пипетки, водяная баня. В колбу помещают 5 мл концентрированной серной кислоты и прибавляют 2,5 мл толуола. Толуол постепенно растворяется в серной кислоте, реакция протекает с выделением тепла. Смесь нагревают осторожно на водяной бане (10-15 мин) до полного растворения толуола. Реакционную смесь выливают в свежеприготовленный насыщенный раствор хлорида натрия. При охлаждении льдом полученной смеси выпадают кристаллы натриевой соли nтолуолсульфокислоты. Объясняют положение сульфогруппы в продукте реакции. Опыт №6. Отношение толуола к окислению. Реактивы: 0,1%-ный раствор перманганата калия, серная кислота (раствор), толуол. Оборудование: пробирки, пипетки, грелка. В пробирку наливают 2-3 мл толуола и прибавляют 1 мл 0,1%-ного раствора KMnO4 и 1-2 капли раствора H2SO4. При встряхивании и нагревании фиолетовая окраска раствора KMnO4 исчезает, боковая цепь при этом окисляется в карбоксильную группу с образованием бензойной кислоты. Теоретически рассматривают отношение бензола к окислителям. В отчете пишут наблюдения, уравнения всех проделанных реакций, механизмов и называют полученные вещества. Вопросы коллоквиума: 1. Напишите уравнения реакций получения бензола из бензойной кислоты и его нитрования, предложите механизм нитрования бензола. 2. Почему при нитровании толуола образуются o- и n-нитротолуолы. Чем нитрование толуола отличается от нитрования бензола? 3. Правила замещения в бензоле. Приведите примеры заместителей I и II рода. 4. Правило ориентации. Напишите уравнение реакции нитрования хлорбензола. Объясните влияние заместителей на реакционную активность бензола. 5. Нахождение в природе важнейших производных бензола и их применение. 55 Лабораторная работа №9 «Гидроксилпроизводные углеводородов» К данному классу относятся углеводороды, содержащие гидроксильную группу. В случае алифатических углеводородов, данные производные называются спиртами, в случае ароматических – фенолами. Как и в случае алифатических углеводородов среди спиртов различают предельные и непредельные спирты. По количеству гидроксильных групп различают одноатомные и многоатомные спирты. Примерами предельных спиртов являются метанол СН3ОН и этанол С2Н5ОН, непредельных спиртов – виниловый спирт СН2=СН-ОН, многоатомных спиртов – этиленгликоль и глицерин: Этиленгликоль Глицерин Среди ароматических спиртов так же встречаются одно- и многоатомные спирты. Например: Фенол о-Крезол Пирокатехин Пирогаллол Химические свойства спиртов обуславливаются наличием функциональной группы – гидроксильной ОН. Связи углерод – кислород и кислород – водород (СО-Н) поляризованы, причем отрицательным концом диполя является кислород, как наиболее электроотрицательный атом элемента. Это определяет склонность спиртов к реакциям, идущим либо с разрывом связи С-О, либо с разрывом связи О-Н. Атомы водорода гидроксильных групп в спиртах проявляет определенную подвижность (активность). Подвижность водородного атома в гидроксильной группе, т.е. кислотные свойства спиртов, объясняется смещением электронной плотности связи к наиболее электроотрицательному атому кислорода. Подобно воде спирты реагируют со щелочными металлами (натрием, калием). Гидроксильная группа спиртов способна замещаться галогеном (нуклеофильное замещение) с образованием галогенпроизводных углеводородов. Фенолы являются более сильными кислотами по сравнению со спиртами. Кислотность фенола объясняется взаимодействием неподеленной электронной пары атома кислорода гидроксильной группы с π-электронным облаком бензольного ядра. В результате сопряжения электронная пара связи О-Н смещена в сторону кислорода, что способствует легкому протонированию. 56 Прежнее название бензола – «фен». Отсюда «фенол» - соединение, содержащие гидроксильную группу в бензольном ядре. Сам фенол, а также о, м, п-метилфенолы (крезолы) содержатся в каменноугольной смоле. Многоатомные фенолы в виде различных производных встречаются в природе и применяются в медицине, технике и парфюмерии (таннин, ванилин, лакмус, ализарин и др.). Обычно фенолы представляют собой твердые кристаллические вещества с резким характерным запахом. Они плохо растворимы в воде, хорошо - в спирте и эфире. Практическая часть Опыт №1. Обнаружение воды в этиловом спирте и его обезвоживание. Реактивы: этиловый спирт, безводный сульфат меди. Оборудование: пробирка с резиновой пробкой, пипетки. Вносят в сухую пробирку небольшое количество этилового спирта, добавляют немного безводного сульфата меди, взбалтывают содержимое пробирки, дают ему отстояться. Если спирт содержит воду, осадок сульфата меди окрасится в голубой цвет вследствие образования медного купороса (CuSO4·5H2O). Сохраняют обезвоженный спирт для проведения следующего опыта. Опыт №2. Получение и гидролиз этилата натрия. Реактивы: обезвоженный этиловый спирт (опыт №1), металлический натрий, 1%ный раствор фенолфталеина. Оборудование: пробирка, пинцет, пипетки. В пробирку наливают 1-2 мл обезвоженного этилового спирта и помещают небольшой кусочек металлического натрия с предварительно обрезанными корками и просушенного между листами фильтровальной бумаги. Реакция сопровождается выделением тепла и водорода. При охлаждении на дне пробирки остается осадок этилата натрия. При добавлении 2 мл воды осадок растворяется. Добавляют в пробирку 1-2 капли раствора фенолфталеина. Наблюдается ли изменение окраски раствора и почему? Определяют значение рН с помощью универсальной индикаторной бумаги. Опыт №3. Окисление этилового спирта перманганатом калия. Реактивы: этиловый спирт, щелочной раствор перманганата калия. Оборудование: пробирка, пипетки, горелка. В пробирку наливают 2 мл этилового спирта и прибавить 1 мл щелочного раствора перманганата калия. Приготовленную смесь слегка! подогревают. Как изменится окраска раствора KMnO4? Обращают внимание на запах уксусного альдегида (запах прелого яблока). 57 Опыт №4. Получение глицерата меди. Реактивы: 3% -ный раствор сульфата меди, 10%-ный раствор щелочи, глицерин. Оборудование: пробирка с резиновой пробкой, пипетки, стеклянная палочка. В пробирку наливают 0,5 мл 3%-го раствора сульфата меди и добавляют 1 мл 10%-го раствора щелочи. Отмечают, какого цвета образовавшийся осадок? К полученной смеси добавляют каплю глицерина и взбалтывают до растворения осадка. Опыт №5. Взаимодействие сегнетовой соли с гидроксидом меди (образование фелингового раствора). Реактивы: раствор сульфата меди (II), 10%-ный раствор NaOH, 5%-ный раствор сегнетовой соли. Оборудование: пробирка, пипетки. Получают в пробирке голубой осадок гидроксида меди, используя для этой цели 1 мл раствора CuSO4 и небольшой избыток 10%-го раствора NaOH. К осадку гидроксида меди добавляют 5%-ный раствор калий - натриевой соли винной кислоты (сегнетова соль). Гидроксид меди растворяется, при этом образуется прозрачная жидкость голубого цвета – раствор Фелинга. Опыт №6. Растворимость и кислотный характер фенолов и нафтолов. Реакция фенолов и нафтолов с хлорным железом. Реактивы: фенол, пирокатехин, резорцин, гидрохинон, пирогаллол, α- и β-нафтол, универсальная индикаторная бумага, 10%-ный раствор FeCl3. Оборудование: пробирки с пробками, пипетки, горелка. В пробирки помещают по 0,5 г фенола, пирокатехина, резорцина, гидрохинона, пирогаллола, α- и β-нафтола и добавляют в каждую из них по 3-4 мл воды. Пробирки встряхивают. Что происходит? Если вещество растворяется плохо, смесь нагревают до кипения и охлаждают. Отмечают, какие фенолы проявляют в большей степени кислотные свойства? Пишут структурные формулы всех предложенных веществ. Стеклянной палочкой наносят каплю этих растворов на универсальную индикаторную бумагу. Отмечают реакцию среды. В каждую из пробирок добавляют 5-10 капель 10%-го раствора FeCl3. Отмечают различное окрашивание растворов в зависимости от природы фенолов и нафтолов. Результаты заносят в таблицу 3.1.: Таблица 3.1. Название Формула РаствориРастворирН Окрашивафенола фенола мость в воде мость в воде раствора ние смесей при Ткомн. при с хлорным нагревании железом 58 Опыт №7. Образование и разложение фенолятов. Реактивы: раствор фенола (опыт №6), 10%-ный раствор NaOH. Оборудование: пробирки с пробками, пипетки. Полученный в опыте №6 раствор фенола в воде взбалтывают до образования эмульсии и прибавляют к нему 5 мл 10%-го раствора гидроксида натрия. При этом фенол полностью растворяется. Реакция протекает с образованием фенолята натрия. Если к раствору фенолята натрия прибавить соляную кислоту, происходит выделение свободного фенола в виде мельчайших капель, вследствие чего раствор начинает мутнеть. Опыт №8. Взаимодействие фенола с бромом. Реактивы: 2%-ный водный раствор фенола, бромная вода. Оборудование: пробирки с пробками, пипетки. В пробирку наливают 2 мл 2%-го водного раствора фенола и 4-5 мл бромной воды. Наблюдают выделение хлопьевидного осадка 2,4,6-трибром- фенола. Опыт №9. Сульфирование фенола. Реактивы: фенол, концентрированная серная кислота. Оборудование: пробирки с пробками, пипетки, водяная баня. Помещают в пробирку 0,5 г фенола и 1-2 мл концентрированная серной кислоты. При встряхивании кристаллы фенола растворяются. Несколько капель этого раствора смешивают в другой пробирке с 0,5 мл воды. Наблюдают выделение фенола в виде мутной фазы. Оставшийся раствор фенола в серной кислоте нагревают в течение 5 мин в кипящей водяной бане, охлаждают и осторожно выливают в другую пробирку, содержащую 5-7 мл воды. Мутнеет ли раствор и появляется ли запах фенола? Почему? Опыт №11. Свойства пикриновой кислоты. Реактивы: насыщенный водный раствор пикриновой кислоты, концентрированный раствор щелочи. Оборудование: пробирки, пипетки. В пробирку наливают 2 мл насыщенного водного раствора пикриновой кислоты и добавляют несколько капель концентрированного раствора щелочи. Отмечают изменение окраски раствора в результате образования пикрата натрия. В отчете пишут наблюдения, уравнения всех проделанных реакций, механизмов и называют полученные вещества. 59 Вопросы коллоквиума: 1. Напишите уравнение реакции получения диэтилового эфира из этилового спирта под действием серной кислоты. 2. Какая разница в процессе образования алкоголятов одноатомными и многоатомными спиртами? Объясните эту разницу. 3. Напишите возможные изомеры 3-бутанола и назовите их. 4. Какие реакции характерны для фенола, по какому механизму они протекают? Чем бромирование бензола отличается от бромирования фенола? 5. Почему при сульфировании фенола образуется два продукта. Назовите их. Предложите механизм нитрования фенола и nнитрофенола. Как влияет нитрогруппа на кислотные свойства фенола? 6. Нахождение в природе важнейших производных спиртов и фенолов, их применение. Лабораторная работа №10 «Карбонильные соединения и их производные» Группировка С=О называется карбонильной, или оксо - группой. Любые органические соединения, содержащие ее, могут быть, в принципе, названы карбонильными. Однако чаще всего карбонильными называют соединения общей формулы R-C(O)-X, где R и X – атомы водорода (альдегиды) или углеродные радикалы (кетоны). Среди карбонильных соединений также встречаются предельные, непредельные и ароматические представители. Например: Уксусный альдегид Акролеин Ацетон Бензальдегид Бензофенон Низшие члены ряда карбонильных соединений – ацетон, формальдегид, ацетальдегид – растворимы в воде, высшие альдегиды и кетоны хорошо растворимы в большинстве обычных органических растворителей (спирты, эфир и т.п.). Низшие альдегиды имеют резкий запах, у альдегидов с С3 – С6 весьма неприятный запах, высшие альдегиды обладают цветочными запахами и даже применяются в парфюмерии. 60 К основным типам химических реакций альдегидов и кетонов относятся реакции присоединения, конденсации, замещения и окисления. Химическая активность карбонильной группы в реакциях нуклеофильного присоединения обусловлена, с одной стороны, ее высокой полярностью, с другой – пространственной доступностью атома углерода для атаки нуклеофила, так как карбонильная группа плоская (углерод в sp2-гибридном состоянии). Альдегиды в слабощелочной среде подвергаются альдольной конденсации. Реакция идет с образованием альдегидоспиртов (оксоальдегидов), называемых альдолями. В альдольную конденсацию вступают альдегиды и кетоны, содержащие в α-положении водородные атомы. Альдегиды легко окисляются, т.е. проявляют восстановительные свойства. Продукты окисления – кислоты с тем же числом углеродных атомов в молекуле. Кетоны устойчивы к слабым окислителям. Продукты реакции окисления кетонов сильными окислителями – смесь кислот с меньшим числом углеродных атомов, чем исходный кетон. Ароматические альдегиды способны самопроизвольно окисляться кислородом воздуха до кислот. Окисление катализируется действием света и протекает по радикальному цепному механизму. Практическая часть Опыт №1. Качественная реакция на ацетон. Реактивы: 1%-ный раствор иода в 10%-ом растворе иодистого калия, 1%-ный раствор NaOH, ацетон, 5%-ный раствор нитропруссида натрия. Оборудование: пробирки, пипетки. Помещают в пробирку 1 мл раствора иода в иодистом калии и 2 мл 1%-ного раствора NaOH. К обесцвеченному раствору иодноватистокислого натрия (NaIO) добавить 1 мл водного раствора ацетона. Выпадает желтовато-белый осадок с характерным запахом иодоформа (CHI3). В пробирку наливают 1 мл ацетона и столько же воды и добавляют 3 капли раствора нитропруссида натрия и 3 капли 10%-ного раствора NaOH. Появляется ли оранжево-красное окрашивание? Иодоформная проба на ацетон и реакция с нитропруссидом являются очень чувствительными методами и позволяют обнаруживать ацетон в водных растворах при содержании его 0,04%. Опыт №2. Взаимодействие ацетона с сульфитом натрия. Реактивы: насыщенный раствор сульфита фенолфталеина, серная кислота, ацетон. Оборудование: пробирки, пипетки. 61 натрия, 1%-ный раствор В одну пробирку наливают 1-2 мл насыщенного раствора сульфита натрия Na2SO3 и добавляют 1-2 капли фенолфталеина. При появлении розовой окраски (щелочная среда за счет гидролиза соли Na2SO3) нейтрализуют раствор (исчезновение розовой окраски фенолфталеина), добавив 1-2 капли серной кислоты. В другую пробирку помещают 1-2 мл воды и тоже добавлять 1-2 капли фенолфталеина. В обе пробирки вносят 2-3 капли ацетона. В какой пробирке и почему появляется интенсивно красное окрашивание раствора? Опыт №3. Получение фенилгидразона формальдегида. Реактивы: раствор фенилгидразина солянокислого, 10%-ный раствор формальдегида. Оборудование: пробирки, пипетки. В пробирку наливают 1 мл раствора фенилгидразина солянокислого и добавляют 1-3 капли 10%-го раствора формальдегида. Происходит образование ярко-желтого осадка. Опыт №4. Реакция альдегида с фуксинсернистой кислотой. Реактивы: раствор фуксинсернистой кислоты, 1%-ный раствор формальдегида, 10%-ный раствор уксусного альдегида, раствор соляной кислоты. Оборудование: пробирки, пипетки. В две пробирки наливают по 1 мл бесцветного раствора фуксинсернистой кислоты. В одну пробирку добавляют несколько капель 1%-го раствора формальдегида, в другую – тоже количество 10%-го уксусного альдегида. Отмечают оттенок окраски в пробирках. Далее в каждую пробирку добавляют по 0,5 мл концентрированной HCl. Как изменились окраски в пробирках? Окрашенные продукты взаимодействия фуксинсернистой кислоты с альдегидами обычно обесцвечиваются при добавлении минеральных кислот, за исключением соответствующего производного формальдегида, который более устойчив и сохраняет окраску в кислой среде. Кетоны, как правило, такой реакции не дают. Опыт №5. Окисление формальдегида гидроксидом меди в щелочном растворе. Реактивы: 10%-ный раствор формальдегида, 10%-ный раствор гидроксида натрия, 2%-ный раствор сульфата меди. Оборудование: пробирки, пипетки, горелка. В пробирку наливают немного 10%-го раствора формальдегида, 3-4 мл 10%-го свежеприготовленного раствора NaOH и добавляют по каплям 2%-ный раствор сульфата меди до образования мути. Смесь нагревают до образования желтого осадка, переходящего в красный. Формальдегид, в отличие от других альдегидов, восстанавливает оксиды меди (I и II) до металлической меди. 62 Опыт №6. Окисление бензойного альдегида кислородом воздуха. Реактивы: бензойный альдегид. Оборудование: стеклянная пластинка, стеклянная палочка. На стеклянную пластинку помещают каплю бензойного альдегида и оставляют на некоторое время на воздухе. Отмечают, что наблюдается и с чем это связано? Опыт №7. Синтез бензальанилина. Реактивы: бензойный альдегид, анилин, этиловый спирт, лед. Оборудование: стакан вместимостью 100 мл, ледяная баня, стеклянная палочка, воронка Бюхнера. В стакан наливают 2,5 мл бензойного альдегида. Затем при перемешивании приливают 2,5 мл анилина. Сразу же начинается реакция, сопровождающаяся выделением теплоты. По окончании реакции (прекращение разогревания) реакционной смеси дают отстояться 15-20 мин. Далее при перемешивании выливают в стакан 6 мл 95%-ного спирта. Смесь оставляют при комнатной температуре на 10 мин, затем в течение 30 мин охлаждают в ледяной бане с целью более полного осаждения продукта реакции. Полученную кристаллическую массу отфильтровывают на воронке Бюхнера и высушивают. Определяют выход продукта. Опыт №8. Синтез дибензальацетона. Реактивы: гидроксид натрия, бензойный альдегид, ацетон, этиловый спирт, лед. Оборудование: стакан вместимостью 250 мл, водяная баня со льдом, стеклянная палочка, магнитная мешалка, колба коническая 50 мл, термометр. В стакан наливают охлажденный до комнатной температуры раствор 0,7 г NaOH в смеси 7 мл воды с 6 мл спирта. Реакционную смесь перемешивают на магнитной мешалке с водяной баней при температуре 20-25 0С и добавляют половину приготовленной заранее смеси 7,5 мл бензальдегида и 2,8 мл ацетона. Через 2-3 мин начинается помутнение и образуется хлопьевидный осадок. В ходе процесса нельзя допускать разогрев смеси, т.к. при температуре свыше 300С идет побочная реакция, снижающая выход бензальдегида и ацетона. Содержимое перемешивают еще 30 минут. Выпавший осадок отфильтровывают на воронке Бюхнера, промывают водой и высушивают. Определяют выход продукта. В отчете пишут наблюдения, уравнения всех проделанных реакций, механизмов и называют полученные вещества. 63 Вопросы коллоквиума: 1. Напишите уравнение реакции получения ацетона пиролизом ацетата кальция. 2. Напишите уравнение реакции и механизм взаимодействия ацетона с сульфитом натрия 3. Напишите уравнение реакции окисления формальдегида «серебряного зеркала». Какие свойства проявляет формальдегид? 4. Назовите отличия альдегидов и кетонов. 5. Предложите механизм альдольной конденсации. Какова роль среды? 6. Нахождение в природе важнейших производных альдегидов и кетонов, их применение. Лабораторная работа №11 «Карбоновые кислоты и их производные» К классу карбоновых карбоксильную группу кислот относятся соединения, содержащие По количеству карбоксильных групп различают моно- и дикарбоновые кислоты. Например: Уксусная кислота Щавелевая кислота Монокарбоновые кислоты называют также одноосновными, поскольку их молекула может связывать один эквивалент основания. Также различают алифатические и ароматические кислоты. Среди алифатических кислот встречаются предельные и непредельные карбоновые кислоты. Например: Акриловая кислота Бензойная кислота Кислотные свойства карбоновых кислот (легкость отщепления протона от карбоксильной группы) обусловлены не только электроноакцепторным влиянием двух атомов кислорода, но и сравнительной устойчивостью образующегося аниона (RCOO-). 64 Карбоновые кислоты, как и другие протонные кислоты, взаимодействуют с достаточно активными металлами, их оксидами и гидроксидами, а также с аммиаком с образованием соответствующих солей. Карбоновые кислоты в большинстве случаев являются слабыми кислотами и уступают таким кислотам, как соляная, азотная и серная. Двухосновные карбоновые кислоты сильнее соответствующих одноосновных кислот. Первые четыре представителя ряда карбоновых кислот – жидкости, смешивающиеся с водой во всех отношениях. Кислоты, в молекулах которых содержится от пяти до девяти атомов углерода (а также изомасляная кислота), маслянистые жидкости и растворимость их в воде невелика. Высшие кислоты (от С10) – твердые тела, практически нерастворимы в воде, при перегонке в обычных условиях они разлагаются. Из реакций с участием карбонильной группы большое значение имеет реакция этерификации – взаимодействия карбоновой кислоты со спиртом в кислой среде, приводящее к образованию сложного эфира: Одноосновные кислоты, как правило, устойчивы к действию окислителей. Легко окисляется лишь муравьиная кислота ( до углекислого газа и воды) и кислоты с третичным атомом углерода в α-положении. При окислении последних получаются α-оксикислоты: Предельные двухосновные кислоты – твердые кристаллические вещества. При нагревании щавелевой кислоты легко отщепляется углекислый газ: Муравьиная, уксусная и пропионовая кислоты имеют острый запах; средние члены ряда обладают неприятным запахом, высшие кислоты запаха не имеют. Муравьиная кислота (НСООН) содержится в выделениях желез некоторых видов муравьев, отсюда ее название. Практическая часть Опыт №1. Получение уксусной кислоты из ацетата натрия. Реактивы: безводный ацетат натрия, концентрированная серная кислота. Оборудование: пробирки, пипетки, газоотводная трубка, стаканчик со льдом. В пробирку с газоотводной трубкой помещают 4 г обезвоженного ацетата натрия и приливают 4 мл концентрированной серной кислоты. Газоотводную трубку опускают в другую пробирку, охлаждаемую льдом. Смесь в первой пробирке нагревают. Что наблюдается? 65 Опыт №2. Растворимость предельных карбоновых кислот. Реактивы: муравьиная, уксусная, стеариновая кислоты. Оборудование: пробирки, пипетки. В три пробирки помещают небольшие количества следующих кислот: муравьиной, уксусной и стеариновой. В каждую пробирку добавляют по 2 мл воды. Содержимое пробирки взбалтывают. Если кислота не растворяется, пробирку нагревают. После охлаждения отмечают растворимость взятых кислот в воде. Опыт повторяют, используя в качестве растворителя толуол. Как изменяется растворимость карбоновых кислот с увеличением числа атомов углерода? Как влияет природа растворителя на растворимость карбоновых кислот и почему? Опыт №3. Кислотные свойства карбоновых кислот. Реактивы: 10%-ный раствор уксусной кислоты, магний, Na2CO3. Оборудование: пробирки, газоотводная трубка, пипетки, щипцы. А) Кислотные свойства предельных кислот. В две пробирки с газоотводными трубками наливают по 2-3 мл 10%-ного раствора уксусной кислоты. В одну пробирку опускают кусочек магния, в другую помещают немного карбоната натрия. Газ, выделяющийся во второй пробирке, пропускают через известковую воду. Б) Кислотные свойства высших предельных кислот. В сухую пробирку помещают стеарин (на кончике шпателя), добавляют немного диэтилового эфира до растворения (без нагревания), приливают 2-3 капли 1%-ного раствора фенолфталеина. Затем по каплям приливают 10%-ный раствор гидроксида натрия. Появляющаяся вначале малиновая окраска исчезает при встряхивании. Объясняют наблюдаемое явление. Пишут уравнения диссоциации всех соответствующих кислот. Опыт №4. Разложение щавелевой кислоты. Реактивы: щавелевая кислота, концентрированная серная кислота, 5%-ный раствор KМnO4, известковая вода. Оборудование: пробирки с газоотводной трубкой, пипетки, горелка. Работа под тягой! Разложение щавелевой кислоты. В пробирку помещают 1 г щавелевой кислоты и добавляют 1-2 мл концентрированной H2SO4. Пробирку закрывают пробкой с газоотводной трубкой, конец которой опускают в пробирку с известковой водой. Смесь в пробирке осторожно нагревают. Что при этом наблюдается? После чего извлекают газоотводную трубку из пробирки с известковой водой и у ее отверстия поджигают другое газообразное вещество, которое горит характерным пламенем – голубоватыми вспышками. 66 Какие газообразные вещества выделяются при разложении щавелевой кислоты? Опыт №6. Отношение карбоновых кислот к действию окислителей. Реактивы: раствор перманганата калия, муравьиная кислота, лед. уксусная кислота, концентрированная серная кислота, известковая вода, щавелевая кислота, олеиновая кислота. Оборудование: пробирки, газоотводные трубки, пипетки, горелка. А) Отношение муравьиной кислоты. В пробирку наливают 1-2 мл муравьиной кислоты, добавляют 1 мл H2SO4 (1:5), 1-2 мл раствора KМnO4 . Пробирку закрывают пробкой с газоотводной трубкой, конец которой опустить в пробирку с известковой водой. Смесь в пробирке нагревают над пламенем сухого горючего. Что наблюдается и почему? Б) Отношение уксусной кислоты к действию окислителей. В пробирку наливают 0,5 мл лед. уксусной кислоты, 2 мл 5%-ного раствора H2SO4 и 3-4 мл 1%-ного раствора KМnO4. Содержимое пробирки взбалтывают. Происходит ли изменение окраски раствора и почему? В) Отношение олеиновой кислоты. В пробирку помещают 0,5 мл олеиновой кислоты, добавляют 0,5 г 5%-го раствора карбоната натрия и 0,5 мл 2%-го раствора перманганата калия. Встряхивают пробирку несколько раз. Отмечают, какие изменения происходят с первоначальной окраской раствора. Г) Отношение щавелевой кислоты. Работа под тягой! В пробирку наливают 1 мл 10%-ного раствора H2SO4 и 3 мл 5%-ного раствора KМnO4 и 1 мл насыщенного раствора щавелевой кислоты. Пробирку соединяют с газоотводной трубкой, конец которой опускают в пробирку с известковой водой. Реакционную смесь осторожно нагревают. Как изменяется окраска реакционной смеси и что происходит в пробирке с известковой водой? Делают вывод об отношении данных карбоновых кислот к действию окислителей. Опыт №7. Свойства винной кислоты. Реактивы: 15%-ый раствор винной кислоты, 5%-ый раствор гидроксида калия, . 2%-ый раствор сульфата меди (II), 10%-ый раствор гидроксида натрия. Оборудование: пробирки, пипетки, стеклянные палочки. А) Доказательство наличия двух карбоксильных групп в винной кислоте. В пробирку помещают 1 мл 15%-го раствора винной кислоты, 2 мл 5%-го раствора гидроксида калия и встряхивают. Постепенно начинает выделяться белый кристаллический осадок малорастворимой в воде кислой калиевой соли винной кислоты (гидротартрат калия). Если осадок не выпадает, охлаждают пробирку под струей воды и трут внутреннюю стенку пробирки стеклянной палочкой. Добавляют в пробирку еще 4-5 мл раствора гидроксида калия. Кристаллический осадок постепенно растворяется, так как образуется хорошо 67 растворимая в воде средняя калиевая соль винной кислоты (тартрат калия). Раствор тартрата калия сохраняют до следующего опыта. Б) Доказательство наличия гидроксильных групп в винной кислоте. В две пробирки помещают по 2 мл 2%-го раствора сульфата меди (II) и 10%ного раствора гидроксида натрия. Выпадает голубой осадок гидроксида меди (II). В первую пробирку добавляют раствор тартрата калия, полученный в предыдущем опыте. Осадок гидроксида меди (II) растворяется с образованием синего раствора. Жидкости в обеих пробирках нагревают на водяной бане до кипения. Что происходит в каждой из пробирок и почему? Образовавшийся синий раствор носит название реактива Фелинга. Что такое реактив Фелинга и для чего он используется? Опыт №8. Взаимодействие карбоновых кислот с бромом. Реактивы: бромная вода, олеиновая кислота, раствор бензойной кислоты, раствор салициловой кислоты. Оборудование: пробирки, пипетки, резиновая пробка. В три пробирки помещают: в первую - 2-3 капли олеиновой кислоты, во вторую - 2 мл раствора бензойной кислоты, в третью – 2 мл раствора салициловой кислоты. В каждую пробирку добавляют 2-3 мл бромной воды и сильно встряхивают. Что наблюдается? Протекают ли реакции бромирования? Производные карбоновых кислот. Опыт №9. Получение уксусноизоамилового эфира. Реактивы: лед. уксусная кислота, концентрированная серная кислота, изоамиловый спирт. Оборудование: пробирки, пипетки, стеклянная палочка, водяная баня, стаканчик со льдом. Работать под тягой! В пробирку наливают 2 мл лед уксусной кислоты, 2 мл изоамилового спирта и 1 мл концентрированной серной кислоты. Содержимое пробирки перемешивают палочкой и помещают на несколько минут в водяную баню при температуре, близкой к кипению. Затем содержимое пробирки выливают в стакан с холодной водой. Уксусноизоамиловый эфир, всплывающий на поверхность воды, обладает характерным приятным запахом грушевой эссенции. Опыт №10. Растворимость и обменные реакции мыла и синтетических моющих средств (СМС). Реактивы: мыло, синтетическое моющее средство, щавелевая кислота, концентрированная уксусная кислота, 10%-ный раствор серной кислоты, стеарин, диэтиловый эфир, 1%-ный раствор фенолфталеина, 10%-ный раствор гидроксида 68 натрия, 5%-ный раствор CaSO4, раствор хлорида магния, раствор сульфата меди (II), 10%-ный раствор уксусной кислоты, бензол. Оборудование: пробирки, пипетки, горелка, шпатель, резиновая пробка. А) В одну из двух пробирок помещают мелко раздробленные кусочки мыла на кончике шпателя, в другую – синтетическое моющее средство (СМС). В каждую из пробирок добавляют 3-4 мл дистиллированной воды. Смесь нагревают и встряхивают, переводя испытуемые вещества в раствор. Охлаждают содержимое пробирок холодной водой. В результате раствор мыла образует плотный студень, а раствор СМС остается без изменения и в отличие от раствора мыла сильно пенится при встряхивании. Б) Выделение жирных высших кислот из мыла. В пробирку наливают 2-3 мл водного раствора мыла и добавляют по каплям 10%-ный раствор серной кислоты. Что наблюдается? Г) Образование нерастворимых солей высших карбоновых кислот. Загустевший раствор мыла (опыт А) снова нагревают до полного растворения. Растворы мыла и моющих средств разделяют на четыре части (восемь пробирок) и добавляют к каждой из них такие же объемы: водопроводной воды, 5%-ный раствор гипса CaSO4, растворы хлористого магния (морская вода) и сульфата меди. Растворы мыла дают осадки кальциевого, магниевого и медного мыла, а при встряхивании жидкости над осадком пены не образуется. Содержимое пробирки с осадком медного мыла разделяют на две части, одну нагревают до начинающегося кипения, при этом наблюдают, как медное мыло всплывает в виде изумрудно-зеленого кольца (если в растворе находится избыток натриевого мыла, то зеленого кольца не получается). Растворы СМС при взаимодействии с теми же реактивами резких изменений не дают. Опыт №11. Качественная характеристика эмульгирующих свойств мыла и моющих средств. Реактивы: растительное масло, мыло (раствор), синтетическое моющее средство (раствор). Оборудование: пробирки, пипетки, резиновая пробка. В каждую из трех пробирок помещают по 2 капли растительного масла. В одну пробирку доливают 5-6 мл дистиллированной воды, а в другие по 5-6 мл 10%-ных растворов мыла и моющих средств. Сильно встряхивают содержимое пробирок и оставляют на 5 минут до образования эмульсий. В первой пробирке (вода) эмульсия содержит довольно крупные капли масла, которые выделяются сразу при стоянии; эмульсия в водном растворе мыла более устойчива, но уступает эмульсии в растворе СМС. Опыт №12. Омыление жира. Реактивы: раствор растительного мыла, 15%-ный раствор гидроксида натрия. 69 Оборудование: пробирки, коническая колба на 50 мл, электрическая плитка, асбестовая сетка. В коническую колбу вместимостью 50 мл наливают 20 капель растительного мыла и 4-5 мл 15%-ного раствора гидроксида натрия, тщательно перемешивают и кипятят на асбестовой сетке 3-4 мин. Гидролиз жира можно считать законченным, если несколько капель гидролизата (белого цвета), внесенные в пробирку, будут полностью растворяться в дистиллированной воде(без выделения капелек масла). Полное растворение пробы свидетельствует о том, что нерастворимое в воде масло полностью гидролизовано с образованием растворимых в воде глицерина и солей высших жирных кислот. Реакция щелочного гидролиза (омыления) протекает по следующей схеме: H2C O CH O CH2 CO C17H33 CO C17H33 O CO H2C + OH CH OH NaOH CH2 C17H33 +3 C17H33 COONa OH В отчете напишите наблюдения, уравнения всех проделанных реакций, механизмов и дайте названия полученным веществам. Вопросы коллоквиума: 1. Как изменяются физические свойства (Тпл/Ткип, плотность, агрегатное состояние) в гомологическом ряду карбоновых кислот и почему? 2. Напишите уравнение реакции и механизм получения сложного эфира – этилпропионата и пропилформиата. Какова роль среды реакции? 3. Напишите уравнения реакции гидролиза полученных сложных эфиров. 4. Что такое эмульсия. Какие явления лежат в основе действия моющих средств? 5. Особенности малеиновой и фумаровой кислот. 6. Нахождение в природе важнейших производных карбоновых кислот и их применение. Лабораторная работа №12 «Азотосодержащие органические соединения» К азотсодержащим соединениям относятся соединения, содержащие группы, в которые входят азот. К таким группам, например, относятся нитро- (-NO2) и аминогруппы (-NH2). Среди них также различают алифатические и ароматические соединения, например: Этиламин Нитрометан 70 Аминобензол 2,4,6-тринитрофенол (Анилин) (Пикриновая кислота) Производные углеводородов, содержащие аминогруппу –NH2 называются аминами. Различают первичные, вторичные и третичные амины в зависимости от замещения атома водорода аминогруппой: Первичный амин вторичный амин третичный амин Анилин является простейшим представителем ароматических аминов. Ароматические амины в отличие от аминов жирного ряда представляют собой слабые основания, водный раствор ароматических аминов не изменяет окраску лакмуса и фенолфталеина, хотя с сильными кислотами эти амины образуют соли. При действии азотистой кислоты в кислых растворах на холоде на анилин и другие ароматические первичные амины образуются так называемые соли диазония: Азотсодержащие соединения образуют большой класс красителей. Одной из таких групп красителей являются азокрасители. Согласно хромофорно ауксохромной теории цветности, предложенной О. Виттигом в 1887 г., окраска органического соединения обусловлена наличием в его молекуле ненасыщенных группировок, например азогруппы (-N=N-), нитрогруппы (NO2), хиноидной ) и другие. Эти группы были названы хромофорами. группировки ( В азокрасителях хромофорной группой является азогруппа –N=N- , находящаяся между двумя ароматическими радикалами или же между ароматическим и гетероциклическим радикалами, например: Анилиновый желтый Получают азокрасители реакцией сочетания диазотированного ароматического первичного амина с аминами или фенолами. 71 Реакцией азососчетания, приводящей к образованию азокрасителей, называют реакцию между солью диазония и ароматическими соединениями, содержащими электронодорнорные заместители (-NH, -NHR, -NR2, -OH), например: Практическая часть Опыт №1. Образование солей анилина. Реактивы: анилин, концентрированная соляная кислота, концентрированная серная кислота, раствор нитрита натрия. Оборудование: пробирки, пипетки, резиновые пробки. А) В пробирку с 5 мл воды добавляют несколько капель анилина и взбалтывают. Что наблюдается? Добавляют в пробирку несколько капель концентрированной HCl. Записывают уравнение реакции. Далее к полученному солянокислому анилину добавляют раствор нитрита натрия. Что при этом происходит? Б) В пробирке смешивают 1 мл анилина и 0,5 мл разбавленной серной кислоты. Что наблюдается? Опыт №2. Бромирование анилина. Реактивы: анилин, бромная вода. Оборудование: пробирки, пипетки, резиновые пробки. В пробирку наливают 3 мл воды, добавляют 0,5 мл анилина и встряхивают. Затем приливают 0,5 мл бромной воды. Что наблюдается? Опыт №3. Получение хлористого фенилдиазония. Реактивы: раствор солянокислого анилина, иодкрахмальная бумага, 20%-ный раствор нитрита натрия Оборудование: пробирки, пипетки, резиновые пробки, стакан, лед. В пробирку наливают 5 мл раствора солянокислого анилина и охлаждают, помещая пробирку в стакан со льдом. Затем по каплям добавляют 20%-ный раствор нитрита натрия до появления на иодкрохмальной бумажке синего пятна, которое свидетельствует об избытке нитрита натрия и, следовательно, о конце диазотирования. Образовавшийся водный раствор хлористого фенилдиазония используют для получения азокрасителей. 72 Опыт №4. Получение азосоединений. Реактивы: 10%-ный раствор NaOH, фенол, анилин, концентрированная соляная кислота, диметиланилин, 10%-ный раствор HСl, раствор ацетата натрия. Оборудование: пробирки, пипетки, резиновые пробки. А) Получение красителя анилинового оранжевого. В пробирке с 2 мл 10%-ного раствора NaOH растворяют 0,1 г фенола. Затем приливают примерно равный объем соли диазония (опыт 3). В пробирке немедленно появляется интенсивное окрашивание. Б) Получение красителя анилинового желтого. В пробирку помещают 4 капли диметиланилина, 2 мл воды и для растворения добавляют по каплям при встряхивании 10%-ный раствор HCl. Смесь охлаждают и добавляют в нее 3 мл раствора хлористого фенилдиазония (опыт 3), а затем 2 мл насыщенного раствора ацетата натрия (его добавляют для связывания соляной кислоты, т.к. реакция азосочетания в сильнокислой среде не идет). Смесь взбалтывают. Что наблюдается? В) Получение кислотного азокрасителя. К 2 мл водного раствора хлористого фенилдиазония (опыт 3) прибавляют равное количество фенола в щелочи. Появляется интенсивное красное окрашивание (раствор должен быть все время щелочным). Опыт №5. Получение фенолфталеина. Реактивы: фталевый ангидрид, фенол, концентрированная серная кислота, этиловый спирт, раствор щелочи. Оборудование: пробирки, пипетки, резиновые пробки. В пробирку помещают 1 г фталевого ангидрида, 1 г фенола и добавляют 2 капли концентрированной H2SO4 . Смесь нагревают в течение нескольких минут. Затем охлаждают и прибавляют к ней 2 мл этилового спирта. Несколько капель полученного фенолфталеина наливают в другую пробирку с раствором щелочи. Что наблюдается? Раствор подкисляют. Что наблюдается? Опыт №6. Получение флуоресцеина. 73 Реактивы: фталевый ангидрид, резорцин, концентрированная серная кислота, этиловый спирт, водный раствор аммиака. Оборудование: пробирки, пипетки, резиновые пробки. В пробирку помещают 0,5 г фталевого ангидрида, 0,5 г резорцина и несколько капель серной кислоты. Смесь расплавляют в пробирке до получения темнокрасного сплава, После охлаждения сплав растворяют в небольшом количестве спирта. Спиртовой раствор флуоресцеина выливают в колбочку с водным раствором аммиака. Что наблюдается? Какой цвет имеет полученная жидкость в проходящем и отраженном свете? В отчете пишут наблюдения, уравнения всех проделанных реакций и называют веществам. Делают выводы. Вопросы коллоквиума: 1. Восстановление ароматических нитросоединений в нейтральной, щелочной и кислой средах. Привести примеры. 2. Таутомерия нитросоединений и диазосоединений. Привести примеры. 3. Напишите уравнение реакции ацилирования анилина уксусным ангидридом. 4. Напишите схему получения сульфаниловой кислоты. 5. Реакции диазотирования и азосочетания. Механизмы. 74 Лабораторная работа №13 «Гетероциклические соединения» К классу гетероциклов относятся вещества, содержащие циклические группировки атомов, включающие в цикл кроме атомов углерода и другие (гетеро-) атомы. Наибольшее значение имеют гетероциклы, содержащие атомы N, O и S. Они называются соответственно азот-, кислород- и серосодержащие гетероциклы. Свойства гетероциклов определяется не только имеющимися гетероатомами, в значительной степени они зависят от характера связей в цикле. Гетероциклы, не содержащие кратных связей, как правило, по химическим и физическим свойствам похожи на соответствующие ациклические соединения. Но существует и другая, очень обширная группа гетероциклов, имеющих сопряженные системы кратных связей. Такого рада гетероциклы напоминают своей устойчивостью и типами реакций бензол, а его производные получили название ароматических гетероциклов. Гетероциклические соединения по количеству атомов в цикле делятся на пятии шестичленные, например: тетрагидрофуран пирролидин фуран пиррол тиофен пиридин Гетероциклы могут включать несколько гетероатомов, как одинаковых, так и различных, например: имидазол пиримидин 1,3,5-триазин оксазол тиазол Еще различают конденсированные гетероциклические содержащие дополнительный цикл, например: Индол бензимидазол хинолин 75 изохинолин соединения, Практическая часть Опыт №1. Растворимость пиридина в воде и его основные свойства. Реактивы: пиридин, красная лакмусовая бумага. Оборудование: пробирки, пипетки. К 1 мл пиридина приливают 5 мл воды. Каплю полученного раствора наносят на красную или индикаторную бумагу. Какой цвет приобретает индикаторная бумага? Почему? Объясняют значение рН. К капле пиридина осторожно прибавляют концентрированную соляную кислоту. Затем к реакционной смеси добавляют воду. Что при этом наблюдается? Опыт №2. Образование солей пиридина. Реактивы: водный раствор пиридина (опыт №1), 2%-е растворы хлорида железа (III) и сульфата меди (II). Насыщенный раствор пикриновой кислоты. Оборудование: пробирки, пипетки. В две пробирки наливают по 1 мл 2%-ых растворов хлорида железа (III) и сульфата меди (II) и добавляют по 1-2 капли раствора пиридина, полученного в предыдущем опыте. Что при этом происходит? Ко второй пробирке добавляют избыток гидроксида меди (II). Что наблюдается? К 1 мл насыщенного раствора пикриновой кислоты приливают 5-10 капель раствора пиридина и встряхивают. Что при этом наблюдается? Данная реакция используется для обнаружения пиридина. Опыт №3. Устойчивость пиридина к окислителям. Реактивы: пиридин, 1% раствор перманганата калия, 1% раствор гидрокарбоната натрия (NaHCO3). Оборудование: пробирки, пипетки, резиновые пробки, электрическая плитка. В пробирку помещают по одной капле раствора пиридина, 1%-ного раствора перманганата калия, 1% -ного раствора гидрокарбоната натрия. Содержимое пробирки встряхивают и нагревают в течение 2 минут. Изменяется ли цвет раствора? Опыт №4. Основные свойства хинолина. Реактивы: хинолин, концентрированная соляная кислота, 10% раствор NaОH. Оборудование: пробирки, пипетки, резиновые пробки. В пробирку помещают 2 капли хинолина, 3 капли воды и по каплям до полного растворения хинолина, приливают 3-4 капли концентрированной соляной кислоты. К солянокислому раствору хинолина добавляют 4 капли 10% -ного раствора NaОH. Объясняют выделение тяжелой капли свободного хинолина. 76 Опыт №5. Получение фурфурола. Реактивы: отруби, 20%-ый раствор серной кислоты. Оборудование: круглодонная колба, обратный холодильник, пипетки, плитка, аппарат для перегонки. К 3 г отрубей (кукурузные початки, древесные опилки, шелуха семечек) добавляют 30 мл 20%-ой серной кислоты и нагревают реакционную смесь на водяной бане с обратным холодильником в течение 30 минут. Фурфурол образуется из пентозанов, которые вместе с клетчаткой входят в состав отрубей: Реакционную смесь отфильтровывают от отрубей и с фильтратом осуществляют перегонку при атмосферном давлении (см. лаб. раб №1, опыт 4). Фурфурол имеет т. пл. 1620С, а серная кислота 3200С. С помощью мерного цилиндра или пробирки определяют объем полученного после перегонки фурфурола. Для доказательства полученного фурфурола проводят качественную реакцию на фурфурол и его окисление (опыты №6, 7 лабораторной работы № 13) для сравнения проводят эти же реакции с эталоном – реактивом фурфурола, который имеется в лаборатории. Опыт №6. Качественная реакция на фурфурол. Реактивы: анилин, уксусная кислота, фурфурол. Оборудование: часовое стекло, пипетки, фильтровальная бумага. На два часовых стекла наносят каплю анилина и смешивают ее с одной каплей уксусной кислоты. Далее на первое добавляют каплю фурфурола, полученного в опыте №5, а на другое – каплю фурфурола, имеющегося в лаборатории. Описывают свои наблюдения и делают вывод. Почему полученный фурфурол прозрачный и бесцветный, а реактив фурфурола имеет коричневый цвет? Опыт №6. Окисление фурфурола. Реактивы: фурфурол, аммиачный раствор оксида серебра. 77 Оборудование: часовое стекло, пипетки. На два часовых стекла наносят каплю аммиачного раствора оксида серебра и на первое добавляют каплю фурфурола, полученного в опыте №5, а на другое – каплю фурфурола, имеющегося в лаборатории. Наблюдают появление черного пятна свободного серебра. В отчете пишут наблюдения, уравнения всех проделанных реакций и называют веществам. Делают выводы. Вопросы коллоквиума: 1. Напишите структурные формулы следующих соединений: 2,5диметилфуран, 2-хлортиофен, 2,5- диэтилпиррол, 2-аминопиридин. 2. Объясните причину ароматического характера фурана, тиофена и пиррола. 3. Напишите уравнения реакции полного и неполного гидрирования фурана, дайте названия полученным продуктам реакций. 4. Перечислите области применения фурфурола. Список рекомендуемой литературы к разделу «Ознакомительный (малый) практикум» 1. Иванов В.Г., Гева О.Н., Гаверова Ю.Г. Практикум по органической химии. М.: Академия, 2000. 2. Артеменко А.И. Практикум по органической химии. - М.: Высшая школа, 2001. 3. Травень В.Ф. Органическая химия. В 2 т. – М.: Изд. Академкнига, 2001. 4. Реутов О.А., Курц А.Л., Бутин К.П. Органическая химия. – М.: Изд. Московского университета, 1992. 5. Грандберг И.И. Органическая химия. –М.: Дрофа, 2002. 78 3.3. Методы синтеза органических соединений Использование реакций электрофильного замещения. Реакции электрофильного замещения условно обозначаются символом SEмоно записать эти реакции в виде уравнения: А:В + Х+ → A:X + B+ Здесь А:В обозначает молекулу субстрата, две точки — электронную пару связи А—В или А—X, Х+ — электрофильный реагент. Реакции электрофильного замещения наиболее характерны для соединений ароматического ряда. Важнейшие реакции электрофильного замещения в ароматическом ряду: C6H6 + HNO3 + H+ → C6H5NO2 + H2O C6H6 + H2SO4 → C6H5SO3H + H2O Нитрование Сульфирование Галогенирование Алкилирование Ацилирование HOC6H6 + C6H5N2Cl → HOC6H4N=NC6H5 Азосочетание Склонность ароматических соединений к реакциям замещения объясняется тем, что этот тип взаимодействия, в отличие от реакций присоединения, не ведет к нарушению устойчивой ароматической системы и, следовательно, не требует добавочной энергии на преодоление энергии стабилизации этой системы. Реакции SE представляют собой многостадийные процессы, механизм которых можно изобразить следующей схемой. Сначала под воздействием катализатора реагент X—Y переходит в активную форму Х+: X:Y + Катализатор → Х++ [Y : Катализатор]Далее следует взаимодействие катиона Х+ с молекулой ароматического соединения, например, бензола: Первая стадия, образование так называемого π - комплекса, включает взаимодействие между электрофильным реагентом Х+ и делокализованными π электронами ядра. Эта стадия осуществляется быстро и всегда обратимо. При этом происходит лишь незначительное нарушение π - электронного облака бензольного кольца. Вторая стадия состоит в перестройке π - комплекса в карбениевый ион, так называемый π - комплекс, в котором реагент Х+ связан с 79 определенным углеродным атомом ядра. В π - комплексе ароматический секстет нарушен: четыре π - электрона делокализованы в сфере воздействия пяти углеродных атомов. Шестой углеродный атом переходит при образовании π комплекса из состояния sp2 в состояние sp3 гибридизации. Третья стадия реакции — отщепление протона и образование молекулы замещенного бензола. Отщеплению протона помогает основание В-, в роли которого может выступать растворитель или комплекс [Y-Катализатор]-. Обратимыми являются реакции сульфирования и алкилирования ароматических соединений. Если же в σ - комплексе образуются прочные π - связи между ядром и реагентом, обратный процесс, переход от σ-комплекса к π - комплексу, не наблюдается и реакция протекает необратимо. Это имеет место, в частности, при нитровании, галогенировании и ацилировании ароматических соединений. Скорость реакции электрофильного замещения водорода в ароматическом ядре меняется при переходе от бензола к его гомологам и производным: она зависит от числа и вида уже имеющихся в ядре заместителей. Реакция электрофильного замещения осуществляется тем легче, чем больше нуклеофильность ароматического ядра. Заместители электронодонорные, повышающие общую электронную плотность в ядре, облегчают реакцию электрофильного замещения и являются, таким образом, активирующими, а заместители электроноакцепторные, понижающие электронную плотность в кольце, этой реакции препятствуют, т. е. являются дезактивирующими. К электронодонорным (активирующим) заместителям относятся следующие атомы и группы атомов (в скобках указаны основные электронные эффекты, проявляемые этими заместителями): алкилы (+I), —ОН, —OR, —NH2, —NHR, — NR2, (+M, - I), — О-, (+М и +I). К электроноакцепторным (дезактивирующим) заместителям относятся: —COR, —СООН, -COOR, -CN, -NО2 (—М и -I), галогены (+М , - I), NR+ (—I). Пользуясь такой классификацией, можно заранее предсказать, что, например, толуол и фенол будут легче нитроваться и сульфироваться, чем бензол, а для введения в реакцию нитрования нитро- или бромбензола потребуются более жесткие условия, чем для аналогичной реакции с бензолом. X H H X R H X устойчивее, чем R R B случае электронодонорных заместителей с - I и +М - эффектами энергия активации образования σ - комплекса для орто- и паразамещения оказывается ниже, чем для метазамещения, а потому соответствующие продукты (орто- и параизомеры) образуются скорее, чем метаизомеры. Электронодонорные заместители являются, таким образом, орто-, параориентантами. 80 Электроноакцепторные заместители (обладающие —I- и - М-эффектами) дестабилизируют σ - комплекс, и это их влияние в наибольшей степени проявляется тогда, когда заместитель непосредственно связан с положительно заряженным углеродным атомом, поэтому: H X H Y X H X менее устойчивы, чем Y Y Соответственно энергия активации для метазамещения оказывается несколько меньше, чем для орто- и паразамещения и соответствующий изомер образуется с большей скоростью. Электроноакцепторные заместители являются метаориентантами. Заместители, обладающие - I- и +М-эффектами, в положительно заряженном и поэтому сильно электрофильном σ-комплексе проявляют главным образом свой +Мэффект. С его помощью они понижают энергию активации образования σ - комплекса с заместителями в орто- и параположениях. Это справедливо также для случая заместителей типа галогенов. Таким образом, галогены, подобно электронодонорным заместителям, являются орто-, параориентантами. Поскольку они одновременно являются дезактивирующими заместителями, энергия активации образования σ комплекса с галогенбензолами оказывается выше, чем с незамещенным бензолом, но ниже, чем соответствующая энергия σ -комплекса с соединениями, имеющими в ядре заместители с - I- и - М-эффектами. Если в ароматическом ядре имеется не один, а несколько заместителей, место вступления нового заместителя будет определяться суммарным влиянием уже имеющихся. При этом возможны два случая: влияние имеющихся заместителей может быть согласованным или несогласованным. Согласованное влияние оказывают заместители одного рода (орто-,, пара- или метаориентирующие), когда они находятся в метаположении друг к другу, например: H CH3 X X + X CH3 CH3 -H CH3 X O2N CH3 CH3 H NO2 + X X + NO2 NO2 -H + O2N NO2 Если в ядре находятся заместители разного рода (один орто, пара- а другой метаориентирующий), их совместное влияние будет согласованным только при их нахождении в орто- или параположении, например: 81 CH3 H H X X H CH3 X , но не NO2 NO2 CH3 NO2 Несогласованная ориентация имеет место в тех случаях, когда заместители одного рода находятся в орто- или параположениях по отношению друг к другу, или когда заместители разного рода оказываются в метаположении. В этом случае место вступления нового заместителя определяется относительной ориентирующей (активирующей) силой заместителей. Например, при нитровании ометоксиацетанилида под влиянием ориентирующего действия амидной группы образуется в основном 2-метокси-4-нитроацетанилид: NHCOCH3 NHCOCH3 MeO HNO3 , H2SO4 MeO NO2 Рассмотренные правила ориентации справедливы лишь для кинетически контролируемых процессов. В равновесных условиях преимущественно образуются наиболее стабильные изомеры (термодинамический контроль). Так, при алкилировании толуола хлористым алкилом в присутствии А1С13 при низкой температуре образуется в основном параалкилтолуол, а при 60° С — более устойчивый метаизомер: 82 Лабораторная работа №14 «Сульфирование» Реакции, сопровождающиеся замещением водорода ядра сульфогруппой, относятся к сульфированию. Открытие реакции сульфирования принадлежит В. Бранду, который в 1819 г. осуществил синтез сульфопроизводного нафталина. Специфика этого процесса в отличие от других электрофильных реакций заключается в том, что он носит необратимый характер: В зависимости от условий сульфирования в качестве сульфирующих агентов могут выступать различные частицы, однако во всех случаях процесс протекает через стадию образования σ-комплекса: Наиболее распространенным сульфирующим агентом является серная кислота различной концентрации. Для трудно сульфируемых соединений применяют олеум с различным содержанием оксиды серы (VI). В этом случае первой стадией процесса является образование π - комплекса между π- электронной системой бензольного ядра (донор электрона) и электрон-дефицитным атомом серы в молекуле серного ангидрида SO3: + SO3 SO3 В ходе реакции π – комплекс перегруппировывается в σ-комплекс: _ O O O S SO3 H + При дальнейшем течении процесса σ - комплекс с переносом протона от атома углерода к атому кислорода в экзотермической реакции превращается в бензолсульфокислоту: 83 _ O O O S H SO3H + Сульфирующим агентом служит также хлорсульфоновая кислота ClSO3H (монохлорангидрид серной кислоты). Для проведения сульфирования в газовой фазе используют триоксид серы. Сульфирование соединений, требующих мягких условий, осуществляют пиридинсульфотриоксидом C6H5N SO3. Практическая часть Получение 2-нафталинсульфокислоты Реактивы: нафталин, серная кислота концентрированная, соляная кислота концентрированная. Оборудование: пипетки, пробирки, горелка. В колбе на 25 мл расплавляют 2,5г нафталина и нагревают его до 1600С (шарик термометра должен быть погружен в жидкость). К расплавленному нафталину при перемешивании по каплям добавляют 3 мл серной кислоты, следя за тем, чтобы температура не поднималась выше 1700С. Смесь перемешивают при 1600С в течение 20 мин, дают охладиться до комнатной температуры и медленно при перемешивании палочкой, выливают в стакан с 10-15 мл воды. Раствор отфильтровывают от выпавшего осадка ди-2-нафтилсульфона и 0 непрореагировавшего нафталина, охлаждают до 0 С и оставляют до выпадения кристаллов тригидрата 2-нафталинсульфокислоты (Т. пл. 1240С, Выход 60%). 2-Нафталинсульфокислота – бесцветное, очень гигроскопическое вещество, хорошо растворима в воде и спирте, плохо – в бензоле. Отфильтрованный осадок высушивают и определяют выход и температуру плавления. Для доказательства структуры осуществите дальнейшее превращение 2нафталинсульфокислоты в β - нафтол по методике. Для этого необходимо сначала получить натриевую соль 2-нафталинсульфокислоты. 84 Получение натриевой соли 2-нафталинсульфокислоты Реактивы: 2-нафталинсульфокислота, карбонат натрия. Оборудование: коническая колба (стакан), фарфоровая чашка, пипетки, плитка. В термостойкой конической колбе или стакане на 100 мл растворяют в 10-15 мл воды 2,08 г (0,01 моль) 2-нафталинсульфокислоты при нагревании. К полученному раствору добавляют раствор 2,5 г (0,023 моль) карбонат натрия в 10 мл воды до щелочной реакции на лакмус. После охлаждения смесь фильтруют для отделения углекислого газа. Фильтрат упаривают в фарфоровой чашке до появления первых кристаллов. Полная кристаллизация продолжается несколько часов. Вторую фракцию кристаллов можно получить дальнейшим упариванием маточного раствора. Обе фракции сушат в фарфоровой чашке на водяной бане. Натриевая соль 2-нафталинсульфокислоты получается в виде мелких стекловидных пластинок (выход 66% от теоретического). Определите выход продукта. Получение β - нафтола Реактивы: гидроксид натрия, натриевая соль 2-нафталинсульфокислоты, 20%-ая соляная кислота. Оборудование: тигель, фарфоровая чашка, пипетки, плитка, термометр. В тигле расплавляют 2,8 г (0,07 моль) гидроксида натрия в 5 мл воды. Когда температура плава достигнет 2800С, к нему при энергичном перемешивании, добавляют по возможности быстро 0,92 г (0,004 моль) натриевой соли 2нафталинсульфокислоты, причем температура реакционной массы не должна опускаться 2600С. Затем температуру повышают до 3000С; реакционная масса при этом становится очень пористой вследствие выделения из нее водяного пара. Собственно реакция начинается при температуре 310-3200С и продолжается 5 минут. При температуре выше 3200С плав темнеет и становится жидким; его 85 быстро выливают на железный противень. Застывшую массу измельчают в ступке и растворяют в возможно малом количестве воды. Раствор подогревают и подкисляют при перемешивании раствором соляной кислоты (1:1). При охлаждении β - нафтол выделяются в виде бесцветных игл. Кристаллы отфильтровывают и промывают подкисленной водой. Полученное вещество перекристаллизовывают из воды, слегка подкисленной соляной кислотой. Выход β - нафтола составляет около 70%; т. пл. 1220С. После получения проведите идентификацию полученного вещества, используя в качестве эталона β - нафтол (имеющийся реактив). Для этого определите физические константы (температура плавления, растворимость и Rf) и сделайте вывод (см. 3.5.) о структуре полученного вещества: β - нафтола и 2нафталинсульфокислоты. В отчете пишут уравнения реакции и механизм сульфирования нафталина. Лабораторная работа №15 «Нитрование» Введение в молекулу органического соединения нитрогруппы называется реакцией нитрования. Впервые ее осуществил Митчерлих при получении нитробензола. В зависимости от природы субстрата и характера нитрующего агента механизм реакции может быть различным. Так, введение нитрогруппы в ароматическое ядро протекает по электрофильному механизму. Для наиболее распространенного случая нитрования смесью концентрированных азотной и серной кислот на примере бензола можно представить следующей схемой: Электрофильный характер реакции определяется атакой на молекулу бензола нитроний - катиона NO2+ c образованием промежуточного σ - комплекса и последующем отщеплением протона. Если для нитрования используется разбавленная азотная кислота, а в качестве субстрата углеводороды жирного ряда (реакция Коновалова, 1888), то процесс протекает по радикальному цепному механизму, который может осуществляться с участием азотной кислоты: 86 Для ароматического ряда выбор нитрующего агента определяется реакционной способностью нитруемого соединения в процессах электрофильного замещения. Для нитрования применяют азотную кислоту различной концентрации. При этом необходимо учитывать, что нитрование сопровождается образованием воды, которая, разбавляя азотную кислоту, может привести к остановке реакции. Во избежание влияния разбавления в реакционную среду в качестве водоотнимающего средства добавляют концентрированную серную кислоту. Особенно широко применяют заранее приготовленные смеси азотной и серной кислот (нитрующие смеси) в соотношении 1:1. В лабораторной практике часто используется нитрующая смесь из нитрата натрия или калия и концентрированной серной кислоты. Для получения нитросоединений ароматического и алифатического рядов используют также парафазное нитрование оксидами азота. Реакция нитрования экзотермична, т.е. происходит с выделением тепла. Очень много теплоты выделяется также при разбавлении серной кислоты водой. Поэтому во многих случаях проходиться прибегать к внешнему охлаждению содержимого колбы, а также практиковать медленное прибавление реагентов. Практическая часть α - Нитронафталин NO2 HNO3 H2SO4 H2O Реактивы: нафталин, серная кислота концентрированная, азотная кислота концентрированная, этиловый спирт. Оборудование: фарфоровый стакан вместимостью 50 мл; термометр; мешалка. В фарфоровом стакане смешивают 2,5 мл серной кислоты с 2 мл воды и приливают 2 мл азотной кислоты. Температуру реакционной смеси отслеживают термометром, который помещен в смесь и укреплен в штативе. Нитрующую смесь нагревают до 500С и вносят 2,5 г хорошо измельченного нафталина. Реакционную смесь перемешивают на магнитной мешалке в течение 1 часа при 500С. Затем температуру повышают до 600С и перемешивают еще 1 час. После охлаждения реакционной массы полученный нитронафталин всплывает на поверхность кислоты в виде пористой лепешки. Продукт отделяют и промывают в фарфоровой 87 чашке кипящей водой несколько раз. При этом с парами воды отгоняются остатки нафталина. Расплавленный нитронафталин выливают тонкой струей в стаканчик с холодной водой при сильном перемешивании. Застывший в виде мелких шариков продукт фильтруют и сушат на воздухе. После того как нитронафталин высохнет, определяют выход. Выход 86% от теоретического. Проведите очистку α - нитронафталина перекристаллизацией из этилового спирта. Выпавшие кристаллы после перекристаллизации отфильтровывают, высушивают. Затем определяют температуру плавления, растворимость и проводят ТСХ. Полученные данные сравнивают с теоретическими данными. α - Нитронафталин – желтый кристаллический продукт с Т. Пл. 600С. Растворяется в этаноле, эфире, хлороформе, ацетоне. Хроматография: адсорбент: Sorbfil, элюент: ацетон – петролейный эфир – бензол – метанол – водный раствор аммиака (концентрированный) (100:200:50:2). α - Нитронафталин вводится в ацетоновом растворе. Rf=0.8. В отчете пишут все наблюдения, уравнения реакции и механизм нитрования нафталина. Делают вывод о полученном продукте по результатам идентификации. п - Нитроацетанилид и п - нитроанилин Реактивы: ацетанилид, серная кислота концентрированная , 25%-ный раствор серной кислоты, азотная кислота концентрированная , 10%-ный раствор гидроксида натрия. Оборудование: фарфоровый стакан вместимостью 50 мл; термометр; мешалка. В стаканчике на 50 мл растворяют при перемешивании 5 г ацетанилида в 10 мл концентрированной серной кислоте при температуре не выше 400С. Затем в охлажденный до 50С реакционный раствор медленно приливают при постоянном перемешивании смесь 4 мл концентрированной серной и 2,4 мл азотной кислоты, наблюдая за температурой, которая не должна быть выше 150С. Реакционную смесь выдерживают при этой температуре 45 мин и выливают в 250-300 мл охлажденной льдом водой. Отбирают для анализа пробу вещества, которую сушат при температуре 100-1500С. п - Нитроацетанилид – аморфное твердое вещество бледно-желтого цвета, труднорастворимое в большинстве органических растворителей, Т. пл. 2100С. 88 Сырой п - нитроацетанилид помещают в колбу с обратным холодильником и кипятят с 26 мл 25%-ной серной кислоты до полного растворения. Горячий раствор фильтруют и подщелачивают 10%-ным раствором гидроксида натрия. После охлаждения выпавшие кристаллы п - нитроанилина отфильтровывают, промывают холодной водой и перекристаллизовывают из воды. Выход 4 г (78% от теоретического из расчета на ацетанилид). п - Нитроанилин – желтые кристаллы с т. пл. 1470С. Растворяется в этиловом спирте, ацетоне, эфире, плохо в воде. Хроматография: адсорбент: Sorbfil, элюент: ацетон – петролейный эфир – бензол – метанол – водный раствор аммиака (концентрированный) (100:200:50:2). α - Нитронафталин вводится в ацетоновом растворе. Rf=0.7. Определяют выход полученного вещества, температуру плавления, растворимость и проводят ТСХ и сравнивают полученные результаты с теоретическими данными. Делают соответствующий вывод. Лабораторная работа №16 «Галогенирование» Галогенированием называются реакция введения одного или нескольких атомов галогена в молекулу органического соединения. В зависимости от природы галогена различают фторирование, хлорирование, бромирование и иодирование, а в зависимости от количества вводимого – моно-, ди- и полигалогенирование. Методы введения галогена в органическое соединение могут быть разделены на две группы: 1. прямое галогенирование – замещение атома водорода галогеном, а также присоединение галогенов и галогенводородов по месту разрыва кратных связей; 2. непрямое галогенирование – замещение атомами галогена гидроксильной группы, кислорода карбонильной группы и диазагруппы органических соединений. Прямое галогенирование осуществляют с помощью галогенов (Cl2, Br2, I2) или смешанных галогенов (BrCl, FCl, ICl и т.д.) При непрямом галогенировании применяют галогенводородные кислоты и кислородсодержащие кислоты галогенов либо их соли со щелочными металлами. Иодоформ 89 Реактивы: ацетон, иод, иодид калия, 10%-ный раствор гидроксида натрия. Оборудование: стакан вместимостью 150 мл, воронка капельная. В стакан помещают 2 г иодида калия, приливают 4 мл дистиллированной воды и после растворения соли добавляют 1 г иода. К полученному раствору приливают 10 мл воды. Затем в реакционную массу вводят 2,5 мл ацетона и при перемешивании по каплям добавляют из капельной воронки 10%-ный раствор гидроксида натрия до исчезновения красноватой окраски раствора. Иодоформ, который при этом выпадает в виде желтого кристаллического осадка, через 30 мин отфильтровывают, промывают небольшим количеством воды и высушивают на воздухе. Иодоформ (трииодметан) – твердое кристаллическое вещество желтого цвета со специфическим запахом и т. пл. 1190С. Растворяется в этиловом спирте, диэтиловом эфире, хлороформе. В воде практически не растворим. Под действием света быстро гидролизуется раствором щелочи, поэтому следует избегать сильнощелочной среды в процессе синтеза. Хроматография: адсорбент: Sorbfil, элюент: гексан – ацетон – бензол – метанол (20:20:5:1). Иодоформ вводится в ацетоновом растворе. Rf=0.81. Определяют выход полученного вещества, температуру плавления, растворимость и проводят ТСХ и сравнивают полученные результаты с теоретическими данными. Делают соответствующий вывод. Лабораторная работа №17 «Алкилирование» Реакциями алкилирования называют реакции, включающие замену атома водорода органического соединения алкильным радикалом. В зависимости от того, при каком атоме в молекуле происходит замещение водорода, N-, O-, и Салкилирование. Независимо от природы реакционного центра субстрата и его строения, а также от строения алкилирующего агента во всех случаях процессы протекают по электрофильному механизму. Практическая часть Феноксиуксусная кислота Реактивы: фенол, хлоруксусная кислота, 10%-ный раствор соляной кислоты, 25%ный раствор гидроксида натрия, 10%-ный раствор карбоната натрия, соляная кислота, диэтиловый эфир. 90 Оборудование: круглодонная колба вместимостью 50 мл; обратный воздушный холодильник, делительная воронка, стакан на 100 мл, водяная баня, термометр; мешалка. В круглодонной колбе смешивают 1 г фенола, 10 мл 25%-ного раствора гидроксида натрия и 2,4 г хлоруксусной кислоты. Осторожно, хлоруксусная кислота вызывает ожоги кожи! Смесь нагревают на кипящей водяной бане с воздушным обратным холодильником в течение 1 ч, а затем охлаждают 6 мл 10%-ной соляной кислоты до рН 3-5 и экстрагируют дважды эфиром порциями по 5 мл. Эфирные вытяжки осторожно смешивают в стакане с 10 мл раствора карбоната натрия. После прекращения выделения углекислого газа смесь встряхивают в делительной воронке. Эфирный слой отбрасывают, а водный медленно и осторожно при размешивании в стакане подкисляют концентрированной соляной кислотой до рН 3-5. Выпавшую феноксиуксусную кислоту отфильтровывают, промывают водой и высушивают на воздухе. Перекристаллизовывают из воды. Определите выход полученного вещества, температуру плавления, растворимость и проведите ТСХ и сравните полученные результаты с теоретическими данными. Сделайте соответствующий вывод. Феноксиуксусная кислота представляет собой бесцветные пластинки или иглы. Т. пл. 990С, плохо растворима в воде, хорошо – в этиловом спирте, диэтиловом эфире, бензоле, уксусной кислоте. Хроматография: бумага типа «быстрая» или адсорбент: Sorbfil, элюент: изопропиловый спирт – раствор аммиака - вода (6:1:1). Проявление в сушильном шкафу при 140-1500С в течение 30 мин, в результате появляется желтое пятно. Rf=0.58. Определяют выход полученного вещества, температуру плавления, растворимость и проводят ТСХ и сравнивают полученные результаты с теоретическими данными. Делают соответствующий вывод. 91 Использование реакций нуклеофильного замещения. Реакции нуклеофильного замещения. Например, алкилгалогениды с нуклеофилами реагируют по схеме: R-H + Y- → R – Y + HalR – Hal + HY: → R – Y+H + Hal- → R-Y + HHal Нуклеофильное замещение представляет собой разрыв связи R – Hal и образование новой связи R – Y. Эти процессы могут идти или одновременно (синхронно) или последовательно (асинхронно). Замещения галогена на алкоксильную группу: R-X + H – O - Alk → R - O – Alk + HX Простой эфир Замещения галогена на нитро группу: R-X + NaNO2 → R – NO2 + NaX нитроалкан Замещения галогена на аминогруппу: R-X + NaNH2 → R – NH2 + NaX аминоалкан Замещения галогена на нитрильную группу: R-X + NaC≡N → R – C≡N + NaX алкилнитрил Синхронный (SN2) механизм. Замещения галогена на гидроксильную группу: CH3 – CH2 – C*H – CH3 + NaOH → CH3 – CH2 – C*H – CH3 + NaCl Cl втор - бутилхлорид OH втор – бутиловый спирт * - асимметричный атом углерода, то есть атом углерода с четырьмя разными заместителями. Нуклеофил (ОН ) атакует асимметрический атом углерода субстрата с «тыла» по отношению к галогену (так легче подойти, отсутствует межатомное отталкивание от атома галогена). По мере приближения нуклеофила все три заместителя, отталкиваясь от него, «уходят» по другую сторону плоскости, проходящей через асимметрический атом углерода и перпендикулярной линии связи С-Hal. В переходном состоянии три заместителя оказываются в этой плоскости, а атом углерода, атакуемый нуклеофилом, переходит в sр2-гибридное состояние, его р - орбиталь перекрывается одновременно с атомными орбиталями кислорода и хлора. Максимальное перекрывание в этом случае возможно при расположении атомов О, С, О на прямой, перпендикулярной плоскости, в которой располагаются три заместителя. Формирование обычной двухэлектронной двухцентровой связи О - С продукта реакции приводит к перемещению трех заместителей по другую сторону плоскости, и таким образом продукт реакции 92 образуется в виде энантиомера, относящегося к противоположному по отношению к исходному соединению конфигурационному ряду. Происходит обращение конфигурации. Энергетический профиль SN2: (R) – вторбутилхлорид (переходное состояние) (S) – втор - бутиловый спирт Рис. 3.26 . Энергетический профиль реакции, протекающей по механизму SN2. Еакт – энергия (энтальпия) активации; ∆G – энтальпия реакции. Асинхронный (SN1) механизм. Согласно этому механизму реакция протекает в 2 стадии. Первая стадия: диссоциация, которая протекает под действием полярного растворителя, характеризуется максимальной энергией активации, поэтому протекает с минимальной скоростью: RX = [Rδ+ ·····Xδ-] = R+ + X- (лимитирующая) Вторая стадия: образование новой связи RY R+ + Y- → RY Данные процессы относят к мономолекулярным поскольку скорость такой химической реакции будет определяться концентрацией исходного соединения RХ: υx.p. = k [RX] 93 1 - RX 2 – разрыхление связи RX [Rδ+ ·····Xδ-] 3 - R+ + X4 – образование связи RY R+ + Y- → [Rδ+ ·····Yδ-] 5 - → RY Рис. 3.27. Энергетический профиль реакции, протекающей по механизму SN1. Еа1 – энергия (энтальпия) активации первой стадии; Еа2 – энергия (энтальпия) активации второй стадии. Cтереохимия данного процесса. Атака нуклеофила может идти с двух сторон: а и b с равной вероятностью: В случае а – происходит обращение конфигурации продукта относительно исходной конфигурации. В случае b – обращения конфигурации не происходит. Следовательно, продукт химических реакций, протекающих по механизму SN1 – рацемат (смесь двух оптических изомеров, имеющих разную конфигурацию). Лабораторная работа №18 «Ацилирование» Замена атома водорода аминоили гидроксигруппы остатком кислородсодержащей минеральной, карбоновой или сульфокислоты называется ацилированием. В практике ацилирования наиболее широкое применение получили кислоты, среди которых особое значение имеет муравьиная, уксусная щавелевая, реже бензойная кислота. Чаще применяют уксусные и фталевые ангидриды, а из неорганических оксид серы (VI). Большое значение реакциях ацелирования приобрели галогенангидриды, в частности хлорангидриды, например, хлористый бензоил С6Н5СОСl и ряд его замещенных. 94 Практическая часть Фениловый эфир бензойной кислоты Реактивы: фенол, хлористый бензоил, гидроксид натрия, этиловый спирт. Оборудование: колба плоскодонная вместимостью 50, пробка, нутч-фильтр. Фенол, попадая на кожу, может вызывать сильные ожоги! Работы с фенолом проводят в вытяжном шкафу! В плоскодонной колбе готовят раствор 1 г едкого натрия в 10 мл воды и растворяют в нем 0,6 г фенола. Затем туда же добавляют 1-2 мл хлористого бензоила и колбу энергично встряхивают в течение 15-20 мин до исчезновения запаха хлористого бензоила. При встряхивании постепенно выпадают кристаллы фенилового эфира бензойной кислоты. Осадок отфильтровывают, промывают водой и сушат. Перекристаллизацию ведут из этилового спирта, не доводя его до кипения. Т. пл. 700С. Определяют выход полученного вещества, температуру плавления, растворимость и сравнивают полученные результаты с теоретическими данными. Делают соответствующий вывод. Использование реакций окисления и восстановления. Лабораторная работа №19 «Окисление» Процессы, сопровождающиеся отнятием от субстрата электронов, называют окислением. В этих реакциях окислителями являются соединения, обладающие большим сродством к электрону (электрофильностью), а субстратами – соединения, имеющие склонность к отдаче электронов (нуклеофильность). Процессы окисления связаны либо с присоединением кислорода, либо с дегидрированием – отнятием водорода (точнее, двух электронов и двух протонов). Наиболее распространенными окислителями являются вещества с сильно выраженными электрофильными свойствами: азотная кислота, кислород и пероксидные соединения (пероксид водорода, пероксиды металлов, неорганические и органические надкислоты), сера, бром, хлор. Кислородные кислоты галогенов и их соли. К эффективным окислителям относятся соединения металлов в высших степенях окисления: соединения железа (III), перманганат калия, диоксид марганца, хромовая кислота и ее ангидрид. 95 Практическая часть Антрахинон O + 4H2O + 2Cr(CH3COO)3 2CrO3 + 6CH3COOH O Реактивы: антрацен, оксид хрома (VI), уксусная кислота (ледяная). Оборудование: колба двухгорлая вместимостью 50 мл; обратный холодильник, стакан на 100 мл, колба коническая на 100 мл. Собирают прибор (рис.3.28.). В двугорлой колбе, снабженной капельной воронкой и обратным холодильником (или в колбе снабженной двурогим форштосом) (рис. 29), растворяют при нагревании 0.32 г (0,0018 моль) антрацена в 12 мл ледяной уксусной кислоты. Содержимое доводят до легкого кипения и из капельной воронки медленно приливают окислительную смесь. Ее готовят заранее из 1 г (0,01 моль) оксида хрома (VI), 1 мл воды и 4 мл уксусной кислоты. После введения всего раствора окислителя, на что требуется приблизительно 1 ч, реакционную массу выдерживают при температуре кипения 15 мин (на окончание реакции указывает окрашивание массы в зеленый цвет). Смесь выливают в стакан с 6 мл воды и оставляют на ночь. Выпавший за это время осадок отфильтровывают, промывают водой и влажный перекристаллизовывают из ледяной уксусной кислоты. Рис.3.28. Установка окисления антрахинона. 1 – реакционная колба; 2 – двурогий форштос; 3 – капельная воронка с окислительной смесью; 4 – обратный холодильник 96 для Либиха. Антрахинон – кристаллическое вещество в виде светло-желтых ромбов, нерастворимых в воде, трудно растворим в спирте, эфире, хорошо – в анилине, горячем толуоле, концентрированной серной кислоте. Т. пл. 2860С. Хроматография: элюент: гексан – этилацетат (17:1). Rf=0.42. Идентификации полученного соединения провести с помощью инфракрасной спектроскопии (см. раздел 3.5). Определите выход полученного вещества, температуру плавления, растворимость и проведите ТСХ и сравните полученные результаты с теоретическими данными. Сделайте соответствующий вывод. Бензойная кислота Реактивы: толуол, перманганат калия, соляная кислота. Оборудование: круглодонная колба вместимостью 100 мл; обратный холодильник, стакан на 100 мл, песчаная баня, колба коническая на 100 мл. В круглодонной колбе, снабженной обратным холодильником (рис. 3.29.), кипятят на песчаной бане в течение 3 ч 1,2 мл толуола с 40 мл воды и 3,4 г мелко растертого перманганата калия. Для равномерного кипения в колбу бросают несколько «кипелок». После завершения реакции раствор в колбе над осадком диоксида марганца должен быть бесцветным. Если реакционная смесь остается окрашенной, обесцвечивания достигают прибавлением 0,2 мл спирта или 0,1 г щавелевой кислоты при нагревании. Горячий раствор фильтруют через складчатый фильтр, осадок диоксида марганца промывают небольшим количеством горячей воды. Фильтрат упаривают в стакане до объема 10-20 мл и отфильтровывают от вновь выпавшего оксида марганца (IV). Рис.3.29. Установка для синтеза бензойной кислоты. 1 – реакционная колба; 2 - обратный холодильник. 3 – песчаная баня. 97 Промыв осадок 5 мл горячей воды, объединенный фильтрат подкисляют концентрированной соляной кислотой до кислой реакции по индикаторной бумаге. При этом осаждается бензойная кислота, которую отфильтровывают, промывают небольшим количеством холодной воды и сушат. Бензойная кислота – белое кристаллическое вещество в виде пластинок, плохо растворяется в холодной воде, лучше – в горячей. Хорошо растворяется в хлороформе, ацетоне, бензоле. Т. пл. 1220С. Хроматография: адсорбент: Sorbfil; элюент: петролейный эфир – этилацетат – ледяная уксусная кислота (17:2:1). Rf=0.2. Определяют выход полученного вещества, температуру плавления, растворимость и проводят ТСХ и сравнивают полученные результаты с теоретическими данными. Сделайте соответствующий вывод. Монокалиевая соль сахарной кислоты Реактивы: глюкоза, 25%-ная азотная кислота, карбонат калия, уксусная кислота (ледяная); активированный уголь. Оборудование: чашка фарфоровая, стакан на 50 мл – 2 шт., водяная баня. В фарфоровой чашке смешивают 2 г глюкозы с 10 мл 25% раствора азотной кислоты и нагревают на слабо кипящей водяной бане, непрерывно помешивая жидкость стеклянной палочкой. Смесь выдерживают на водяной бане до прекращения выделения оксидов азота (признак завершения реакции окисления) и достижения содержимым консистенции сиропа желтовато-коричневого цвета. Затем к сиропообразной массе для растворения добавляют 1 мл воды. Нагревая на водяной бане, смесь нейтрализуют порошком карбоната калия до щелочной реакции по лакмусу. При этом получается хорошо растворимая средняя калиевая соль сахарной кислоты. Раствор охлаждают и при перемешивании стеклянной палочкой прибавляют по каплям ледяную уксусную кислоту до тех пор, пока раствор не станет пахнуть уксусной кислотой, это указывает на то, что вся средняя соль перешла в кислую. После чего раствор оставляют стоять на ночь. Выпавшие окрашенные кристаллы монокалиевой соли сахарной кислоты отфильтровывают, переносят в небольшой стакан, растворяют в возможно меньшем количестве воды, добавляют мелкий активированный уголь и кипятят 13 минуты. Горячую смесь фильтруют через складчатый фильтр в маленькую колбу или стаканчик. При охлаждении образуются бесцветные кристаллы сахарнокислого калия. Их отделяют фильтрованием и сушат на воздухе. Сахарная кислота растворима в воде, спирте, не растворима в эфире. Т.пл. 1250С (с разложением). 98 Определите выход полученного вещества, температуру плавления, растворимость и сравните полученные результаты с теоретическими данными. Сделайте соответствующий вывод. Идентификацию полученного соединения проведите с помощью инфракрасной спектроскопии (см. пункт 3.5.). Лабораторная работа №20 «Восстановление» Реакции, состоящие в присоединении электронов атомами или ионами, т.е. сопровождающиеся понижением степени окисления, называют восстановлением. В органической химии, однако, по традиции эти широко распространенные процессы связывают с присоединением водорода к молекуле органического соединения. Если присоединение водорода приводит к частичному или полному насыщению кратных связей, то такие реакции называют гидрированием, а удаление кислорода из органических молекул (элиминирование) – собственно восстановлением. Восстановление осуществляют различными по характеру веществами: водородом в присутствии катализаторов; действием металлов в присутствии соединений, содержащих подвижный атом водорода; электролитически; соединениями, способными изменять степень окисления одного из своих атомов. Наиболее распространенным восстановителем является водород в момент выделения (атомарный водород), а также молекулярный водород в присутствии катализаторов (никель Ренея, платиновая чернь, палладий на угле и др.). Для восстановления карбонильных соединений используют гидриды металлов (LiAlH4, NaBH4 и др.). Группу восстановителей, содержащих серу в степени окисления -2 (H2S, Na2S, K2S, NaHS и др.) используют главным образом для восстановления в динитросоединениях одной из нитрогрупп. В лабораторной практике чаще всего в качестве восстановителей применяют металлы (Zn, Sn, Fe) в кислой, щелочной и нейтральной средах. Практическая часть Фенилгидроксиламин 99 Реактивы: нитробензол, цинковая пыль, хлорид аммония, хлорид натрия. Оборудование: стакан фарфоровый вместимостью 500 мл, мешалка, баня водяная, термометр. В фарфоровом стакане, снабженном мешалкой и термометром, помещают раствор 4,5 г хлорида аммония в 135 мл воды и 7,1 мл нитробензола. Смесь перемешивают и в течение 15 мин к ней добавляют 15,5 г цинковой пыли. Если цинковая пыль активная, температура самопроизвольно повышается до 60-650С. В противном случае реакционную массу нагревают до этой температуры на водяной бане. После добавления последней порции цинковой пыли содержимое стакана перемешивают еще 15 мин до окончания реакции, о чем судят по исчезновению запаха нитробензола и снижению температуры реакционной массы. Теплый раствор фильтруют, осадок промывают 20 мл горячей воды. Затем фильтрат насыщают 50 г хлорида натрия и охлаждают в смеси льда и соли. Фенилгидроксиламин выделяется в виде длинных светло-желтых игл, которые отфильтровывают и сушат на фильтровальной бумаге при температуре 40-500С. Для очистки фенилгидроксиламина от содержащихся в нем примесей минеральных солей его перекристаллизовывают из бензола. Фенилгидроксиламин – бесцветные иглы, т.пл. 820С (разл.), легко растворяется в эфире, спирте, хлороформе, горячем бензоле, умеренно растворяется в воде. Хроматография: элюент: ацетон - петролейный эфир – бензол - метанол – водный раствор аммиака (концентрированный) (100:200:50:5:2). Проба в растворе ацетона. Rf=0.68. Идентификации полученного соединения провести с помощью инфракрасной спектроскопии (см. раздел 3.5) Список рекомендуемой литературы к разделу «Методы синтеза органических соединений» 1. Гиттис С.С., Глаз А.И., Иванов А.В. Практикум по органической химии. Органический синтез. – М.: Высшая школа, 1991. 2. Гинзбург О.Ф. Практикум по органической химии. Синтез и идентификация органических соединений. - М.: Высшая школа, 1989. 3. Травень В.Ф. Органическая химия. В 2 т. – М.: Изд. Академкнига, 2001. 4. Реутов О.А., Курц А.Л., Бутин К.П. Органическая химия. – М.: Изд. Московского университета, 1992. 5. Грандберг И.И. Органическая химия. –М.: Дрофа, 2002. 100 3.4. Идентификация органических соединений Приступать к идентификации и установлению строения вещества можно только после установления его индивидуальности. Следует особо подчеркнуть, что независимо от метода получения, степени апробированности методики синтеза полученное вещество необходимо подвергнуть тщательной очистке. Это требование обусловлено спецификой органических реакций, неизбежным, как правило, протеканием побочных реакций, которые в некоторых случаях даже при весьма незначительных отклонениях от стандартных условий проведения синтеза могут стать главными и тем самым привести к получению совершенно другого вещества или смеси веществ. Идентификация органических соединений обычно проводится сопоставлением физико-химических констант, растворимости, хроматограмм или спектров полученных веществ с табличными константами, растворимостью, хроматограммами и спектрами эталонов с использованием физико-химических методов анализа. • Предварительные испытания Прежде всего отмечают агрегатное состояние вещества, окраску и запах. Для твердых веществ устанавливают состояние (кристаллическое или аморфное) и форму кристаллов (призмы, плстинки и т.д.). Затем определяют физико-химические константы: температуры плавления или кипения, коэффициента преломления, плотности (см. лабораторную работу №2). • Исследование растворимости Издавна известно, что подобное растворяется в подобном, т.е. вещество лучше растворяется в тех растворителях, к которым оно ближе по своей природе. Вследствие этого изучение растворимости органических соединений в растворителях различной природы может дать первоначальные сведения о его структуре. Все измерения проводят при комнатной температуре с 0,02-0,03 мл жидкости или 4-6 мг твердого тонкоизмельченного вещества и 0,2 мл растворителя, при этом смесь растирают палочкой и сильно встряхивают. Затем делаю вывод о растворимости исследуемого вещества. • Установление индивидуальности Чистоту и индивидуальность контролируют с помощью тонкослойной хроматографии (см. лабораторную работу №5). Для этого находят экспериментальный коэффициент подвижности (Rf) исследуемого вещества и сравнивают со справочными данными. • Установка структуры Общее число физико-химических методов анализа довольно велико – оно составляет несколько десятков. Наиболее практическое значение среди них имеют следующие: 1. спектральные и другие оптические методы; 2. электрохимические методы; 101 3. хроматографические методы анализа; Среди указанных трех групп наиболее обширной по числу методов и важной по практическому значению является группа спектральных и других оптических методов анализа. Он включает в себя методы инфракрасной спектроскопии, спектрофотометрии, люминесценции и другие методы, основанные на измерении различных эффектов, возникающих при взаимодействии вещества и электромагнитного излучения. В группу хроматографических методов входят методы тонкослойной хроматографии, масс-спектральные и многие другие. Инфракрасная спектроскопия Облучение исследуемого вещества инфракрасным излучением (отсюда и название) приводит к изменению колебательно-вращательных движения свободных или взаимодействующих молекул, а также отдельных связей в сложных молекулах. В спектрограмме это проявляется в виде набора полос, положения которых может быть охарактеризовано значениями волновых чисел, длин волн или частот. Экспериментальные исследования большого числа веществ, в молекулы которых входят одни и те же группы атомов, показали, что независимо от различий в остальной части молекул одинаковые группы поглощают в достаточно строгом диапазоне частот. Эти частоты называют характеристическими. Характеристические полосы важнейших органических соединений образуют большие таблицы. Задача экспериментатора провести анализ характеристичных полос и определить наличие в соединениях тех или иных структурных фрагментов. Особенно это касается случаев, когда анализируют вещество с неустановленной структурой. В том случае, когда необходимо подтвердить структуру синтезированного вещества, полученного по известной и отработанной методике, нужно сравнить экспериментальный ИК-спектр полученного вещества с ИК-спектром эталона. Далее приведены ИК-спектры (рис. 3.30. – 3.33.) эталонов, синтез которых осуществляется в разделе «Методы синтеза органических соединений». 102 Рис.3.30. ИК-спектр м-нитроанилина. Рис.3.31. ИК-спектр антрахинона. 103 Рис.3.32. ИК-спектр сахарной кислоты. Рис.3.33. ИК-спектр фенилгидроксиламина. 104 Масс-спектрометрия Данным методом изучают поток ионов, полученных при ионизации атомов или молекул исследуемого вещества. Этот поток делится по скоростям, которые в свою очередь делятся по массам. В результате можно зафиксировать распределение по массам, т.е. можно узнать массу иона. Ионизация молекул в газе происходит под действием потока электронов. Наиболее вероятными являются процессы образования однозарядных положительных ионов: М + е- = М+ + 2е Образование двух- и более высокозаряженных ионов, а также захват электрона с образованием отрицательных ионов является менее вероятным. Масс-спектральный анализ основан на способности газообразных ионов разделяться в магнитном поле в зависимости от отношения m/e, где m – масса, е – заряд иона. По величине m/e определяют массовое число иона, а по интенсивности соответствующего сигнала судят о концентрации ионов. Для создания потока ионов, необходимо испарить вещество. Ионизированные ионы разгоняются по определенной скорости и фиксируются в определенном направлении, что приводит к образованию пучка. Затем из пучка по разным скоростям разделяют ионы, которые детектируются и расшифровываются. Идентификация веществ с помощью масс-спектрометрии осуществляется сопоставлением молекулярной массы образующегося при ионизации молекулярного иона с теоретической молекулярной массой предполагаемого исследуемого вещества. Например, молекулярная масса о-нитротолуола составляет 137 г/моль, поэтому в его масс-спектре, изображенном на рис. 42, имеется пик молекулярного иона 137 m/z. Рис.3.34. Масс-спектр о-нитротолуола. Также идентификацию веществ можно осуществить с помощью химических методов анализа. Данные методы заключаются в использовании химических превращений. 105 К химическим методам анализа относится «встречный» синтез, который заключается в получении анализируемого вещества другим способом. Далее проводят сопоставление физических констант полученных веществ различными способами. Если константы совпадают, то делают вывод об их идентичности, что является доказательством структуры. Также для доказательства структуры анализируемого вещества можно осуществить дальнейшее его превращение в соединение с доказанной или известной структурой. Далее проводится идентификация или сравнение полученного вещества с эталоном. Например, для доказательства синтезируемой β-нафталинсульфокислоты используют ее превращение в β-нафтол. Затем полученный продукт анализируют и сопоставляют физические константы либо с литературными данными либо эталоном, в данном случае с β-нафтолом. Список рекомендуемой литературы к разделу «Идентификация органических соединений» 1. Гиттис С.С., Глаз А.И., Иванов А.В. Практикум по органической химии. Органический синтез. – М.: Высшая школа, 1991. 2. Гинзбург О.Ф. Практикум по органической химии. Синтез и идентификация органических соединений. - М.: Высшая школа, 1989. 3. Артеменко А.И. Практикум по органической химии. - М.: Высшая школа, 2001. 4. Шрайнер Р. Идентификация органических соединений. Под ред. Б.А. Руденко. – М.: Госхимиздат, 1956. 106 Библиографический список 1. Артеменко А.И. Практикум по органической химии. - М.: Высшая школа, 2001. 2. Гинзбург О.Ф. Практикум по органической химии. Синтез и идентификация органических соединений. - М.: Высшая школа, 1989. 3. Гиттис С.С., Глаз А.И., Иванов А.В. Практикум по органической химии. Органический синтез. – М.: Высшая школа, 1991. 4. Грандберг И.И. Органическая химия. –М.: Дрофа, 2002. 5. Животовская Г.П., Тихонов С.С. Органическая химия и биохимия: учебное пособие. – Челябинск: Изд. ЮУрГУ, 2003. 6. Иванов В.Г., Гева О.Н., Гаверова Ю.Г. Практикум по органической химии.М.: Академия, 2000. 7. Прянишников Н.Д. Практикум по органической химии для вузов/под ред. Успенского А.Е. – М.: Госхимиздат, 1956. 8. Реутов О.А., Курц А.Л., Бутин К.П. Органическая химия. – М.: Изд. Московского университета, 1992. 9. Реутов О.А., Курц А.Л., Бутин К.П. Органическая химия. – М.: Изд. Московского университета, 1992. 10. Тикунова И.В., Артеменко А.И., Малеванный В.А. Справочник молодого лаборанта-химика. – М.: Высшая школа, 1985. 11. Травень В.Ф. Органическая химия. В 2 т. – М.: Изд. Академкнига, 2001. 12. Тюкавкина Н.А. Руководство к лабораторным занятиям по биоорганической химии. – М: Медицина, 1985. 107 Приложение 1. МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ ФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ОБРАЗОВАНИЮ ЮЖНО-УРАЛЬСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ КАФЕДРА «ОРГАНИЧЕСКАЯ ХИМИЯ» Отчеты по лабораторному практикуму «Органическая химия» Выполнил: Ф.И.О. Студент (ка) _______группы Проверил: Ф.И.О. преподавателя г. Челябинск, 200_г. 108 Приложение 2. Методика приготовления реактивов для проведения лабораторных работ по органической химии. Аммиачный раствор оксида меди (I). К раствору 1 г медного купороса в 50 мл воды прибавляют 4 мл концентрированного раствора аммиака, затем раствор 3 г солянокислого гидроксиламина в 50 мл воды и хорошо перемешивают. Полученный раствор можно хранить несколько дней в хорошо закрытой склянке (в темноте). Срок хранения можно увеличить, добавив в раствор медную стружку. Раствор можно приготовить другим способом. Для этого предварительно готовят два раствора: а) 1,5 г хлорида меди (П) и 3 г хлорида аммония растворяют в 20 мл концентрированного раствора аммиака и добавляют воды до общего объема 50 мл; б) 5 г солянокислого гидроксиламина растворяют в 50 мл воды. Перед употреблением растворы (а) и (б) смешивают в объемном соотношении 1 : 2. Аммиачный раствор оксида серебра. К 100 мл 0,1 н. раствора нитрата серебра добавляют 0,5 мл 25%-ного раствора аммиака. Полученный раствор хранят в темной склянке не более 1 месяца. По истечении этого срока реактив, если он полностью не израсходован, осторожно нейтрализуют азотной кислотой и готовят новую порцию. Нельзя допускать испарения реактива и готовить его впрок в больших количествах. Аммиачный раствор хлорида меди (I). 25 г кристаллического сульфата меди (II) растворяют при нагревании в 80 мл воды. К раствору прибавляют 14 г хлорида натрия. К смеси медленно при перемешивании доливают раствор 12,6 г кристаллического сульфита натрия в 20 мл воды. Смеси дают охладиться, затем промывают белый осадок хлорида меди (I) водой (декантацией), растворяют его в 30—40 мл концентрированного раствора аммиака и добавляют 200 мл воды. Бесцветный раствор хранят в плотно закрывающейся склянке над очищенной медной проволокой. Анилин. Для очистки перегоняют (т. кип. 184 °С) с воздушным холодильником из колбы Вюрца, в которую добавляют 0,5—1 г цинковой пыли. Полученную бесцветную жидкость со слабым запахом хранят в плотно закрытой склянке (желательно заполненной почти до пробки). Добавка небольшого количества цинковой пыли стабилизирует анилин. Анилиновая вода. Для проведения опытов по экстракции анилина его перемешивают с водой в объемном соотношении 1 : 20 и оставляют, не разделяя слои. Для исследования химических свойств анилина достаточно его растворить в воде в объемном соотношении 1 : 100. Баритовая вода, насыщенный раствор. 70 г кристаллического гидро-ксида бария Ва (ОН)2 • 8Н20 растворяют при кипячении в 200 мл воды. Затем разбавляют водой до 1 л и дают отстояться. Через несколько часов прозрачный раствор осторожно сливают. Бензол. Бензол марки ч. д. а. достаточно чист для большинства работ. Для удаления следов воды бензол перегоняют, отбрасывая первые 10—20% дистиллята. Т. кип. 80,2 °С. 109 Бром, раствор в хлороформе или тетрахлориде углерода. В склянку с притертой пробкой вносят 5 мл брома (под тягой!) и осторожно добавляют 95 мл растворителя. Склянку хранят в эксикаторе. Работать с бромом нужно в резиновых перчатках и защитных очках. Случайно разлившийся бром засыпают сульфитом или тиосульфатом натрия. Бромид-броматная смесь. 2,8 г бромата калия в 12 г бромида калия растворяют в воде и доводят объем до 1 л (в мерной колбе). Бромоводородная кислота. Раствор, образующийся при поглощении бромоводорода, перегоняют с дефлегматором, собирая фракцию, кипящую при 122— 126 °С. Эта фракция (р 1,47—1,49) представляет собой примерно 47%-ную бромоводородную кислоту. При перегонке сильно разбавленных растворов может получаться кислота меньшей плотности, которую при необходимости повышают повторной перегонкой. Кислоту хранят в хорошо закупоренной склянке в темноте. Бромная вода. Для получения насыщенного раствора брома в воде в склянку с притертой пробкой вместимостью 1 л вносят 6 мл брома, добавляют 500 мл воды и энергично перемешивают. Небольшое количество брома при этом остается нерастворенным (при длительном хранении он исчезает). Для опытов используется раствор, разбавленный водой (1 : 2). Необходимо соблюдать меры предосторожности. Глюкоза, 5%-ный раствор. Готовят не менее чем за 12 ч до начала работы с раствором. Раствор необходимо прокипятить. 2,4-Динитрофенилгидразин солянокислый. 0,2 г 2,4динитрофенилгидразина растворяют при нагревании на водяной бане в 100 мл 2 н. раствора соляной кислоты. Диэтиловый эфир, содержащий пероксиды. В склянку из белого стекла вместимостью 0,5 л помещают 100 мл диэтилового эфира. Добавляют примерно 10 мл концентрированной серной кислоты, взбалтывают смесь и ставят в светлое место (на окно). Ежедневно встряхивают склянку и открывают пробку для доступа воздуха. Через 8—10 дней в эфире можно обнаружить гидропероксид и уксусный альдегид. На склянке делают надпись «Диэтиловый эфир, загрязненный пероксидом. Осторожно!». Железа (III) хлорид, 0,1 н. раствор. 9 г FeCl3 ·6H2О растворяют в 1 л воды. При помутнении добавляют несколько капель концентрированной соляной кислоты. Банку с кристаллическим хлоридом железа необходимо снова залить парафином. Известь натронная. Насыщенный раствор гидроксида натрия перемешивают с негашеной известью (массовое отношение 2 : 1), выпаривают досуха в железном сосуде, затем прокаливают при температуре около 500 °С и измельчают. Препарат хранят в плотно закупоренных банках. Готовую натронную известь перед использованием высушивают при 115 °С (при значительном содержании карбонатов прокаливают в муфельной печи). Иода раствор в иодиде калия (раствор Люголя). 6 г иодида калия растворяют в 6 мл воды, размешивают в этом растворе 2 г иода и разбавляют водой до 100 мл. 110 Известковая вода. Гашеную известь (гидроксид кальция) перемешивают с водой. Хранят вместе с нерастворенным остатком, чтобы раствор был насыщен гидроксидом кальция. Перед использованием прозрачный раствор сливают с осадка в небольшие склянки. Иод крахмальная бумага. 0,5 г крахмала тщательно перемешивают с 10 мл холодной воды. Полученную массу вливают в 100 мл кипящей воды и продолжают кипячение до образования однородного клейстера. После охлаждения добавляют 0,5 г иодида калия и 0,5 г кристаллического карбоната натрия, растворенных в небольшом количестве воды. Полученным раствором пропитывают фильтровальную бумагу и сушат ее на воздухе, избегая попадания солнечных лучей. Высушенную бумагу режут на полоски и хранят в плотно закрытых банках. Калия гидроксид, 0,5 н. спиртовой раствор. Этиловый спирт кипятят 30 мин с гидроксидом калия (10 г КОН в 1 л спирта) в колбе с обратным холодильником, а потом спирт перегоняют (над той же щелочью). Затем в этот спирт при перемешивании добавляют 30—35 г гидроксида калия и через сутки быстро сливают прозрачную жидкость с осадка. Раствор хранят в темной склянке с хлоркальциевой трубкой, заполненной натронной известью. Калия карбонат (поташ). Перед использованием прокаливают при 200 °С. Калия перманганат, кислый раствор. К 100 мл 1%-ного раствора перманганата калия осторожно при перемешивании добавляют 5 мл концентрированной серной кислоты. Кальция карбид. Технический продукт разбивают молотком на кусочки размером около 3—5 мм. Продукт обычно хранят в банке, залитой парафином. Кальция хлорид. Отработанный хлорид кальция регенерируют, расплавляя его на железном противне. Слой должен быть не толще 2 см (пары воды, испаряясь, вызывают разбрызгивание, особенно большое, если слой соли толстый). После испарения всей воды нагревание продолжают еще некоторое время (при температуре 250—300 °С). Спекшуюся соль разбивают на куски и еще теплой кладут в сухую банку. Для заполнения хлоркальциевых трубок используют кусочки размером 2—5 мм. Конго красный. Готовят 0,1%-ный раствор в воде. Раствор конго красного (для крашения). Растворяют 1 г конго красного,1 г карбоната натрия и 10 г сульфата натрия в 400 мл воды. Крахмал, 1%-ный раствор. 1 г сухого крахмала взбалтывают с 5 мл воды, после отстаивания воду сливают. Промывание крахмала повторяют 2—3 раза. Добавив новую порцию воды, взмучивают крахмал и выливают при помешивании в 100 мл кипящей воды Крахмальный клейстер. 2 г растворимого крахмала смешивают с 10 мл холодной воды и вливают эту кашицу в 100 мл кипящей воды. Кипятят 5—10 мин, охлаждают, дают отстояться и, если образуется хлопьевидный осадок, сливают с него раствор (или фильтруют). Для стабилизации добавляют 0,1 г КОН или салициловой кислоты (можно также 0,01 г иодида ртути). Лактоза, 1%-ный раствор. После приготовления кипятят несколько минут. 111 Меди сульфат (безводный). Медный купорос нагревают в фарфоровой или никелевой чаше на песчаной бане при постоянном перемешивании. Если температура не превышает 220 °С, получается белый или слегка желтоватый продукт (при нагревании свыше 220 °С сульфат меди частично разлагается, приобретая серый цвет; при этом осушающая способность понижается). Высушенный сульфат меди растирают в ступке и хранят в сухой, плотно закрытой банке. Меди (I) хлорид. Реактив можно приготовить двумя способами: а) 25 г медного купороса и 65 г хлорида натрия растворяют при нагревании в 80 мл воды, фильтруют, а затем к фильтрату добавляют раствор 7 г кристаллического сульфита натрия (или 3,5 г безводного) в 40 мл воды. С выпавшего осадка сливают воду и несколько раз промывают (декантацией) 2 н. раствором соляной кислоты до исчезновения сульфатиона (проба с хлоридом бария), а затем спиртом. Соль сушат при 100 °С и хранят в плотно закрытых банках. Препарат очень чувствителен к окислению. Позеленевший препарат, содержащий основные соли меди (П), вновь светлеет при промывке раствором ОД н. соляной кислоты; б) к насыщенному раствору хлорида меди (II) добавляют кусочки меди. В склянке, которую оставляют плотно закрытой на несколько дней, постепенно выпадает белый осадок. Металлическую медь выбирают пластмассовым пинцетом, а осадок обрабатывают, как описано выше. Метиловый красный (метилрот). 0,1 г красителя растворяют в 30 мл этанола, затем добавляют воду до 50 мл. Получают 0,2%-ный раствор индикатора. Переход от красного цвета к желтому происходит при рН= 4,2-6,2. Метиловый оранжевый. 0,1 г индикатора растворяют в 100 мл воды. Натрий металлический. Банку с натрием распаивают с помощью паяльника, натрий режут на куски (работать в резиновых перчатках!) и помещают их в банку с керосином или вазелиновым маслом (можно также использовать ксилол, декалин, тетралин). Для работы куски натрия извлекают из-под керосина пинцетом, быстро осушают фильтровальной бумагой и сухим ножом отрезают кусочек нужной величины. Остальную часть снова помещают в банку. С отрезанного кусочка тщательно удаляют фильтровальной бумагой следы керосина, срезают тонкие корочки, а обрезки помещают в банку с натрием. Брать натрий нужно только пинцетом. Ни в коем случае нельзя работать с натрием поблизости от водопроводного крана. Натрия ацетат (безводный). Кристаллическую соль CH3COONa-3H20 нагревают в фарфоровой или никелевой чашке. Соль плавится в кристаллизационной воде, а по испарении большей части воды застывает. Усиливая нагревание, снова доводят соль до плавления (т. пл. 324 °С). При этом следует избегать сильного перегрева (может начаться разложение). Расплавленную соль выливают на металлическую пластинку с загнутыми краями и после остывания (теплую) измельчают в фарфоровой ступке. Хранят в плотно закрытой банке. Натрия гидроксид (спиртовой раствор). 30 г гидроксида натрия растворяют в 30 мл воды. После охлаждения раствора добавляют этанол (ректификат) до 200 мл. 112 Натрия гидросульфит (натрия бисульфит) NaHS03. Гидрокарбонат или кристаллический карбонат натрия заливают водой так, чтобы она чуть покрывала кристаллы, и пропускают диоксид серы до почти полного растворения кристаллов. Полученный зеленовато-желтый раствор хранят в склянке с притертой пробкой. Диоксид серы получают, добавляя по каплям серную кислоту к сульфиту натрия в колбе Вюрца с высоким отводом. В отвод желательно вложить стекловату для задержки уносимых током газа следов жидкости. В лабораториях бывает гидросульфит натрия технический — белый слеживающийся порошок со слабым запахом сернистого ангидрида. Он обладает сильными восстанавливающими свойствами даже после длительного хранения, но истинным гидросульфитом не является. Его формула Na2S2О4 ·2H2О, а правильное название дитионит натрия. С ацетоном и альдегидами осадков не образует. Натрия гидроксид, 2 н. раствор. 84 г едкого натра (из расчета, что в нем 95% основного вещества) растворяют при перемешивании в 500 мл воды и доводят объем до 1 л. Натрия сульфат (безводный). Реактив высушивают при 200—300 °С. Возможна регенерация отработанного осушителя. n-Нитрофенилдиазонийхлорид (раствор). В стакане смешивают 12,5 мл концентрированной соляной кислоты и 150 мл воды и растворяют при нагревании 7 г п-нитроанилина. Раствор охлаждают в воде со льдом до 00С и затем быстро при энергичном перемешивании приливают охлаж денный раствор 3,5 г нитрита натрия в 20 мл воды. Реакционную смесь оставляют в ледяной воде на 30—40 мин. Раствор диазотированного п -нитроанилина (желтого цвета) должен быть прозрачным. В холодиль нике он может храниться в течение 10—12 дней. Нитрующая смесь. Смешивают 2 объема концентрированной азотной кислоты (р 1,42 г/см3) с 3 объемами концентрированной серной кислоты (р 1,84 г/см3), медленно добавляя серную кислоту к азотной при охлаждении и перемешивании. Орциновый реактив. 1 г орцина растворяют в 500 мл 25%-ного раствора соляной кислоты и добавляют 20 капель 3%-ного раствора хлорида железа (III). Реактив Селиванова. 50 мл концентрированной соляной кислоты смешивают с 50 мл воды. В 100 мл полученного раствора соляной кислоты (1:1) растворяют 0,5 г резорцина. Используют в реакциях на кетогексозы. Реактив Толленса. К 20 мл 5%-ного раствора нитрата серебра прибавляют 10 мл 10%-ного раствора гидроксида натрия. К выпавшему осадку оксида серебра добавляют 5 мл разбавленного раствора аммиака (10 мл концентрированного раствора аммиака разбавляют 100 мл дистиллированной воды). Колбу закрывают пробкой и встряхивают. Повторяют операцию до полного растворения осадка, избегая избытка аммиака. Затем добавляют воду до объема 100 мл. Сахароза, 1%-ный раствор. Растворяют 10 г сахарозы (или пищевого сахара) в 1 л воды, фильтруют и кипятят несколько минут. Серебра нитрат, 0,2 н. раствор. 3,4 г нитрата серебра растворяют в 20 мл воды и доводят объем до 100 мл. Для приготовления раствора используется бидистиллят. Посуда должна быть очень тщательно отмыта от возможных 113 органических загрязнений. Хранят раствор в темной склянке с притертой пробкой. Серебра остатки, регенерация. Все растворы и осадки, содержащие серебро, подлежат сбору и переработке. Ацетиленид серебра и «серебряное зеркало» растворяют в разбавленной азотной кислоте. К остаткам серебра прибавляют раствор гидроксида натрия до щелочной реакции по фенолфталеину, нагревают на водяной бане и добавляют формалин. Через 5—15 мин выделяется темно-серый рыхлый порошок металлического серебра. Его отсасывают, промывают водой до удаления щелочи и хлорид-ионов и сушат при 100 °С. К навеске высушенного осадка прибавляют избыток азотной кислоты (1 мл азотной кислоты плотностью 1,42 г/см3 или 1,5 мл плотностью 1,31 г/см3 на 1 г серебра) и нагревают при 50 °С до растворения. Жидкость фильтруют и упаривают на водяной бане до появления кристаллической пленки. Если за это время оксиды азота не удалились полностью, добавляют воду и упаривают вновь. Упаренный раствор охлаждают, кристаллы нитрата серебра отсасывают, промывают небольшим количеством ледяной воды и сушат при 110 °С. Маточный раствор и промывные воды можно упарить и выделить дополнительное количество нитрата серебра или же слить в склянку с остатками серебра. Серная кислота 2 н. 56 мл концентрированной серной кислоты (р 1,84 г/см3) медленно прибавляют к 944 мл воды. Соляная кислота 2 н. 167 мл концентрированной соляной кислоты (р 1,19 г/см3) разбавляют водой до 1 л. Углеводороды насыщенные жидкие. Образцом смеси жидких предельных углеводородов может служить петролейный эфир (tкип ~ +60 °С), который представляет собой смесь углеводородов, главным образом смесь пентана и гексана. Перед работой проверяют петролейный эфир на отсутствие непредельных углеводородов реакциями с бромной водой и с раствором перманганата калия. Насыщенные жидкие углеводороды можно выделить из бензина или керосина. Для отделения насыщенных углеводородов от ненасыщенных (и от других примесей) бензин (или керосин) обрабатывают 3—4 раза концентрированной серной кислотой в делительной воронке (на 100 мл бензина или керосина берут 10 мл серной кислоты). Вначале углеводороды с серной кислотой встряхивают осторожно, затем, когда смесь перестанет разогреваться, сильно. Пары и газы выпускают, как обычно, приоткрывая кран (воронка в этот момент должна быть перевернута краном вверх). Обработку углеводородов каждой порцией серной кислоты ведут в течение 5—10 мин. После серной кислоты углеводороды обрабатывают в той же делительной воронке 1%ным раствором перманганата калия, к которому добавлен 5%-ный раствор карбоната натрия. В заключение их промывают водой. После отделения воды (нижнего слоя) углеводороды сливают в колбу и сушат гранулированным хлоридом кальция. Приготовленная таким способом смесь углеводородов состоит из алканов и циклопарафинов (нафтенов), которые по многим свойствам похожи на алканы; в приведенных в практикуме опытах присутствие циклопарафинов не обнаруживается. 114 Углеводороды ненасыщенные жидкие. Для опытов с жидкими ненасыщенными углеводородами можно использовать бензин или керосин: в них содержится некоторое количество смеси алкенов. Больше всего алкенов в крекинг-бензине. Если бензин (керосин) не бесцветен, его перегоняют, собирая бесцветный дистиллят в широких температурных пределах. Уксусная кислота 2 н. 116 мл ледяной уксусной кислоты разбавляют водой до 1 л. Фелингова жидкость (реактив Фелинга). Готовят два раствора. Раствор I. 34,6 г чистого перекристаллизованного сульфата меди растворяют в воде, содержащей несколько капель серной кислоты, и разбавляют до 500 мл. Раствор II. 173 г сегнетовой соли (тартрат натрия-калия) растворяют в 200 мл воды, добавляют раствор 70 г гидроксида натрия (или 85 г гидроксида калия) в 100 мл воды и разбавляют до 500 мл. Фелингову жидкость готовят непосредственно перед каждой лабораторной работой, смешивая равные объемы растворов I и П (исходные растворы могут храниться неограниченно долго). Если отсутствует сегнетова соль, то для приготовления раствора П растворяют 121 г гидроксида натрия и 93,1 г чистой винной кислоты в 400 мл воды и доводят объем до 500 мл. Фенилгидразин уксуснокислый (раствор). 10 г солянокислого фенилгидразина и 20 г ацетата натрия растворяют при небольшом нагревании в 200 мл воды. Теплый раствор фильтруют. (Фенилгидразин вызывает экзему, поэтому работа с ним требует соблюдения правил техники безопасности.) Фенолфталеин. 0,2 г фенолфталеина растворяют в 165 мл спирта и добавляют 135 мл воды. Фишера реактив. В сухую склянку из темного стекла вносят 200 мл пиридина, растворяют в нем 100 г иода и добавляют 600 мл метанола. Склянку плотно закрывают и оставляют на двое суток. Затем склянку взвешивают и помещают в баню со льдом, а через раствор пропускают диоксид серы из баллона до увеличения массы на 45—50 г. Приготовленный таким образом реактив выдерживают перед использованием не менее недели. Хранят, хорошо защищая от влаги. Флороглюцин (раствор). 0,4 г флороглюцина растворяют в 200 мл 30%-ной соляной кислоты. Фруктоза, 0,5%-ный раствор. После приготовления раствор кипятят. Фурфурол. Соломенно-желтая жидкость, быстро темнеющая на воздухе. Очищают перегонкой, собирая фракцию, кипящую при 159—163 °С (т. кип. чистого фурфурола 162 °С). Окисление (потемнение) фурфурола замедляют добавкой 0,1% гидрохинона или пирогаллола. Фуксинсернистая кислота (раствор). 0,2 г фуксина растворяют в 200 мл дистиллированной воды и вводят 2 г гидросульфита натрия и 2 мл концентрированной соляной кислоты. Если через 15—20 мин жидкость не обесцветится, то добавляют немного активированного угля, встряхивают смесь до обесцвечивания и затем фильтруют. Реактив хранят в плотно закрывающейся 115 склянке из темного стекла. Чем меньше избыток оксида серы (IV) в реактиве, тем он чувствительнее. Хромовая смесь (смесь Бекмана). В 100 мл воды растворяют 20 г дихромата калия и добавляют 10 мл концентрированной серной кислоты. Такая хромовая смесь (смесь Бекмана) предназначена для окисления спиртов и анилина и не годится для мытья посуды. Щавелевая кислота, 0,01 н. В мерной колбе вместимостью 1 л растворяют в 100 мл воды 0,63 г свежеперекристаллизованной щавелевой кислоты. Затем добавляют 1 мл 30%-ной H2S04 и разбавляют водой до метки. Швейцера реактив (медно-аммиачный реактив). К раствору 10 г медного купороса в 100—200 мл воды добавляют 100 мл 2 н. раствора гидроксида натрия. Образовавшийся осадок отсасывают, промывают несколько раз водой до отрицательной реакции на сульфат-ион и растворяют в минимальном количестве 25%-ного раствора аммиака. Часть гидроксида меди при этом должна остаться нерастворенной. Раствору дают отстояться и сливают декантацией. Эмпирическая формула реактива [Cu(NH3)4](OH)2. Эфир абсолютный. Эфир проверяют на наличие пероксидов, встряхивая его с равным объемом 2%-ного раствора иодида калия, подкисленного разбавленной соляной кислотой. Присутствие пероксидов определяют по синей окраске водного слоя при добавлении раствора крахмала. (Подкисленный серной кислотой раствор ванадата аммония с эфиром, содержащим пероксиды, окрашивается в красный цвет, а такой же раствор дихромата калия — в синий.) Если пероксиды отсутствуют, приступают к осушке, если они есть, от них избавляются встряхиванием раствора с порошкообразным гидроксидом калия (70 г на 1 л). После отстаивания эфир сливают, добавляют 100 г хлорида кальция и через сутки фильтруют. Затем в эфир вносят около 5 г металлического натрия в виде тонко нарезанных листочков или проволоки, выдавливаемой из пресса. Если через 24 ч не наблюдается выделения пузырьков водорода, то осушка считается законченной, если же водород выделяется, добавляют еще 2—3 г натрия. Эфир можно перегнать на водяной бане над натрием, предохраняя его от атмосферной влаги, но можно обойтись и без перегонки, лишь слив его в сухую склянку. Склянку с эфиром закрывают корковой пробкой с хлоркальциевой трубкой. Для предотвращения окисления можно внести несколько крупинок дифениламина или фосфорного ангидрида, или, еще лучше, несколько гранул гидроксида калия, который действует одновременно и как осушитель. 116 Приложение 3. Характеристические частоты колебаний в ИК-спектрах основных классов органических соединений Соединение Алканы Частота Интен- Отнесение и характер колебаний υ, см-1 2975— 2950 сивность* с. υ as CH3 2885— 2860 с. υ as CH3 1470— 1435 1385— 1370 ср. δas сн3 с. δs сн2 расщепляется в дублет для гемдиметильной группы 2940— 2915 2870— 2845 1480— 1440 с. υ as CH2 с. υ s CH2 ср. δ CH2 2830—2815 в ОСН3 2820—2730 в NCH3 117 Соединение Частота υ, см-1 750—720 2900—2880 Интенсивность* с. сл. Отнесение и характер колебаний Маятниковое -СН2υ CН 1340 сл. δсн ср. ср. ср. с. с. ср. ср. с. ср. ср. ср. ср. υ as CH2 υ s CH2 υ C=C δСН неплоские δСН2 неплоские δсн2 υ C=C 5δСН2 неплоские υ CН υ C=C δСН неплоские υ CН 1675—1665 980—960 3040—3010 1675—1665 850—790 1690—1670 1650 1600 ~1625 1660—1580 ср. с. ср. п. и. с. сл. с. сл. с. с. υ C=C δСН неплоские υ CН υ C=C δСН неплоские υ C=C υ C=C υ C=C υ C=C υ C=C 3310—3300 2140—2100 2260—2190 3080—3030 1625—1575 1525—1475 1465—1440 1590—1575 с. сл. сл. ср. п. и. п. и. п. и. п. и. υ CН υ C≡C υ C≡C υ CН 770—730 710—690 с. Алкены R—СН=СН2 3095—3010 2975 1645—1640 995—985 915—905 RR'C=CH2 3095—3075 1660—1640 895—885 RCH=CHR' (цис) 3040—3010 1665—1635 730—665 RCH=CHR' (транс) 3040—3010 RR'C=CHR" RR'C=CR"R" Диены С=С—Ar С=С—С=О Алкины RC≡CH RC≡CR' Ароматические соединения Монозамещенные Колебания кольца Появляется в сопряженных соединениях δСН неплоские Соединение 1,2-Замещенные 1,3-Замещенные 1,4- и 1,2,3,4Замещенные 1,2,3-Замешенные 1,2,4-Замещенные 1,3,5-Замешенные Спирты и фенолы Частота υ, см-1 770—735 900—860 810—750 725—680 860—800 Интенсивность* с. ср. ср. ср. с. 800—770 720—685 860—800 900—860 с. ср. с. ср. Кетоны R—CO—R' С=С—СО—R С=С—СО—С=С Циклопентаноны Аr—СО—R Аr—СО—Аr CHal—CO—R —СО—СН2—СО— енольная форма кетонная форма Кислоты R—СООН δСн неплоские δсн неплоские δСН неплоские 900—860 ср. 865—810 c. 730—675 3670—3580 п. и. δсн неплоские 3590—3420 п. и. υ 0H, свободная группа ОН, узкая полоса υ OH, межмолекулярная водородная связь димеров, узкая полоса υ 0H, межмолекулярная водородная связь полиассоциатов, широкая полоса υ 0H. внутримолекулярная водородная связь, узкая полоса 3200—2500 cл. υ 0H, хелаты, 1740—1720 с υ C=O 1705—1685 с. 1715—1695 с. 2880—2650 ср. υ C=O υ C=O υ CH, во всех альдегидах; могут быть две полосы 1725—1700 1695—1660 1670—1660 1750—1740 1700—1680 1670—1650 1745—1725 1640—1535 3200—2700 -1720 υ υ υ υ υ υ υ υ υ υ 3400—3200 с. С=С—СНО Аr—СНО δсн неплоские δСн неплоские 3550—3450 п. и. Альдегиды R—СНО Отнесение и характер колебаний с. с с. с. с. с. с. с. п. и. с. 1725—1700 с. υ 119 C=O C=O C=O C=O C=O C=O C=O C=O 0H C=O C=O очень широкая полоса Соединение Частота υ, см-1 1715—1680 1700—1680 1740—1715 3300—2500 1610—1550 1420—1300 Интенсивность* с. с. с. п. и. с. ср. υ C=O υ C=O υ C=O υ 0H, широкая, для всех кислот υ as COOυ s COO- 1750-1735 1730—1715 с. с. υ υ 1800—1770 с. υ 1840—1800 1780—1740 1820—1780 с. с. с. υ υ υ 1760—1720 с. υ 1870—1830 1800—1760 1850—1810 1795—1740 с. с. с. с. υ υ υ υ 1815—1785 1800—1770 с. с. υ υ R—CONH2 3540—3200 с. υNH2, две полосы в разбавленных растворах R—CONHR' 1690—1650 1650—1590 3460—3100 с. с. п. и. Полоса «Амид I» Полоса «Амид II» υ NH, одна полоса в разбавленных растворах R—CONR2 —NH—CO—NH— —СО—NH—СО- 1680—1630 1570—1510 1670—1630 1660 1790—1720 1720—1670 с. с. с. с. с. с. Полоса «Амцд I» Полоса «Амид II» Полоса «Амид I» Полоса «Амид I» С=С—СООН Ar—COOH CHal—СООН Соли кислот Сложные эфиры R—COOR С=С—COORН Ar —COOR —СООС=С и —СООАr Ангидриды: ациклические насыщенные ациклические сопряженные ангидриды с пятичленным циклом: предельные непредельные Галогенангидриды R—CO—Hal С=С—СО—Hal Отнесение и характер C=O C=O C=O C=O C=O C=O C=O C=O C=O C=O C=O C=O C=O Амиды кислот 120 Соединение Частота υ, см-1 Интенсивность* Отнесение и характер Амины R—NH2 3500—3300 п. и. υNH2 , в разбавленных растворах две полосы R2NH 1650—1580 п. и. 900—650 ср. 3600—3300 п.и. δNH2, неплоские δNH2, плоские υNH, в разбавленных растворах одна полоса R—NH3 1650—1550 ел. 3350—3150 ср. δNH υNH3 , широкая полоса или группа полос δ NH3+ -1600 ср. R2NH2 -1300 ср. 2700—2250 с. 1620—1560 ср. υNH2+ δNH2+ R3NH 2700—2250 ср. υNH+ Непредельные азотсодержащие соединения R2C=N С=С—C=N C=N в цикле R—C=N C=C—C=N Ar—C=N Ar—NT=N Нитросоединения R—N02 1690—1635 1665—1630 1660—1480 2260—2240 2235—2215 2240—2220 2300—2230 п. и. п. и. п. и. п. и. с. с. с. υC=N υC=N υC=N υC=N υC=N υC=N υ CN2 1565—1545 1385—1360 1530—1510 1360—1335 1550-^1510 1365—1335 860—840 -750 с. с. с. с. с. с. с. с. υas N02 υs N02 υas N02 υs N02 υas N02 υs N02 C=C—N02 Ar—N02 121 . Соединение R—0—N02 Частота υ, см-1 1655—1610 1300—1255 Серосодержащие соединения R—SH 2590—2550 R2SO 1070—1030 R2S02 1350—1300 1160—1120 R—S03H 1260—1150 1080—1010 700—600 R—S02NR2 1370—1300 1180—1140 Гетероциклические соединения фуран 3175—3137 3137—3112 1615—1639 1500 1274—1253 1109—1092 880—859 842—812 тиофен 3125—3050 1520 1040 750—640 пиррол, индол 3440—3400 1565 1500 пиридин 3070—3020 900—670 1650—1580 1580—1550 1570—1480 Кремнийорганические соединения Si—H ~2100 Si—H ~2100 Интенсивность* с. с. Отнесение и характер υas N02 υs N02 cл. с. оч. с. оч. с. с. с. с. с. с. υSH υSO υas S02 υs S02 υas S02 υs S02 с. с. с. с. с. с. с. υCH с. с. υCH Колебания кольца оч. с. ср. п. и. п. и. с. Самая сильная из полос υNH В растворах Колебания кольца ср. сл. ср. υas S02 υs S02 Колебания кольца υC-O Наиболее характерны для фуранов υCH и δСН Колебания кольца с. υSi-H с. υSi-H 122 Соединение Частота υ, см-1 ИнтенОтнесение и характер колебаний сивность* Si—СН3 1260 800 с. δсн3 Si(CH3)2 1260 815—800 1250 840 755 1430—1425 1135—1090 1090—1020 810 3690 с. с. с. с. с. с. с. с. с. с. δсн3 Si(CH3)3 Si—С6Н5 Si—О—Si Si—О—С— Si—CI Si—OH δсн2 υSi-O υOH * с.—сильная; ср.—средняя; cл.—слабая; п. и.—переменной интенсивности. 123 СОДЕРЖАНИЕ 1. ОСНОВНЫЕ ПРАВИЛА И ОРГАНИЗАЦИЯ РАБОТЫ В ЛАБОРАТОРИИ ОРГАНИЧЕСКОЙ ХИМИИ .................................................................................. 3 1.1. Техника безопасности .................................................................................. 3 1.2. Правила оформления, ведения рабочего (лабораторного) журнала и составление отчета............................................................................................ 12 2. ЛАБОРАТОРНАЯ ХИМИЧЕСКАЯ ПОСУДА .............................................. 13 2.1. Перечень и краткое описание лабораторной посуды ............................. 13 2.2. Правила сборки установок для выполнения органических синтезов ... 19 3. ПРАКТИЧЕСКАЯ ЧАСТЬ ............................................................................... 20 3.1. Изучение состава органических соединений, их очистка и определение физических констант ......................................................................................... 20 Лабораторная работа №1 «Методы очистки и выделения органических веществ» .......................................................................................................... 20 Лабораторная работа №2 «Определение физических констант органических соединений» ........................................................................... 29 Лабораторная работа № 3 «Качественный элементный анализ» .............. 36 Лабораторная работа № 4 «Функциональный анализ» .............................. 40 Лабораторная работа №5 «Тонкослойная хроматография» ...................... 42 3.2. Ознакомительный (малый) практикум. .................................................... 47 Лабораторная работа №6 «Алифатические углеводороды» ...................... 47 Лабораторная работа №7 «Галогеналканы» ................................................ 50 Лабораторная работа №8 «Ароматические углеводороды и их производные».................................................................................................. 53 Лабораторная работа №9 «Гидроксилпроизводные углеводородов»....... 56 Лабораторная работа №10 «Карбонильные соединения и их производные».................................................................................................. 60 Лабораторная работа №11 «Карбоновые кислоты и их производные».... 64 Лабораторная работа №12 «Азотосодержащие органические соединения» .......................................................................................................................... 70 Лабораторная работа №13 «Гетероциклические соединения» ................. 75 3.3. Методы синтеза органических соединений ............................................. 79 Лабораторная работа №14 «Сульфирование» ............................................. 83 Лабораторная работа №15 «Нитрование» ................................................... 86 Лабораторная работа №16 «Галогенирование» .......................................... 89 Лабораторная работа №17 «Алкилирование» ............................................. 90 Лабораторная работа №18 «Ацилирование» ............................................... 94 Лабораторная работа №19 «Окисление» ..................................................... 95 Лабораторная работа №20 «Восстановление» ............................................ 99 3.4. Идентификация органических соединений ........................................... 101 Список используемой литературы ............ Ошибка! Закладка не определена. Приложение 1. ..................................................................................................... 108 Приложение 2. ..................................................................................................... 109 Приложение 3. .......................................................................................................................117 124 Дмитрий Гымнанович Ким, Анастасия Владимировна Журавлева, Татьяна Владимировна Тюрина ЛАБОРАТОРНЫЙ ПРАКТИКУМ ПО ОРГАНИЧЕСКОЙ ХИМИИ ДЛЯ НЕХИМИЧЕСКИХ СПЕЦИАЛЬНОСТЕЙ Под редакцией Д.Г. Кима Техн.редактор Издательство Южно-Уральского государственного университета 125