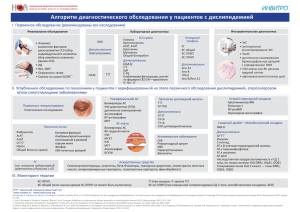
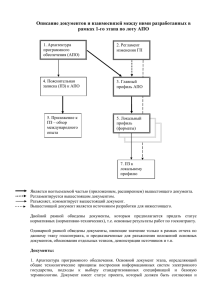
практическая липидология с методами медицинской генетики
реклама

УДК 616.1 ББК 54.11 К 76 В.А. Кошечкин, П.П. Малышев, Т.А. Рожкова ПРАКТИЧЕСКАЯ ЛИПИДОЛОГИЯ С МЕТОДАМИ МЕДИЦИНСКОЙ ГЕНЕТИКИ Учебное пособие Утверждено РИС Ученого совета Российского университета дружбы народов Р е ц е н з е н т ы: кандидат медицинских наук, доцент кафедры общей практики медицинского факультета РУДН Н.В. Стуров кандидат медицинских наук, научный сотрудник отдела проблем атеросклероза НИИ клинической кардиологии им А.Л. Мясникова, РКНПК, Министерства здравоохранения и социального развития Российской Федерации В.С. Тутунов К 76 Кошечкин, В.А. Практическая липидология с методами медицинской генетики [Текст] : учеб. пособие / В.А. Кошечкин, П.П. Малышев, Т.А. Рожкова. – М. : РУДН, 2012. – 00 с. : ил. ISBN 978-5-209-04851-0 Изучение основных принципов метаболизма липидов является главной задачей липидологии – науки о нарушениях обмена липидов и липопротеидов в плазме крови. Среди нарушений липидов и липопротеидов особое значение имеют гиперлипидемии, характеризующиеся повышением содержания холестерина в липопротеидах плазмы крови. В последние годы выявлены многочисленные генетические механизмы, детерминирующие нарушения метаболизма липидов. Практическое применение накопленных знаний в области генетики может быть реализовано с использованием методов медико-генетического консультирования в семьях с наследственными дефектами метаболизма липидов. Предназначено для студентов, ординаторов, аспирантов медицинских вузов, терапевтов, кардиологов и др. УДК 616.1 ББК 54.11 ISBN 978-5-209-04851-0 Москва Российский университет дружбы народов 2012 1 © Кошечкин В.А., Малышев П.П., Рожкова Т.А., 2012 © Российский университет дружбы народов, Издательство, 2012 2 4.9. Обсуждение диагноза и генетического состояния с пациентом 4.10. Этико-правовые вопросы обследования медико-генетического консультирования ОГЛАВЛЕНИЕ 78 79 ПРИЛОЖЕНИЕ 80 ВВЕДЕНИЕ 5 ЛИТЕРАТУРА 83 СПИСОК СОКРАЩЕНИЙ 7 СПИСОК ТЕРМИНОВ 85 Глава 1. Характеристика структур, участвующих в метаболизме липопротеидов крови 1.1. Липопротеиды 1.2. Липиды 1.3. Аполипопротеины 1.4. Метаболизм липопротеидов плазмы крови в норме Глава 2. Классификация семейных гиперлипопротеидемий 2.1. Чистая гиперхолестеринемия (Код МКБ-9/10: 272.0/ E78.0) 2.2. Чистая гипертриглицеридемия (Pure hypergly-ceridemia) (номер фенотипа OMIM 144600) 2.3. Смешанная (комбинированная) гиперлипидемия (Mixed hyperlipidemia) (код МКБ-9/10: 272.2/ E78.2) 2.4. Гиперхиломикронемия (МБТ 9/10: 272.3 / Е78.3) Глава 3. Основные принципы профилактики и лечения семейных гиперлипопротеидемий 9 9 14 17 34 ОПИСАНИЕ И ПРОГРАММА КУРСА «?????????» 43 44 48 50 55 60 Глава 4. Основы медико-генетического консультирования при семейных гиперлипопротеидемиях 65 4.1. Общий алгоритм медико-генетического консультирования (МГК) 66 4.2. Обоснование составления родословной 68 4.3. Анализ родословной и семейной истории 71 4.4. Практические рекомендации составления родословной 72 4.5. Показания для проведения медико-генетического тестирования 74 4.6. Семейная чистая гиперхолестеринемия (МКБ-10:Е78.0) как пример использования медико-генетического консультирования 75 4.7. Алгоритм диагностики гипертриглицеридемий 76 4.8. Алгоритм диагностики гиперхиломикронемий (МКБ-10:Е78.3) 78 3 4 106 ВВЕДЕНИЕ Метаболизм липидов является чрезвычайно сложным, жизненно важным процессом в организме человека. Гиперлипидемия (ГЛП), гиперлипопротеинемия, гиперлипопротеидемия (общее название – дислипидемия/дислипопротеидемия) представляет собой повышенный уровень одного или нескольких классов липопротеидов в плазме крови, в составе которых содержится холестерин. В развитии дислипидемий взаимодействуют многочисленные генетические и средовые факторы. Условно дислипидемии разделяют на дислипидемии, обусловленные мутациями генов с самостоятельным эффектом, и дислипидемии, обусловленные мультифакториально. Дислипидемии, обусловленные мутациями генов с самостоятельным эффектом, проявляются в раннем возрасте и характеризуются нарушениями функционирования конкретных структур, обеспечивающих метаболизм липопротеидов. К ним относятся мутации генов, кодирующие рецепторы липопротеидов, ферменты, транспортные белки и др. Дислипидемии, обусловленные мультифакториально, являются результатом взаимодействия многочисленных генетических факторов, каждый из которых имеет небольшой эффект, но на фоне неблагоприятного сочетания средовых факторов (таких как сверхкалорийное питание, малоподвижный образ жизни, избыточное потребление алкоголя, сопутствующие заболевания и др.) приводит к нарушениям метаболизма липидов. 5 Дислипидемии, обусловленные мутациями генов с самостоятельным эффектом, и дислипидемии, обусловленные мультифакториально, по сути являются семейными заболеваниями, имеющими тенденцию к накоплению в отдельных семьях, так называемые болезни с наследственным предрасположением. Наличие генетических факторов, детерминирующих развитие дислипидемий, являющихся промежуточными фенотипами ишемической болезни сердца, инфаркта миокарда, инсульта, внезапной смерти, переводит эти заболевания в разряд генетических заболеваний. С учетом этого, лечение и коррекция дислипидемий должна базироваться на основах медико-генетического консультирования. «Медико-генетическое консультирование – специализированный вид медицинской помощи и является медицинской функцией, при которой пациенты или родственники, имеющие риск наследственного заболевания, получают информацию о возможных последствиях и о природе этого заболевания, возможности его развития или наследования» (Бочков Н.П., 2009). Вместе с тем в нашей стране отсутствует специализированная медико-генетическая служба, функции которой заключались бы в обеспечении медикогенетическим консультированием членов семей, имеющих накопления сердечно-сосудистых заболеваний. Данная книга является попыткой восполнить этот пробел. В книге представлены описания нормального метаболизма липидов, а также генетические и средовые факторы, влияющие на его нарушения. В ней также содержатся современные знания о дислипидемиях, их классификации, принципах диетической и медикаментозной коррекции. Особое внимание уделено принципам медико-генетического консультирования пациентов и членов их семей с генетически детерминированным проявлением дислипидемии. Учебное пособие предназначено для студентов, ординаторов, аспирантов медицинских вузов, терапевтов, кардиологов и др. 6 СПИСОК СОКРАЩЕНИЙ Апо – белковая часть липопротеидов ABC – adenosine triphosphate binding cassette АСТ – аспартатаминотрансфераза АТФ – аденозинтрифосфат АХАТ – ацил-холестерин-ацилтрансфераза БПЭХ – белок, переносящий эфиры холестерина БСЖК – белки, связывающие жирные кислоты БПФЛ – белок, переносящий фосфолипиды ГЛП (гиперлипопротеидемия) – повышенное содержание липопротеидов в плазме крови ГМГ-КoA – 3-гидрокси-3-метилглутарил-КoA ГХС – гиперхолестеринемия – повышенное содержание общего холестерина в плазме крови ГТГ (гипертриглицеридемия) – повышенное содержание триглицеридов в плазме крови ЖК – жирные кислоты ИБС – ишемическая болезнь сердца ЛП – липопротеиды ЛП(а) – липопротеид (а) ЛПЛ – липопротеидлипаза ЛПВП – липопротеиды высокой плотности (альфа-липопротеиды) ЛПНП – липопротеиды низкой плотности (бета-липопротеиды) ЛПОНП – липопротеиды очень низкой плотности (пре~бета~ липопротеиды) ЛХАТ – лецитин-холестерин-ацилтрансфераза 7 МКБ-10 – Международная классификация болезней 10-го пересмотра. МРТ – магнитно-резонансная томография MTПБ – микросомальный триглицерид-переносящий белок ОМИМ – международный интернет-каталог генов (online mendelian inheritance in man) ПЛ – печеночная липаза РМЛП – ремнанты липопротеидов СГЛП – семейная гиперлипидемия СД – сахарный диабет СЖК – свободные жирные кислоты ТГ – триглицериды ФЛ – фосфолипиды ФХ – фосфатидилхолин ФР – факторы риска ХМ – хиломикроны ХС – холестерин ХС-ЛПВП – холестерин липопротеидов высокой плотности ХС-ЛПНП – холестерин липопротеидов низкой плотности ХС-ЛПОНП – холестерин липопротеидов очень низкой плотности ХС-ЛППП – холестерин липопротеидов промежуточной плотности Английские: The International Statistical Classification of Diseases and Related Health Problems, 10th Revision (known as «ICD-10») OMIM – каталог генов и генетических заболеваний (Online Mendelian Inheritance in Man) 8 Глава 1 ХАРАКТЕРИСТИКА СТРУКТУР, УЧАСТВУЮЩИХ В МЕТАБОЛИЗМЕ ЛИПОПРОТЕИДОВ КРОВИ в нормальном организме быстро поглощаются рецепторным путем печенью или превращаются в еще более мелкие липопротеиды низкой плотности под действием печеночной липазы. Физико-химические характеристики ЛППП находятся в пределах таковых ЛПОНП и ЛПНП. Таблица 1.1 Физико-химическая характеристика основных классов липопротеидов Физикохимические свойства и состав 1.1. Липопротеиды Гидратированная плотность, г/мл Размер частиц, А Электрофоретическая подвижность Состав (%): Белки Триглицериды Холестерин Фосфолипиды Содержание апо (%) А1 А2 B-48 В-100 C1 C2 C3 E D Всего Классы липопротеидов ХМ ЛПОНП ЛПНП ЛПВП 0,93-0,94 750-12000 0,94-1,006 280-750 1,006-1,063 1,063-1,21 215-220 75-150 старт пре-бета бета альфа 0,5-2 84-87 5-7 4-7 7-13 50-60 13-18 12-19 21-25 10-12 35-45 22-24 45-55 3-7 17-22 27-30 7,4 4,2 22,4 – 15 15 36 – – 100,0 следы следы 36,9 следы 10 6,7 39,9 12 – 100,0 – – – 98 следы следы следы – следы 100,0 67 22 следы 1-3 1-3 3-5 – – – 100,0 Липопротеиды (липопротеины) (ЛП)представляют собой комплексы, состоящие из белков (аполипопротеинов; сокращенно – апо) и липидов, связь между которыми осуществляется посредством гидрофобных и электростатических взаимодействий. Выделяют пять основных классов липопротеидов: хиломикроны (ХМ); липопротеиды очень низкой плотности (ЛПОНП); липопротеиды промежуточной плотности (ЛППП); липопротеиды низкой плотности (ЛПНП); липопротеиды высокой плотности (ЛПВП) (табл. 1.1). Циркулируя в крови, липопротеидные частицы обмениваются между собой поверхностными липидами и апопротеинами. При этом апопротеины поддерживают структурную целостность липопротеидов, участвуют в процессах обмена между липопротеидами и отвечают за взаимодействие липопротеидов с рецепторами. В табл. 1.1 указаны физико-химические характеристики ХМ, ЛПОНП, ЛПНП и ЛПВП. Липопротеиды промежуточной плотности (ЛППП) образуются при липолитической деградации ЛПОНП под действием липопротеидлипазы. Они характеризуются коротким временем жизни в крови, так как Каждый класс липопротеидов выполняет специфические функции по поддержанию гомеостаза (табл. 1.2). 9 10 Таблица 1.2 Функции липопротеидов Класс липопротеидов ХМ ЛПОНП ЛППП ЛПНП ЛПВП Функции Транспорт холестерина и жирных кислот, поступающих с пищей, из кишечника в периферические ткани и печень Транспорт холестерина, триглицеридов и фосфолипидов от печени к периферическим тканям Транспорт холестерина, триглицеридов и фосфолипидов от печени к периферическим тканям Транспорт холестерина, триглицеридов и фосфолипидов от печени к периферическим тканям Транспорт холестерина от периферических тканей к печени Структура липопротеидов Сердцевина сферической липопротеидной частицы состоит из двух неполярных липидов – триглицеридов и эфиров холестерина, количество которых в разных липопротеидах различно, также белковых молекул (апопротеинов) на поверхности частицы (рис. 1.1). Типичная липопротеиновая частица Хиломикроны (ХМ) Самые крупные липопротеидные комплексы, они содержат 85-92% ТГ, 6-12% фосфолипидов, 1-3% белков; основной белок ХМ – апо В-48. Насцентные ХМ формируются в энтероцитах тонкого кишечника. Липопротеиды очень низкой плотности (ЛПОНП) Транспортируют продукты эндогенного происхождения в отличие от ХМ, которые транспортируют ХС и ТГ экзогенного (диетического) происхождения. ЛПОНП продуцируются печенью в виде насцентных форм, содержат апо В-100, апо C1 и апо E, ХС, эфиры ХС и ТГ. В кровотоке насцентные ЛПОНП захватывают aпо C2 и aпо E, содержащиеся в ЛПВП, в результате чего становятся зрелыми ЛПОНП. Последние могут взаимодействовать с ЛПЛ в капиллярных сетях жировой ткани, сердечной и скелетных мышц. ЛПЛ отщепляет ТГ от ЛПОНП для накопления в клетках и выработки энергии. По мере того как ТГ отщепляются от ЛПОНП при участии ЛПЛ и белка, переносящего эфиры ХС, состав ЛПОНП изменяется, и они превращаются в липопротеиды промежуточной плотности (ЛППП). Липопротеиды промежуточной плотности (ЛППП) Отличие ЛППП от ЛПОНП состоит в том, что ЛППП содержат небольшое количество ТГ, при этом сохраняют в своем составе эфиры ХС. Некоторые ЛППП захватываются печенью, другие остаются в кровотоке, где оставшиеся ТГ подвергаются гидролизу и ЛППП превращаются в ЛПНП. Апопротеин Неэстерифицированный холестерин Липопротеиды низкой плотности (ЛПНП) Рис. 1.1. Схематическое изображение структуры липопротеидной частицы Являются главным представителем группы ЛП, богатых ХС. Их размер позволяет им пересекать эндотелий сосуда и проникать в тканевую жидкость, снабжая ткани ХС. Исключение составляет центральная нервная система, посколь- 11 12 Фосфолипид ку ЛПНП не пересекают гематоэнцефалический барьер. ЛПНП имеют липидное ядро, почти целиком состоящее из эфиров ХС (приблизительно 1500 молекул на одну ЛПНПчастицу). На поверхности этих ЛП содержится единственный апобелок – апо В100. ту в печень для выделения в виде желчных кислот и с калом. В норме около 30% ХС от общего его содержания в крови содержится в ЛПВП. Подклассы ЛПНП ЛП(а) как особая ЛП-частица характеризуется наличием уникального гликопротеина (а), связанного с апо В100 дисульфидными связями. Плотность ЛП(а) занимает промежуточное положение между ЛПНП и ЛПВП. Гетерогенность aпo (a) определяет изменения концентрации ЛП(а) в плазме; между молекулярным весом aпo (a) и концентрацией ЛП(а) в плазме имеется отчетливая обратная корреляция. Нативный ЛП(а), в отличие от ЛПНП, является слабым лигандом для ЛПНП-рецептора, а гиполипидемическая диета и гиполипидемические препараты, которые обычно влияют на уровень ЛПНП, не действуют на ЛП(а). Гомология апо (а) с плазминогеном человека дает повод для гипотезы, что повышенный риск раннего атеросклероза и тромботических осложнений, ассоциирующийся с повышенным уровнем ЛП(а), возникает вследствие молекулярной идентичности плазминогена и апо (а). Физиологические и сосудистые эффекты ЛП(а) окончательно не выяснены, однако показано, что у человека эта частица способна проникать в артериальную стенку. Концентрация ЛП(a) в плазме предопределена генетически и находится под контролем, главным образом, гена apolipoprotein(a) [LPA], локализованного на хромосоме 6q26-27. Доказана гетерогенность циркулирующих в плазме ЛПНП-частиц; они включают два подкласса, отличающихся по ряду физических свойств: размеру частиц, показателю флотации (плавучести) и плотности. Фенотип подкласса А характеризуется преобладанием более крупных и плавучих частиц, тогда как фенотип подкласса В – преобладанием мелких и более плотных ЛПНП-частиц. Большинство (8590%) лиц могут быть идентифицированы как имеющие либо тот, либо иной подкласс ЛПНП-частиц, тогда как у оставшейся части будет промежуточный фенотип. Распространенность фенотипа В составляет приблизительно 30% среди взрослых мужчин, 5-10% среди подростков мужского пола и женщин моложе 20 лет и примерно 15-25% среди женщин постклимактерического периода. Липопротеиды высокой плотности (ЛПВП) Гетерогенный класс ЛП, включающий несколько подклассов, различающихся по размеру и заряду. ЛПВП синтезируются в печени и содержат в основном aпo A1 и aпo A2. Печень синтезирует ЛПВП в виде комплексов, состоящих из аполипопротеинов и фосфолипидов, напоминающих сплющенные сферические структуры, не содержащие холестерина. ЛПВП играют решающую роль в обратном транспорте ХС из периферических клеток и его доставку в печень и стероидогенные клетки для последующего катаболизма. Понятие обратного транспорта ХС описывает процесс, в котором ЛПВП способствуют захвату ХС на периферии и его возвра13 Липопротеид (а) 1.2. Липиды К липидам относят жирные кислоты и их производные. Липиды играют важную роль в жизнедеятельности живых организмов. Они являются одним из основных компонентов 14 биологических мембран, влияя на их проницаемость, участвуют в передаче нервного импульса, создании межклеточных контактов. Липиды входят в состав липопротеидов, которые участвуют в транспорте липидов в организме. Классификация липидов В упрощенном виде (на основании структурных особенностей) можно выделить следующие основные классы липидов: жирные кислоты; нейтральные жиры (триглицериды); фосфолипиды; стероиды. Жирные кислоты (ЖК) В организме либо могут находиться в свободном состоянии (в следовых количествах), либо выполнять роль строительных блоков для большинства классов липидов. Жирные кислоты могут быть насыщенными (только с одинарными связями между атомами углерода), мононенасыщенными (с одной двойной связью между атомами углерода) и полиненасыщенными (с двумя и более двойными связями). Они различаются по количеству углеродных атомов в цепи, а также, в случае ненасыщенных кислот, по положению, конфигурации (как правило цис-) и количеству двойных связей. Жирные кислоты можно условно делить на низшие (до семи атомов углерода), средние (восемь – двенадцать атомов углерода) и высшие (более двенадцати атомов углерода). Нейтральные жиры (триглицериды – ТГ) Это эфиры глицерина и жирных кислот. Если ЖК эстерифицированы, то такое соединение называется триглицеридом (триацилглицеролом), если две – диглицеридом (диацилглицеролом), если одна – моноглицеридом (моноацилглицеролом). Основную массу природных нейтральных жиров составляют триглицериды. ЖК, входящие в состав ТГ, практи15 чески определяют их физико-химические свойства. В одной молекуле ТГ обычно содержатся остатки двух или трех разных ЖК. Последние в ТГ могут быть насыщенными и ненасыщенными. Фосфолипиды (ФЛ) Представляют собой сложные эфиры многоатомных спиртов глицерина или сфингозина с высшими ЖК и фосфорной кислотой. В состав некоторых ФЛ входят также азотсодержащие соединения – холин, этаноламин или серин. ФЛ делят на две группы – глицерофосфолипиды и сфингофосфолипиды в зависимости от того, какой многоатомный спирт (глицерин или сфингозин) они содержат. Наиболее распространенными в тканях животных являются глицерофосфолипиды. Они в свою очередь подразделяются на фосфатидилхолины (ФХ) или лецитины, фосфатидилэтаноламины и фосфатидилсерины в зависимости от характера азотистого основания, присоединенного к фосфорной кислоте. Около половины всех ФЛ животного организма составляют ФХ. На это соединение приходится подавляющая часть ФЛ содержимого тонкой кишки; основная масса ФХ поступает в кишечник с желчью (11-12 г/сут), меньшая (1-2 г/сут) – с пищей. Функция ФЛ в организме многообразна и до конца не выяснена. Они играют важную роль в структуре и функции клеточных мембран, активации мембранных и лизосомальных ферментов, проведении нервных импульсов, свертывании крови, иммунных реакциях, процессах клеточной пролиферации и регенерации тканей, переносе электронов в реакциях дыхательной цепи, всасывании продуктов расщепления жиров, формировании липопротеидных частиц. Стероиды. Стерины или стеролы 16 Стероиды. Стерины или стеролы (холестерин (ХС) и эфиры холестерина) Стероиды – широко распространенные в природе соединения. К ним относятся гормоны коркового вещества надпочечников, половые гормоны, желчные кислоты. В организме человека важное место среди стероидов занимают стерины (стеролы), т.е. стероидные спирты, главным представителем которых является холестерин (ХС). Он содержит стероидное ядро из четырех колец и гидроксильную группу. Последняя может быть эстерифицирована высшей ЖК, при этом образуются эфиры ХС. Основные функции холестерина представлены в табл. 1.3. Таблица 1.3 Основные функции холестерина Основной компонент 3) являются лигандами для некоторых рецепторов на поверхности клеток, определяя, таким образом, деградацию других компонентов ЛП-частиц, например, ХС. Основные аполипопротеины семейства апо А (А1, A2, A4, A5) Апо семейства A – апо А1 и A2 – это основные белковые компоненты ЛПВП плазмы. Основные функции ЛПВП состоят в связывании липидов, удалении ХС из периферических клеток, активации ЛХАТ и узнавании рецепторов в печени и стероидогенных тканях. Апо А-I составляет около 70% от общей массы белка в ЛПВП, что указывает на его важную структурную роль. Апо А-I участвует в обратном транспорте ХС из периферических тканей в печень и является кофактором ЛХАТ. Предшественник Мембран клеток (трансмембранный транспорт) Липопротеидов плазмы (транспорт липидов) Желчных кислот (всасывание жиров) Стероидов надпочечников (гидрокортизон, альдостерон) Половых гормонов (эстрогены, андрогены) 1.3. Аполипопротеины Генетика аполипопротеина А-I Апо А-I кодируется геном APOA1, который локализуется в области длинного плеча 11-й хромосомы (11q23-q24). Мутации APOA1 связаны с дефицитом ЛПВП, включая болезнь Танжера и системный не-нейропатический амилоидоз. OMIM Символика гена 107680 APOA1 Цитогенети- Геномные координаты ческая лока- (NCBI) лизация 11q23.3 11:116,706,468 – 116,708,337 Белки, входящие в состав липопротеидов (ЛП), получили название аполипопротеинов (апо), наиболее важными из них являются: апо А (А1, A2, A4, A5), апо B, апо C (C1, C2, C3, C4), апо D, апо E. Апопротеины выполняют три основные функции: 1) способствуют растворимости неполярных липидов (ТГ, эстерифицированного ХС), взаимодействуя с ФЛ; 2) регулируют взаимодействие липидов с ферментами, такими как липазы и лецитин-холестерин-ацилтрансфераза (ЛХАТ); Апо А2 – другой важный апопротеин ЛПВП, на его долю приходится около 20% от всех белков этих частиц. Апо А-II кодируется геном APOA2, который локализован на хромосоме 1. Мутации APOA2 могут быть причиной дефицита апо A-II или гиперхолестеринемии (ГХС). 17 18 Генетика аполипопротеина А-II OMIM Символика гена 107670 APOA2 ЦитогенетиГеномные координаты ческая лока- (NCBI) лизация 1q23.3 1:161,192,082 – 161,193,417 Генетика аполипопротеина А-III Апо А-III синтезируется и секретируется печенью, в меньшей степени – кишечником, является структурным компонентом ЛПВП, а также липопротеидов, содержащих апо В. Апо А-III влияет на катаболизм и захват печенью частиц ЛПВП. OMIM Символика гена 107720 APOА3 Цитогенетиче- Геномные координаты ская локализа- (NCBI) ция 11q23.3 11:116,700,623 – 116,703,786 Генетика и функции аполипопротеина А-IV Aпо A-IV секретируется из печени в кровоток, располагаясь на поверхности свежесинтезированных ХМ. Абсорбция жиров из тонкого кишечника значительно увеличивает синтез и секрецию apo A-IV. Известно, что апо A-IV: 1) активирует лецитин-холестерин-ацилтрансферазу (ЛХАТ) и белок, переносящий эфиры холестерина (БПЭХ); 2) участвует в регуляции аппетита и чувства насыщения, что было выявлено на моделях животных; 3) проявляет антиоксидантные и антиатерогенные функции; 4) изменяет эффективность транспорта липидов энтероцитами и клетками печени in vitro. 19 Aпо A-IV – плазменный белок, является продуктом гена APOA4, локализованного на хромосоме 11; сцеплен с генами APOA1 и APOC3. OMI M Символика гена 10769 0 APOA4 Цитогенетическая локализация 11q23.3 Геномные координаты (NCBI) 11:116,691,417 – 116,694,010 Генетика и функции аполипопротеина А-V Апо A-V – белок, является составной частью нескольких фракций липопротеидов, включая ЛПОНП, ЛПВП, ХМ. Предполагается, что aпо A-V влияет на метаболизм ЛП, взаимодействуя с семейством рецепторов ЛПНП-Р (LDL-R). Является важным регулятором гидролиза ТГ плазмы, одновременно служит важным стимулятором aпo C-II и ЛПЛ и ингибитором продукции ТГ ЛПОНП в печени. Способен активировать ЛХАТ. Aпо A-V у человека кодируется геном APOA5. Данный ген локализован в проксимальном отделе кластера аполипопротеиновых генов на хромосоме 11q23. OMIM Символика гена 606368 APOA5 Цитогенетическая локализация 11q23.3 Геномные координаты (NCBI) 11:116,660,085 – 116,663,135 Генетика и функции аполипопротеина В Апо В – ключевой белок, вовлеченный в метаболизм ЛП и поддержание нормального гомеостаза уровней ХС плазмы крови. Апо В существует в виде двух изоформ: апо В-48 и апо В-100. Апо В является структурным компонентом 20 нескольких ЛП: ХМ, ЛПОНП, ЛППП, ЛПНП и частиц липопротеида (а). Он необходим для сборки и секреции ХМ, поступающих из тонкой кишки, и ЛПОНП, поступающих из печени, и поддержания структурной целостности частиц ЛПОНП и ЛПНП. Этот белок также важен как лиганд ЛПНП-рецептора, опосредующего поглощение ХС. Aпо B-48 синтезируется исключительно в тонком кишечнике, а апо B-100 – в печени. Повышенные концентрации апо B в плазме крови связаны с сердечно-сосудистыми заболеваниями. Апо В-48 и апо В-100 кодируются одним геном – APOB, однако апо В-48 образуется в результате редактирования мРНК апо В-100, что и приводит к синтезу более короткого полипептида. Мутации APOB могут детерминировать гипобеталипопротеидемию, гипобеталипопротеидемию в сочетании с нормальными уровнями ТГ в плазме крови, а также гиперхолестеринемию, обусловленную дефектом связывающих функций апо B. OMIM Символика гена +107730 APOB Цитогенетическая локализация 2p24.1 Геномные координаты (NCBI) 2:21,224,300 – 21,266,944 Основные аполипопротеины семейства апо C (C-I, С-II, C-III, C-IV) Апо С плазмы человека относят к так называемым регуляторным белкам. Они составляют 40-80% от общего содержания белка ХМ и ЛПОНП и содержатся также в ЛПВП. Апо С-I является составной частью ЛП, богатых ТГ. Обнаружено, что апо С-I ингибирует печеночную липазу и препятствует клиренсу ЛП с участием: 1) ЛПНП-рецептора; 2) белка, родственного ЛПНП-рецептору (LDL receptorrelated protein, LRP); 3) ЛПОНП-рецептора. 21 Aпо C-I кодируется геном APOC1, который расположен в хромосоме 19, внутри кластера аполипопротеиновых генов. Этот ген экспрессируется, главным образом, в печени. OMIM Символика гена 107710 APOC1 Цитогенетическая локализация 19q13.32 Геномные координаты (NCBI) 19:45,417,920 – 45,422,605 Апо С-II является специфическим физиологическим активатором, необходимым для активности ЛПЛ – фермента, ответственного за удаление ТГ из ЛПОНП; следовательно, он играет центральную роль в метаболизме ТГ плазмы. In vivo апо С-II обнаружен в основном в составе ХМ и ЛПОНП. Апо С-II кодируется геном APOC2. OMIM Символика гена 608083 APOC2 Цитогенети- Геномные координаты ческая лока- (NCBI) лизация 19q13.32 19:45,449,238 – 45,452,821 Апо С-III в основном секретируется печенью и в меньшей степени тонким кишечником. Апо С-III является белковым компонентом ЛПОНП, который ингибирует ЛПЛ и печеночную липазу, и, полагают, замедляет катаболизм ЛП, богатых ТГ. Повышение уровня апо С-III сопровождается гипертриглицеридемией. OMIM Символика гена 107720 APOC3 Цитогенетическая локализация 11q23.3 22 Геномные координаты (NCBI) 11:116,700,623 – 116,703,786 Апо C-IV кодируется геном APOC4, который является членом семейства генов апо С. Экспрессия гена проявляется в клетках печени. В геноме человека гены APOA1, APOC3 и APOA4 тесно сцеплены. OMIM Символика гена 600745 APOC4 Цитогенетическая локализация 19q13.32 Геномные координаты (NCBI) 19:45,445,494 – 45,448,752 Аполипопротеин Е Апо Е характеризуется значительной распространенностью в тканях и широким спектром функций. Хотя печень служит главным источником апо Е плазмы, другие типы клеток также продуцируют этот белок и вносят вклад в его уровень в плазме. Основным местом клиренса апо Е-содержащих ЛП служит печень. Апо Е – основа для связывания ЛПНП-рецептора и липидов. Апо Е вовлечен во многие стадии гомеостаза ЛП, способствуя эндоцитозу ЛП плазмы, особенно ЛПОНП и ремнантных ЛП. Апо Е кодируется геном APOE. OMIM Символика гена 107741 APOE Цитогенети- Геномные координаты ческая лока- (NCBI) лизация 19q13.32 19:45,409,038 – 45,412,649 Генетическая вариабельность аполипопротеина Е Полиморфизм апо Е обусловлен неоднородностью локуса гена AРОЕ, который может присутствовать в одной из трех форм (аллелей): ε2, ε3 («дикий» тип), ε4. Таким образом, в популяции возможно существование шести фенотипов апо Е, отличающихся как по своей распространенности, так и по функциональной значимости. Рецепторы поверхностных мембран клеток Выделяют три основные группы рецепторов наружных клеточных мембран: – рецепторы ЛППП (LRP1, LRP1B, LRP2, LRP3, LRP4, LRP5); – рецепторы ЛПНП (LDLR, LRPAP1); – рецепторы ЛПВП (SCARB1). Рецепторы ЛППП и ЛПНП Рецепторы ЛППП и ЛПНП объединяют в группу белков под общим названием LRP. В эту группу относят также LRP1, известный как α-2-макроглобулиновый рецептор (A2MR), аполипопротеин Е-рецептор (APOER) или кластер дифференциации 91 (CD91), является белком-рецептором, обнаруженным в плазматических мембранах клеток, обеспечивающим рецептор-опосредуемый эндоцитоз. У человека белок LRP1 кодируется геном LRP1. OMIM Символика гена 107770 LRP1 Цитогенетическая локализация 12q13.3 Геномные координаты (NCBI) 12:57,522,281 – 57,607,141 Апо Е человека, в противоположность другим видам, существует в одной из трех основных изоформ, обозначаемых Е2, Е3 и Е4, отличающихся между собой по аминокислотному составу в положениях 112 и 158. LRP1B относят к генному семейству рецепторов ЛПНП. Эти рецепторы выполняют разнообразные функции в жизнедеятельности и развитии клеток, поскольку они взаимодействуют с большим количеством лигандов. LRP1B у человека кодируется геном LRP1B. 23 24 LRP2 (мегалин) у человека кодируется геном LRP2. LRP2 является мультилигандным рецептором, обнаруживается в плазматической мембране многих абсорбтивных эпителиальных клеток. LRP2 является членом семейства рецепторов со структурными сходствами рецептора ЛПНП (LDLR). LRP2 участвует в качестве медиатора в эндоцитозе лигандов, приводя к их распаду или трансцитозу. LRP2 активен в эпителиальных клетках щитовидной железы (тироцитах), где функционирует в качестве рецепторов тироглобулина. Мутации гена LRP2 связаны с синдромом DonnaiBarrow. LRP3 у человека кодируется геном LRP3. LRP4 у человека кодируется геном LRP4. LRP5 у человека кодируется геном LRP5. Показано, что LRP5 взаимодействует с AXIN1. ЛПНП-рецептор содержит ~840 аминокислот и участвует в эндоцитозе ЛПНП-частиц. ЛПНП-Р располагается на поверхности мембран и распознает апо В-100, расположенные во внешнем слое этих частиц. Данный рецептор также узнает белок aпo E, содержащийся в ХМ и ремнантах ЛПОНП (ЛППП). У человека ЛПНП-Р кодируется геном LDLR, принадлежащим к семейству рецепторов ЛПНП. ЛПНП-рецептор (ЛПНП-Р) Ген, кодирующий ЛПНП-рецепторы, состоит из 18 экзонов. Экзон 1 содержит сигнальную последовательность, которая локализует рецептор в эндоплазматическом ретикулуме для последующего транспорта на поверхность клетки. Экзоны 2-6 кодируют лигандсвязывающий участок. Экзоны 7-14 кодируют домен, гомологичный эпидермальному фактору роста. Экзон 15 кодирует участок белка, богатый олигосахаридами. Экзон 16 (и в некоторых случаях 17) кодирует трансмембранный домен; экзон 18 (и частично экзон 17) кодируют цитоплазматический домен. ЛПНП-рецептор относят к химерным белкам, поскольку он состоит из функционально самостоятельных участков (доменов), действующих независимо друг от друга. В 1985 г. Brown и Goldstein получили Нобелевскую премию за идентификацию ЛПНП-Р в результате изучения семейной гиперхолестеринемии. Рис. 1.2. DiPiro JT, Talbert RL, Yee Gc,Matzke GR, Wells BG, Posey Lm: Pharmacotherapy: A pathophysiologic Approach, 7 th Edition: http://www.accesspharmacy.com Copyright © The McGraw-Hill Companies, Inc. All reserved OMIM Символика гена 606945 LDLR Цитогенетическая локализация 19p13.2 Геномные координаты (NCBI) 19:11,200,037 – 11,244,505 Структура гена LDLR и белка ЛПНП-Р ЛПВП-рецепторы-скэвенджеры (очистители) Scavenger receptor class B member 1 (SRB1/SR-BI/ SCARB1) Скэвенджер-рецепторы класса В типа I (Scavenger receptor class B member 1 (SRB1), также известные как SR-BI 25 26 (SCARB1), входят в состав клеточных мембран и присутствуют во многих типах клеток и тканей, включая печень и надпочечники. Наиболее изучена их роль в обеспечении захвата эфиров ХС из ЛПВП в печени, в результате обеспечивается перенос ХС из периферических тканей в печень для дальнейшей их экскреции. Этот процесс переноса ХС известен как обратный транспорт ХС и защитный механизм от развития атеросклероза. Скэвенджер-рецепторы класса В типа I кодируются геном SCARB1. OMIM Символика гена 601040 SCARB1 ЦитогенетиГеномные координаты ческая лока- (NCBI) лизация 12q24.31 12:125,262,173 – 125,348,518 Белок, переносящий эфиры холестерина (БПЭХ) Сholesteryl ester transfer protein (CETP) БПЭХ секретируется в печени и переносит эфиры ХС от ЛПВП к ЛП, богатым ТГ, и к ЛПНП, а также ТГ от ЛП, богатых ТГ, к ЛПВП. БПЭХ модулирует уровни ЛП плазмы, транспортируя неполярные липиды между разными классами ЛП. Выявлены мутации в гене БПЭХ и их связи с регуляцией липидов плазмы и ИБС. БПЭХ кодируется геном CETP. OMIM Символика гена 118470 CETP Цитогенетическая локализация 16q13 27 Белок, связывающий жирные кислоты (БСЖК) Fatty acid-binding protein 1 (FABP1) Длинноцепочечные ЖК являются гидрофобными молекулами; свои функции они выполняют с помощью специфических внутриклеточных липид-связывающих белков, которые обозначают как белки, связывающие жирные кислоты (БСЖК). В настоящее время установлено, что БСЖК играют важную роль: а) в контроле процессов поглощения ЖК клетками и их последующего метаболизма, б) распределении/хранении ЖК внутри клеток, в) модуляции внутриклеточной сигнальной трансдукции, г) переносе сигнальных ЖК к ядерным рецепторам, таким как PPAR. Большое количество типов этих белков свидетельствует об их особых функциях в специфических тканях. Множеством экспериментальных данных показано, что отдельные БСЖК обладают как уникальными, так и перекрестными функциями, обусловленными специфическими элементами их белковой структуры. БСЖК не только модулируют внутриклеточный липидный гомеостаз, регулируя транспорт ЖК в ядерные и неядерные области клетки, но влияют и на системный энергетический гомеостаз. Геномные координаты (NCBI) OMIM Символика гена 16:56,995,834 – 57,017,755 134650 FABP1 Цитогенетическая локализация 2p11.2 28 Геномные координаты (NCBI) 2:88,422,500 – 88,427,649 Микросомальный триглицерид-переносящий белок (МТПБ) Microsomal triglyceride transfer protein (MTTP) OMIM Символика гена Является внутриклеточным липид-переносящим белком, обнаруженным в эндоплазматическом ретикулуме, отвечающим за перенос липидных молекул (в частности, эфиров ХС, ТГ и ФЛ) из цитозоля и/или мембраны эндоплазматического ретикулума к насцентным апо В для образования апо В-содержащих ЛП. Этот перенос является частью сборки ЛП, богатых ТГ, таких как ХМ в кишечнике и ЛПОНП в печени. У пациентов с генетическим нарушением вследствие мутаций с утратой функции белка в гене MTTP отмечаются экстремально низкие концентрации ХС и ТГ в плазме и отсутствие ХМ, ЛПОНП и ЛПНП. 172425 PLTP OMIM Символика гена 157147 MTTP Цитогенетическая локализация 4q23 Геномные координаты (NCBI) 4:100,485,239 – 100,545,153 Белок, переносящий фосфолипиды (БПФЛ) Phospholipid transfer protein (PLTP) Ускорение переноса ФЛ между митохондриями, микросомами, клеточными мембранами и ЛП плазмы обеспечивается с помощью белка под названием «белок, переносящий фосфолипиды» (БПФЛ). БПФЛ взаимодействует с апо А-I и апо А-II, усиливает связывание на поверхности клетки и ремоделирование ЛПВП-частиц, что повышает их способность усиливать отток ХС и ФЛ. БПФЛ кодируется геном PLTP. 29 Цитогенетическая локализация 20q13.12 Геномные координаты (NCBI) 20:44,527,258 – 44,541,002 АВС-транспортеры Кассетные белки-транспортеры, связывающие аденозинтрифосфат (АТФ) (adenosine triphosphate binding cassette (ABC) transporters), являются одним из самых крупных белковых семейств, представленных в широком диапазоне живых организмов, от бактерий до человека. Несмотря на свое разнообразие, с точки зрения воздействия на субстрат все они похожи в обеспечении вследствие гидролиза АТФ клеток энергией, необходимой для активного транспорта субстратов через биологические мембраны. В метаболизм клеточных липидов вовлечено значительное число транспортеров АВС. Тот факт, что мутации этих генов ассоциируются с патологическими фенотипами или даже клинически значимыми заболеваниями, связанными с липидным метаболизмом, подчеркивает их решающую роль в гомеостазе клеточных липидов. Насчитывается 48 АВС-транспортеров, которые классифицированы на 7 семейств. АВСА1-транспортер (АТФ-связывающий кассетный белок-транспортер типа 1) является ключевым «игроком» в гомеостазе липидов клетки, опосредуя отток ХС и ФЛ из клеток к апо А-I и контролируя скорость образования ЛПВП. OMIM Символика гена 600046 ABCA1 Цитогенетическая локализация 9q31.1 30 Геномные координаты (NCBI) 9:107,543,282 – 107,690,526 Стеролрегуляторный элемент-связывающий транскрипторный фактор Sterol regulatory element-binding transcription factor 1 (SREBF1) Стеролрегуляторный элемент-связывающий транскрипторный фактор (SREBP1) и SREBP2 (600481) – структурно сходные белки, которые обеспечивают контроль гомеостаза холестерина с помощью активирования транскрипции стерол-регуляторных генов. OMIM 184756 600481 Символика гена SREBF1 SREBF2 Цитогенетическая локализация 17p11.2 22q13.2 лоновую кислоту. Его активность снижается конечными продуктами реакции, в том числе ХС, а также метаболитами, такими как 26-гидрокси-ХС. Эндогенный синтез ХС снижается при экспозиции клеток с ЛПНП, которые обеспечивают доставку к клетке ХС, тогда как ЛПВП, которые осуществляют акцепцию ХС из клеток, оказывают обратный эффект. Фармакологические агенты, которые конкурентно ингибируют ГМГ-КоА-редуктазу, блокируют эндогенный синтез ХС и посредством этого стимулируют активность ЛПНПрецепторов, в результате чего уровень ХС ЛПНП в плазме крови снижается. Геномные координаты (NCBI) OMIM 17:17,714,662 – 17,740,324 Символика гена 142910 HMGCR 22:42,229,105 – 42,302,374 3-Гидрокси-βметилглутарил-коэнзим А-редуктаза Цитогенетическая локализация 5q13.3 Геномные координаты (NCBI) 5:74,632,992 – 74,657,925 Внеклеточные ферменты ЛХАТ (LCAT), ПЛП (LIPC), ЛПЛ (LPL) Лецитин-холестерин-ацилтрансфераза (ЛХАТ) ГМГ-КоА-редуктаза катализирует лимитирующую раннюю стадию синтеза холестерина. Этот микросомальный фермент превращает ГМГ-КoA (3-гидрокси-3-метилглутарил-КoA EC 1.1.1.88) в мевалонат путем двухступенчатого восстановления за счет НАДФH (на первом этапе синтеза холестерина из ацетил-КoA ). Предполагается, что эта реакция является скорость-лимитирующей стадией на пути синтеза холестерина. ГМГ-КоА-редуктаза представляет собой гликопротеин, который находится в эндоплазматическом ретикулуме всех клеток, обладает способностью синтезировать ХС, в частности, клеток печени, тонкого кишечника, надпочечников и гонад. Фермент катализирует превращение ГМГ-КоА в мева- Лецитин-холестерин-ацилтрансфераза, или фосфатидилхолинстерол O-трансфераза (ЛХАТ, КФ 2.3.1.43, англ. Lecithin-cholesterol acyltransferase, LCAT) – фермент, превращающий свободный холестерин ЛПВП в эфиры холестерина, являющиеся более гидрофобной формой холестерина. Таким образом, ЛХАТ является ферментом метаболизма липопротеидов. ЛХАТ связана с поверхностью липопротеидов высокой плотности, которые содержат апо A-I – активатор этого фермента. ХC, превращенный в эфиры ХС, благодаря высокой гидрофобности перемещается с поверхности липопротеида в ядро, освобождая место на поверхности частицы для захвата нового свободного ХС. Таким образом, эта 31 32 реакция является исключительно важной для процесса освобождения периферических тканей от ХС (обратный транспорт ХС). OMIM Символика гена 606967 LCAT Цитогенетическая локализация 16q22.1 Геномные координаты (NCBI) 16:67,973,786 – 67,978,014 Триацилглицероллипаза печеночная (ПЛП) Печеночная липаза (печеночная триглицеридная липаза, триглицеридная липаза, ПЛ; англ. hepatic lipase, hepatic triglyceride lipase, LIPC), КФ 3.1.1.3) – один из ферментов липидного метаболизма. Печеночная липаза синтезируется в печени и секретируется в кровь. После секреции фермент связывается со стенкой сосуда (почти исключительно в печени) и расщепляет липиды липопротеидов, участвует в регенерации ЛПНП. ПЛ работает в кровотоке в тандеме с ЛПЛ. Последняя расщепляет липопротеиды, богатые ТГ (ЛПОНП и ХМ), до их ремнантов. Ремнанты липопротеидов являются, в свою очередь, субстратом для печеночной липазы. Таким образом, в результате действия печеночной липазы образуются атерогенные ЛПНП, которые поглощаются печенью посредством рецепторного эндоцитоза. OMIM Символика гена 151670 LIPC Цитогенетическая локализация 15q21-q23 33 Геномные координаты (NCBI) 15:58,724,174 – 58,861,072 Липопротеидлипаза (ЛПЛ) (Lipoprotein lipase, LPL), EC 3.1.1.3) Липопротеидлипаза – многофункциональный белок, вовлеченный в различные аспекты метаболизма ЛП и липидов. Фермент располагается на поверхности эндотелия, обращенного в просвет сосуда, и катализирует гидролиз ФЛ и ТГ ХМ и ЛПОНП. ЛПЛ вовлечена в перенос липидов между различными типами ЛП и играет важную роль в формировании ЛПВП. В дополнение к гидролизу ТГ плазмы до диглицеридов ЛПЛ также участвует во взаимодействии ЛП с клеточными рецепторами. OMIM Символика гена 609708 LPL Цитогенетическая локализация 8p21.3 Геномные координаты (NCBI) 8:19,796,581 – 19,824,769 1.4. Метаболизм липопротеидов плазмы крови в норме Выделяют три основных пути продукции и транспорта липидов в организме. Эти пути включают в себя экзогенный, эндогенный и обратный транспорт холестерина. Экзогенный (диетарный/диетический) путь Жиры, содержащиеся в пище после переваривания в желудке, поступают в двенадцатиперстную кишку, где подвергаются эмульгированию под воздействием желчных кислот, после чего они попадают в тонкий кишечник. Длинноцепочечные жирные кислоты в тонком кишечнике упаковываются в мицеллы (рис. 1.3). 34 Мицеллы состоят из наружного слоя жирных кислот, который окружает гидрофильные и гидрофобные липиды ды проникают в ближайшие клетки различных тканей, где они утилизируются или накапливаются. Ремнанты зрелых ХМ попадают в печень благодаря способности апо В-48 связываться с рецептором печеночной клетки. Таким образом, происходит эндоцитоз ремнантов зрелых ХМ в гепатоциты. В результате этого процесса ХМ транспортируют экзогенные липиды в печень, жировую ткань, сердечную мышцу и скелетные мышцы, где под воздействием ЛПЛ из них высвобождаются ТГ (рис. 1.4, А). Эндогенный путь Рис. 1.3. Структура мицелл Мицеллы захватываются клетками слизистой оболочки тонкого кишечника. В этих клетках содержимое мицелл (желчные кислоты, гидрофильные и гидрофобные липиды) используются для синтеза ХМ. Синтез ХМ стимулируется поступлением мицелл в клетки ворсинок тонкой кишки (рис. 1.4, А). Выделяют три стадии жизненного цикла ХМ: насцентные ХМ, зрелые ХМ и ремнанты ХМ. Насцентные ХМ синтезируются в энтероцитах тонкого кишечника. В составе насцентных ХМ находятся: апо В-48, ХС, ТГ, ФЛ. Насцентные ХМ во время циркуляции в лимфатических сосудах и крови взаимодействуют с ЛПВП, от которых они получают апо С-II и апо Е. В результате этого взаимодействия формируются зрелые ХМ или просто ХМ. Aпо С2 является кофактором фермента ЛПЛ. Зрелые ХМ, освобождаясь от ТГ под воздействием ЛПЛ, расположенной на поверхности эндотелия капилляров, превращаются в ремнанты (остатки) ХМ. Ремнанты зрелых ХС содержат только апо В-48 и апо Е. Освободившиеся от зрелых ХМ свободные жирные кислоты (СЖК) и диглицери35 Эндогенный путь отличается от экзогенного тем, что ЛПОНП в виде насцентных форм образуются в печени и поступают в кровь. Насцентные формы ЛПОНП содержат: апопротеины В100, C1 и E, ХС, эфиры ХС и ТГ. В крови ЛПОНП подвергаются воздействию ЛПЛ, в результате чего от ЛПОНП отщепляются жирные кислоты и глицериды. Одновременно от ЛПВП в насцентные ЛПОНП перемещаются апо С-II и апо Е, в результате чего насцентные ЛПОНП становятся зрелыми. Зрелые ЛПОНП содержат апобелки Е, С2 и В100 (примерно 8% от общего состава), 5-15% ХС, 55-80% ТГ, 10-20% ФЛ. Наряду с ХМ, ЛПОНП относят к триглицерид-богатым ЛП (рис. 1.4, Б). Таким образом, в отличие от ХМ, которые переносят экзогенные липиды, полученные организмом с пищей, ЛПОНП транспортируют эндогенные липиды (синтезированные в печени). Зрелые ЛПОНП взаимодействуют с ЛПЛ в капиллярных сетях жировой, сердечной и скелетных мышц. ЛПЛ, активированная апо С-II, способствует отщеплению ТГ от ЛПОНП. СЖК и диглицериды, возникшие во время этого процесса, захватываются клетками прилегающих тканей, где они утилизируются для выработки энергии или накаплива36 ются в них. При взаимодействии ЛПВП со зрелыми ЛПОНП aпo C-II из ЛПОНП переходят в ЛПВП. В свою очередь ЛПВП также переносят эфиры ХС в ЛПОНП. Перенос ФЛ и ТГ от ЛПОНП в ЛПВП происходит с помощью БПЭХ. По мере того как ТГ отщепляются от ЛПОНП под воздействием ЛПЛ и БПЭХ, состав ЛПОНП изменяется, и они превращаются в ЛППП. Около 50% ЛППП распознается рецепторами печени и подвергается эндоцитозу. Постепенно ЛППП теряют апо Е, и, когда содержание в них ХС становится больше, чем ТГ, они превращаются в ЛПНП, содержащие апо В-100. ЛПНП – основные переносчики ХС в организме во внепеченочные клетки, где они используются для формирования клеточных мембран и синтеза стероидных гормонов. Большая часть ЛПНП захватывается рецепторами ЛПНП печени, их остатки перемещаются через скэвенджер-рецепторы на клеточном уровне. А – экзогенный путь Зрелые ХМ, освобождаясь от ТГ под воздействием ЛПЛ, расположенной на поверхности эндотелия капилляров, превращаются в ремнанты (остатки) ХМ. Ремнанты зрелых ХМ попадают в печень благодаря способности апо В-48 связываться с рецептором печеночной клетки. В – эндогенный путь В крови ЛПОНП подвергаются воздействию ЛПЛ и превращаются в ЛППП. При взаимодействии с ЛПВП, ЛППП превращаются в ЛПНП. ЛПНП захватываются печенью и периферическими тканями посредством рецепторного механизма. Транспорт ЛПНП в соматическую клетку Клетки захватывают ЛПНП, циркулирующие в плазме крови, с помощью рецептор-опосредованного эндоцитоза, который происходит в следующем порядке. Если клетка нуждается в ХС, то она начинает синтезировать ЛПНПрецепторы, которые включаются в плазматическую мембрану. ЛПНП-рецепторы накапливаются в зоне окаймленных ямок (clathrin-coated pits). К ЛПНП-рецепторам присоединяются ЛПНП. Окаймленные пузырьки, содержащие ЛПНП, формируют везикулы, которые поступают внутрь клетки, где они попадают в лизосомы. Там эфиры ХС, содержащиеся в ЛПНП, подвергаются гидролизу, а освобождающиеся ЛПНП-рецепторы снова встраиваются в мембрану клетки. 37 Рис. 1.4. Упрощенная схема метаболизма липопротеидов плазмы крови в норме С – обратный путь Насцентные ЛПВП под воздействием апо А-I и АВСА1 превращаются в ЛПВП2. Более крупные ЛПВП2, которые образуются из ЛПВП3 при дальнейшем присоединении ХС, способны к липидации от гидролиза ЛП, богатых ТГ, дающего ФЛ, которые могут затем переноситься к ЛПВП, – процесс, регулируемый ЛПЛ и белком, переносящим фосфолипиды (БПФЛ). 38 Регуляция активности ЛПНП-рецепторов Активность ЛПНП-рецепторов подавляется высокими концентрациями ХС внутри клетки. Этот процесс включает снижение выделения мембранного белка SREBP (sterol regulatory element binding protein). Подклассы семейства SREBP активируют транскрипцию генов ЛПНП-рецептора, а также такие необходимые для синтеза ХС, как ген HMG-CoA Reductase. Снижение синтеза рецепторов ЛПНП ограничивает избыточное поступление ХС в соматическую клетку. Роль ЛПВП в метаболизме липидов, обратный транспорт холестерина ЛПВП являются ключевыми структурами в обратном транспорте ХС в печень и переносе эфиров ХС между ЛП. ЛПВП синтезируются в насцентной форме, включающей апо А-I (составляющий 70% белка ЛПВП), который образуется в печени и тонком кишечнике, и апо А-II (составляющий 20% белка ЛПВП), который образуется только в печени. Оставшиеся 10% белка приходятся на другие апопротеины, такие как апо А-IV, апо E и апо J (рис. 1.4, С). Насцентные ЛПВП приобретают ФЛ и ХС; этот процесс называется липидацией. Центральную роль в ранней липидации играют апо А-I и АВСА1-транспортер (АТФсвязывающий кассетный белок-транспортер типа 1), содержащийся в клетках печени и кишечника. В периферических тканях липидация в значительной степени происходит благодаря оттоку ХС от скелетных мышц, жировой ткани, кожи и макрофагов. Для созревания ЛПВП и образования гидрофобного липидного ядра необходима эстерификация ХС; этот процесс опосредован действием фермента ЛХАТ. Окончательный состав зрелых ЛПВП (обозначаемых ЛПВП3), имеющих сферическую форму, включает монослой ФЛ, не39 большие количества свободного ХС и ядро из эстерифицированного ХС. Более крупные ЛПВП2, которые образуются из ЛПВП3 при дальнейшем присоединении ХС, способны к липидации от гидролиза ЛП, богатых ТГ, дающего ФЛ, которые могут затем переноситься к ЛПВП, – процесс, регулируемый ЛПЛ и БПФЛ. Таким образом, ЛПВП играют решающую роль в обратном транспорте ХС, который обеспечивает удаление избытка ХС из периферических клеток, и его доставке в печень и стероидогенные клетки для последующего выделения в виде желчных кислот и с калом. ЛПВП удаляются из плазмы с помощью скэвенджеррецепторов (scavenger receptor BI – SR-BI), которые обеспечивают селективный отбор ХС из ЛПВП. Возможно, наиболее важным является непрямой путь с участием БПЭХ. Вместе с тем ЛПВП имеют разновидности, отличающиеся составом липидов и белков; некоторые из этих частиц, несмотря на низкие концентрации, являются биологически очень активными. Они участвуют в подавлении окисления, воспаления, активации эндотелия, свертывания крови и aгрегации тромбоцитов. Транспорт ХС из нагруженных липидами макрофагов, находящихся в атеросклеротических артериях, так называемых пенистых клеток, обеспечивается в несколько этапов метаболизма ЛПВП. Этот путь называют обратным транспортом ХС, который считается классической защитной функцией ЛПВП против атеросклероза. Каждый день организм среднего человека синтезирует от 1 до 5 г холестерина. Самая большая доля холестерина (80%) синтезируется в печени, некоторая часть производится клетками организма, а 300-500 мг поступает с пищей. В результате сложного метаболизма липидов и ЛП в организме формируются плазменный (экзогенный) и внутриклеточный объемы (пулы) ХС. ХС плазменного пула обменивается с внутриклеточным, причем скорости установления равновесия между пулами сильно различаются в разных органах и системах. Например, быстрый обмен плазменногоХС с внут40 риклеточным, имеет место в клетках печени, кишечника, эритроцитах и некоторых других внутренних органов. Более медленный обмен плазменного и внутриклеточного ХС происходит в периферических тканях, таких как кожа и жировая ткань. Медленный обмен наблюдается в скелетных мышцах и стенках сосудов. Кроме того, имеется необменивающийся пул ХС клеток центральной нервной системы с плазменным ХС. В стационарном состоянии метаболизма поступление синтезируемого и всасываемого ХС в быстро обменивающийся пул сбалансировано выведением ХС путем фекальной экскреции. ЛПВП играют защитную роль, они забирают лишний ХС из крови, собирают его с поверхности клеток и несут к печени, где он разрушается (условно, ХС ЛПВП – «хороший» ХС). ЛПНП, напротив, отдают ХС клеточным мембранам, приводя к отложению ХС в стенках артерий и образованию атеросклеротических бляшек (условно, ХС ЛПНП – «плохой» ХС). Хороший и плохой ХС го человека зависит от наличия у него тех или иных факторов риска сердечно-сосудистых заболеваний или осложнений. В целом в соответствии с современными представлениями о липидном обмене показатель ХС ЛПНП у пациентов с ИБС или высоким риском фатальных сердечно-сосудистых осложнений не должен превышать 100 мг/дл, а для пациентов с факторами риска и умеренным и низким риском фатальных осложнений – не более 115 мг/дл. Практика доказательной медицины показывает, что снижение уровня ХС ЛПНП до названных значений ведет к замедлению или стабилизации атеросклеротического процесса. В результате многочисленных эпидемиологических исследований, направленных на изучение связи концентраций липидов плазмы крови с сердечно-сосудистыми заболеваниями, получены эпидемиологические показатели относительно того, какие уровни липидов плазмы крови желательны. В частности, по мнению Всероссийского научного общества кардиологов, уровень общего ХС в плазме крови не должен превышать 5,0 ммоль/л; ТГ – 1,7 ммоль/л; ХС ЛПНП – 3,0 ммоль/л; ХС ЛПВП – в пределах 1-1,89 ммоль/л. Про «хороший» и «плохой» ХС необходимо знать по двум причинам. Во-первых, показатели «хорошего» и «плохого» ХС необходимы для рациональной организации питания. Известно, что некоторые продукты могут понизить «плохой» ХС, а другие – повысить «хороший». Во-вторых, чтобы правильно сориентироваться в показателях анализа крови, который нужен для диагностики заболевания и оценки эффективности проводимого лечения по коррекции нарушенного липидного обмена. Считается, что уровень хорошего ХС должен быть не менее 40 мг/дл, а повышение ХС ЛПВП до 60 мг/дл и более позволяет даже нивелировать один из основных факторов риска развития сердечнососудистых заболеваний. В отличие от «хорошего» ХС, уровень верхней границы «плохого» ХС для каждого конкретно41 42 Глава 2 КЛАССИФИКАЦИЯ СЕМЕЙНЫХ ГИПЕРЛИПИДЕМИЙ Таблица 2.1 Диагностические коды нарушений метаболизма липидов международной классификации МКБ 9-го и 10-го пересмотров Рубрики МКБ-9 МКБ-10 Название Чистая гиперхолестеринемия (Pure hypercholesterolemia) Чистая гипертриглицеридемия (Pure hyperglyceridemia) Смешанная гиперлипидемия (Mixed hyperlipidemia) Гиперхиломикронемия (Hyperchylomicronemia) Генетическая предрасположенность к сердечнососудистым заболеваниям в большой мере связана с содержанием липидов и липопротеидов в плазме крови, а их нарушения изучаются различными методами, включая классические исследования моногенных мутаций; ресеквенсирование (определение участка мутации); исследование ассоциаций генома (широкого) и характеристик метаболизма с фенотипом. Результаты таких исследований позволяют оценивать значение разнообразия, последовательностей ДНК в регуляции содержания липопротеинов в плазме крови. Понимание механизмов генетической регуляции метаболизма липопротеинов открывает новые возможности классифицировать, диагносцировать и лечить дислипидемии. В соответствии с диагностическими кодами международной классификации МКБ 9-го и 10-го пересмотров нарушения метаболизма липидов относятся к группе метаболических заболеваний, которые отнесены, соответственно, к рубрикам 272 и E78 «Нарушения метаболизма липопротеинов и другие липемии». Наиболее важное практическое значение среди указанных нарушений метаболизма липидов относительно их связи с ишемической болезнью сердца являются следующие наследственные заболевания (табл. 2.1). 43 272.0 E78.0 272.1 E78.1 272.2 E78.2 272.3 E78.3 Каждый тип из гиперлипидемий, указанных в табл. 2.1, обусловлен различными генотипами. При этом разные генотипы (мутации) могут формировать сходные фенотипы (сходные клинические полиморфизмы). Конкретные генетические дефекты представлены в международном онлайнкаталоге «Менделевское наследование у человека» (OMIM – Online Mendelian Inheritance in Man, http://www.omim.org/). В результате становится необходимым описание фенотипов нарушений метаболизма липидов в соответствии с МКБ-9 и МКБ-10 с учетом генетических факторов, их формирующих. Такое описание является актуальным, поскольку от конкретного генетического фактора, формирующего фенотип, зависит тактика его диагностики и коррекции. 2.1. Чистая гиперхолестеринемия (Код МКБ-9/10: 272.0/ E78.0) Чистая гиперхолестеринемия (ЧГХС) сопровождается повышением в плазме крови концентрации общего ХС (преимущественно за счет ЛПНП) в сочетании с нормальными концентрациями триглицеридов в плазме крови. 44 Синонимы: семейная гиперхолестеринемия типа II а (по классификации Фредриксона); гипербеталипопротеинемия; гиперлипидемия группы А; гиперлипопротеинемия липоидного типа, обусловленная повышенной концентрацией ЛПНП в плазме крови. Клиническая симптоматика. Концентрации ХС в плазме крови (мг/дл): у гетерозигот 350–500; у гомозигот 650–1000. Соотношение ХС/ТГ в плазме крови ХС/ТГ > 1,5. Повышенные концентрации ХС в плазме крови, наиболее выражены у гомозиготных больных, у которых в раннем детском возрасте характерно появление ярко-желтых кожных ксантом. Наиболее типичной локализацией ксантом являются области сухожилий, ахилловых, разгибателей пальцев верхних и нижних конечностей, коленных и локтевых суставов. Липоидная дуга роговицы и периорбитальные ксантелазмы у гомозигот появляются в детстве, а у гетерозигот после 30 лет. Таблица 2.2 Взаимосвязь фенотипа с генотипом чистой гиперхолестеринемии Название по OMIM Номер фенотипа OMIM ПозиГен/ ция локус локуса (MIM) Дефект Гиперхолестеринемия семейная Измененная струк143890 19p13.2 LDLR тура рецептора ЛПНП Гиперхолестеринемия аутосомнодоминантная, тип В 144010 2p24.1 603776 Мутация гена PCSK9, кодирую1p32.3 PCSK9 щего пропротеин конвертазу субтилизин/гексин Гиперхолестеринемия аутосомнодоминантная, 3 45 ЛигандАРОВ дефектный апо В-100 Ген/локус Номер MIM 606945 107730 607786 Фенотип ЧГХС обусловлен различными генетическими нарушениями. Наиболее распространенными мутациями, приводящими к формированию фенотипа ЧГХС, являются: мутации гена рецептора ЛПНП (LDLR; OMIM 606945); гена апо В-100 (APOB, OMIM* 107730); гена PCSK9 607786) см. табл. 2.2). 2.1.1. Чистая гиперхолестеринемия, детерминированная мутацией гена рецептора ЛПНП (номер фенотипа OMIM 143890) Основной генетический дефект состоит в патологии рецепторов ЛПНП. Насчитывают пять основных классов мутаций гена рецептора ЛПНП (MIM 606945). Класс 1 – мутации поражают синтез рецепторов ЛПНП в эндоплазматическом ретикулюме (ЭР). Класс 2 – мутации прерывают транспорт рецепторов в аппарат Гольджи, где должна происходить их модификация. Класс 3 – мутации останавливают связывание ЛПНП с рецепторами. Класс 4 – мутации подавляют формирование рецепторлиганд комплекса. Класс 5 – мутации влияют на синтез таких рецепторов, которые не рециклируются нормально. Это приводит к такому состоянию, что в клеточной мембране имеются только вновь синтезированные рецепторы. Распространенность. По оценкам, частота гомозигот составляет 1 на 500, что является самой распространенной мутацией. Среди лиц, перенесших инфаркт миокарда гетерозигот распространенность мутаций гена рецептора ЛПНП составляет 1 на 20. 46 2.1.2. Чистая гиперхолестеринемия, аутосомнодоминантная тип В (номер фенотипа OMIM #144010) Синонимы: аполипопротеин В-100, семейная лиганд – дефектная гиперхолестеринемия; семейный дефект аполипопротеина В-100. Фенотип аутосомно-доминантной ЧГХС типа В детерминируется мутациями в участке связывания апо-В с рецептором ЛПНП. Наиболее важными из выявленных мутаций являются зарегистрированные MIM под номерами: 107730.0009 и 107730.0017. Мутация 107730.0009 – характеризуется заменой ARG3500GLN в гене APOB. Мутация 107730.0017 характеризуется заменой ARG3531CYS в гене АРОВ. Обе мутации характеризуются заменой ARG3500GLN в гене APOB, главного белка ЛПНП. Измененный белок затрудняет связывание ЛПНП с рецепторами на поверхности клетки. В результате снижается выведение ХС-ЛПНП из кровотока по сравнению с нормой. Избыток ХС скапливается в коже, сухожилиях, артерий, в том числе снабжающих сердечную мышцу, повышая риск ИБС. Распространенность чистой гиперхолестеринемии, обусловленной аутосомно-доминантным типом В, по различным оценкам составляет 1 на 1000. 2.1.3. Чистая гиперхолестеринемия, аутосомнодоминантная 3 (ГХС-3) (номер фенотипа OMIM 603776) ГХС-3 является аутосомно-доминантным заболеванием, детерминированным множественными мутациями гена PCSK9 (MIM 607786), кодирующего «пропротеин конвертаза субтилизин/куксин тип 9», расположенного на участке 1p32.3 (см. табл. 2.2). 47 Ген PCSK9 детерминирует продукцию белка, регулирующего концентрации ХС в кровотоке. Предполагают, что белок PCSK9 контролирует количество ЛПНП-рецепторов. У больных с мутацией гена PCSK9 проявляется фенотип ЧГХС, однако функции рецепторов ЛПНП и апо В не изменены. Наиболее тяжелым осложнением этого генетического дефекта являются преждевременное атеросклеротическое поражение коронарных артерий, грудного и брюшного отделов аорты. Атеросклеротические поражения часто обнаруживаются на аортальном клапане и эндокардиальной поверхности сердечной мышцы, симулируя клинику клапанного стеноза. Для гомозиготных больных характерно развитие стенокардии, инфаркта миокарда на 2–3 десятилетии жизни, они редко переживают возраст 30 лет. В гетерозиготном состоянии клиническая симптоматика выражена слабее, осложнения развиваются на десятилетие позднее. Распространенность чистой гиперхолестеринемии, ассоциированной с мутациями в гене PCSK9, пока неизвестна. 2.2. Чистая гипертриглицеридемия (Pure hypergly-ceridemia) (номер фенотипа OMIM 144600) Чистая гипертриглицеридемия ЧГТГ характеризуется изолированным подъемом в плазме крови эндогенных триглицеридов в составе ЛПОНП и пониженной элиминацией ЛПОНП, на фоне обычной диеты. Концентрации триглицеридов плазмы постоянно повышены, в то время как концентрации ХС и фосфолипидов остаются в пределах нормы. Распространенность состояния 1 на 100 (табл. 2.3). Синонимы: эндогенная гипертриглицеридемия; гиперлипопротеинемия тип IV по классификации Фредриксона; гиперлипидемия группы В; гиперпребеталипопротеинемия; гипертриглицеридемия эссенциальная; гиперлипопротеинемия липоидный тип [ЛПОНП]. 48 Таблица 2.3 Взаимосвязь фенотипа с генотипом чистой гипертриглицеридемии Название по OMIM Семейная гипертриглицеридемия Номер Позифенотипа ция OMIM локуса 144600 Ген/ локус - Дефект Ген/ локус Номер MIM Избыточная продукция ЛПОНП и недостаточность их утилизации - Характеристика фенотипа. Минимальные критерии: а) один из родителей и половина ближайших родственников имеют гипертриглицеридемию; б) если уровень ТГ в плазме крови не выше 400 мг/дл, то содержание ХC в плазме крови нормальное; в) содержание ТГ в плазме крови обычно ниже 1500 мг/дл; г) частые случаи диабета в семье; д) исключение фенокопий. Лабораторная диагностика. Основным биохимическим проявлением патологии является высокое содержание ЛПОНП в плазме крови, вследствие чего значительно повышено содержание ТГ (200–1000 мг/дл). Уровень ХС нормальный или слегка повышен. Отношение ХС:ТГ всегда выше 2,5. При электрофорезе выявляется интенсивная полоса пре-бета-ЛП. Клиника. ЧГТГ сопровождается атеросклеротическим поражением коронарных артерий с клиническими проявлениями ИБС. Возможно развитие атеросклероза периферических артерий. При исследовании глазного дна обнаруживается липемия сетчатки. Частым сопутствующим клиническим проявлением заболевания бывают панкреатит, абдоминальные колики. Ксантоматоз встречается редко. Эруптивные ксантомы появляются только при значительном нарастании концентрации ТГ в плазме крови (до 1500–2000 мг/дл). 49 Обычно у больных с ЧГТГ снижена толерантность к углеводам, иногда наблюдается гиперинсулинемия, гиперурикемия. Эти проявления заболевания часто сочетаются с ожирением. В ряде случаев при ЧГТГ выявляются патологические значения тимоловой пробы и повышение активности аминотрансфераз, в связи с чем нередко диагностируются паренхиматозные заболевания печени. Генетика. Чистая гипетриглицеридемия является гетерогенным заболеванием, об этом свидетельствует тот факт, что фенотип находится под сильным влиянием средовых факторов, таких как избыточное потребление углеводов или алкоголя. Вне зависимости от причины заболевания гипертриглицеридемия связана с преждевременным развитием сердечно-сосудистых заболеваний. Гипертриглицеридемия может обнаруживаться среди лиц с сахарным диабетом II (OMIM 125853), а также при семейной комбинированной гиперлипидемии (HYPLIP1; 602491), детерминированной вариабельностью USF1 gene (191523) on chromosome 1q21-q23. Некоторые мутации гена ABCA1 (600046), детерминирующие семейный дефицит ЛПВП или гипо-альфалипопротеинемию (604091), сопровождаются гипертриглицеридемией. Отдельными исследованиями показано, что предрасположенность к гипертриглицеридемии связана с мутациями гена аполипопротеина A5 (APOA5; 606368) и липазы 1 (LIPI; 609252), а также с полиморфизмом гена RP1 (603937.0005). 2.3. Смешанная (комбинированная) гиперлипидемия (Mixed hyperlipidemia) (код МКБ-9/10: 272.2/ E78.2) Смешанная гиперлипидемия генетически гетерогенна. В соответствии с классификацией OMIM фенотип СГЛП может быть обусловлен генетическими мутациями, нарушающими структуры липопротеид липаз, приводящими к 50 повышению содержания ХС и/или ТГ в плазме крови (семейная комбинированная гиперлипидемия (OMIM # 144250) и смешанная гиперлипидемия (болезнь широких бета), преимущественно обусловленная дефектом синтеза белка апо Е. Синонимы: гиперхолестеринемия с эндогенной гипертриглицеридемией; липопротеинемия широких или флотирующих бета; комбинированная гиперлипидемия; повышенное содержание холестерина в сочетании с повышенным содержанием триглицеридов; гиперлипопротеинемия тип IIб или III по классификации Фредриксона; гиперохолестеринемия с эндогенной гипертриглицеридемией; гипер-беталипопротеинемия в сочетании с пре-бета-липопротеинемией; гиперлипидемия группы С; узловатая эруптивная ксантома; туберозная ксантома. При смешанной гиперлипидемии (СмГЛП) плазма крови после отстаивания в течение 16–24 часов при + 4 °С прозрачная или слегка мутная без сливкообразного слоя. Концентрация ХС в крови обычно выше 300 мг/дл, ТГ – более 200 мг/дл. На электрофорезе и улътрацентрифугировании определяется повышенное содержание ЛПНП и ЛПОНП. СмГЛП отличаются от семейной гиперхолестеринемии (OMIM 143 890) и семейной гипертриглицеридемии (OMIM 145750) по следующим причинам: 1) у лиц с СмГЛП активность рецепторов ЛПНП не изменена; 2) среди родственников пациентов СмГЛП наблюдаются только гиперхолестеринемия, только гипертриглицеридемия и гиперхолестеринемия в сочетании с гипертриглицеридемией в результате увеличения концентраций ЛПОНП, ЛПНП или их сочетания; 3) в отличие от семейной гиперхолестеринемии, СмГЛП проявляется лишь у 10–20% пациентов в детском возрасте, как правило, в виде гипертриглицеридемии. Ксантомы редки. У гомозигот может наблюдаться тяжелая гипертриглицеридемия. 51 2.3.1. Семейная комбинированная гиперлипидемия (номер фенотипа OMIM # 144250) Семейная комбинированная гиперлипидемия является наиболее распространенной генетической формой гиперлипидемии среди людей, переживших инфаркт миокарда. Предполагается, что до 10% преждевременной коронарной болезни сердца обусловлена СмГЛП. В настоящее время насчитывается три генетических локуса, формирующих фенотип семейной комбинированной гиперлипидемия СКГЛП (см. табл. 2.4). 1. Фенотип СКГЛП сцепленный с локусом 8p21.3, кодирующий ген LPL 609708. Выявлены четыре мутации в гене LPL 609708.0032, -0033, -0035, -0038, все формируют фенотип, соответствующий СмГЛП. 2. Фенотип СКГЛП1 связан с участком на хромосоме 1q21-q23 (HYPLIP1-602491), ассоциируется с геном, кодирующим ген USF1 (191523) (кодирующий транскрипционный фактор). 3. Фенотип СКГЛП2 связан с участком на хромосоме 11 (HYPLIP2; 604499). Таблица 2.4 Связь фенотипа СКГЛП с мутациями гена ЛПЛ Название фенотипа Номер Позиция фенотипа локуса OMIM СКГЛП СКГЛП1 СКГЛП2 Ген/ локус Повышение концентрации апo B 609708 Ассоциируется с геном кодируюHYPLIP1 щим (USF1; 191523) 602491 8p21.3 144250 1q21-q23 11:0 – 53,700,000 Дефект Ген/ локус Номер MIM LPL Детали молекулярHYPLIP2 ного дефекта неиз- 604499 вестны 52 Распространенность фенотипа СКГЛП с мутациями гена ЛПЛ по разным оценкам составляет от 0,2 до 2%. Клиника. Заболевание проявляется в возрасте 30–50 лет, сопровождается различными формами ксантоматоза. Наиболее типичны туберозные и туберо-эруптивные ксантомы с локализацией на локтях, коленях, ягодицах, плоские и туберо-эруптивные ксантомы между пальцев, в складках кожи ладоней – иногда с выраженной ксантохромией. У больных обычно увеличена масса тела. Из осложнений характерно атеросклеротическое поражение коронарных артерий с клиническими проявлениями ИБС. У большинства больных отмечается облитерирующий атеросклероз периферических артерий, чаще нижних конечностей, иногда с клиникой перемежающейся хромоты, нередко диагностируется сахарный диабет или пониженная толерантность к углеводам, гиперурикемия. Характеристика фенотипа. Главные критерии: а) наличие липопротеинемии широких или флотирующих бета в плазме крови у ближайших родственников. Минимальные критерии: а) плоские и тубероэруптивные ксантомы, б) повышенный уровень ХС и ТГ в плазме крови быстро снижается при уменьшении калорийности рациона и ограничении потребления углеводов; в) наличие липопротеинемии широких или флотирующих бета в плазме крови у ближайших родственников; г) исключение фенокопий. Распространенность 1 на 10 000. Лабораторная диагностика. Плазма крови (натощак) обычно мутная. При ее отстаивании в течение 16–24 часов при +4 °С иногда образуется тонкий слой ХМ. Содержание ХС в крови обычно выше 300 мг/дл. Уровень ТГ в плазме крови колеблется от 200 до 900 мг/дл. Отношение ХС/ТГ – 0,5–2,0. Характерно повышенное содержание в плазме крови ЛППП, которые отличаются от нормальных по белковому составу и повышенному содержанию в них эфиров ХС. Указанные особенности состава ЛПОНП определяют такую их электрофоретическую подвижность, которая в норме свойственна ЛПНП. При электрофорезе ЛПОНП образуют широкую полосу – от бета до пре-бета положения. Выявление таких бета-ЛПОНП – широких бета является типоспецифическим диагностическим критерием, а само заболевание называют также болезнью широких бета. Генетика. Фенотип (наличие липопротеинемии широких или флотирующих бета в плазме крови) обнаруживается среди мужчин и женщин одинаково часто, заболевание проявляется редко в возрасте до 20 лет. Почти у половины родственников пробандов с наличием липопротеинемии широких или флотирующих бета в плазме крови также выявляется это нарушение. Генетические исследования выявили семьи как с аутосомно-доминантным, так и с аутосомно-рецессивным характером наследования. Одна группа пациентов (большинство) являются гомозиготами апо Е2. Другая группа пациентов с болезнью широких бета обычно имеют признаки диабета, ожирения или гипотиреоидизма. Белок апо Е кодируется геном APOE, расположенным на 19 хромосоме в одном кластере с APOC1 и APOC2 (табл. 2.5). Полиморфизм апо Е обусловлен неоднородностью локуса гена ApоЕ, который может присутствовать в одной из трех форм (аллелей): апо E2, Апо E3 («дикий» тип), апо E4. Таким образом, в популяции возможно существование шесть фенотипов апо Е, отличающихся как по своей распространённости, так и функциональной значимости. 53 54 2.3.2. Смешанная гиперлипидемия (болезнь «широких» бета) (номер фенотипа OMIM 107741) (табл. 2.5). Таблица 2.5 Связь фенотипа cмешанная гиперлипидемия (болезнь «широких» бета) с мутациями гена ЛПЛ Название фенотипа Номер фенотипа OMIM Позиция локуса Ген/ локус OMIM Дефицит апо Е или дефект апо Е, гиперлипопротеинемия тип 3 107741 19:45,409,038 19q13.32 107741 Дефект Комбинация E2/E2 Комбинацию E2/E2 связывают с генетическим нарушением гиперлипопротеинемии широких бета, а также с повышенным риском атеросклероза. Распространенность генотипа E2/E2 в различных популяциях составляет 1–5%. 2.4. Гиперхиломикронемия (МБТ 9/10: 272.3 / Е78.3) У больных с гиперхиломикронемией (ГХМ) в плазме крови натощак резко повышено содержание ХМ. Фенотипически различают два варианта ГХМ. 1. Чистая ГХМ характеризуется повышенными концентрациями хиломикронов (ХМ) при пониженных концентрациях других классов липопротеидов. При этом выделяются три подтипа (OMIM-фенотипы): 238600; 207750; 118830. 2. Смешанная ГХМ – сочетание повышенных концентраций ХМ и триглицеридов (ОМIМ-фенотипы): #144650 и 145750. Распространенность. Оба варианта ГХМ наблюдаются с одинаковой частотой среди мужчин и женщин. Встречаются крайне редко (по расчетам – 1 на 1 млн человек). Характеристика фенотипа. Главные критерии: а) гиперхиломикронемия; б) наличие у родителей или ближайших родственников пробанда чистой гипертриглицеридемии или смешанной гиперхиломикронемии. 55 Минимальные критерии: а) наличие у одного из родителей или взрослых сибсов чистой гипертриглицеридемии; б) случаи панкреатита, абдоминальных колик, сахарного диабета среди родственников; г) исключение фенокопий. Лабораторная диагностика. При отстаивании в течение 16–24 часов при +4 °С плазма крови мутная, определяется сливкообразный слой ХМ. В плазме крови повышено содержание ХМ и/или ЛПОНП в сочетании с пониженным содержанием ЛПВП. Концентрация ТГ в крови резко увеличена – более 400–500 мг'/дл. Уровень ХС достигает 300– 500 мг/дл. 2.4.1. Чистая гиперхиломикронемия Клиника. Заболевание обычно проявляется в раннем детском возрасте. Часто выявляются гепатоспленомегалия, приступы панкреатита, абдоминальных болей. Заболевание характеризуется выраженными кризами – нарастанием болей в животе, общим недомоганием, появлением лихорадки, лейкоцитоза. В таком состоянии с недиагностированной гиперлипидемией больные иногда попадают на операционный стол по поводу острого живота. Возможно периодическое появление папулоэруптивных ксантом. При высоком уровне ТГ у больных может развиваться липоидная дуга роговицы. Генетика. Выделяют три подтипа чистой ГХМ. Подтип 1а обусловлен дефицитом липопротеид липазы D, а подтип 1b – дефицитом апопротеина С2. Наследуемость подтипов 1а и 1b аутосомно-рецессивная. Подтип 1с обусловлен низкой активностью, постогепариновой липолитической липазы, при этом наследуемость подтипа 1с аутосомно-доминантная (табл. 2.6). 56 Таблица 2.6 Связь фенотипа с генотипом чистой гиперхиломикронемии (МКБ 9/10 – 272.3/ E78.3) Характеристики Фенотип Синонимы OMIM (фенотип) MIM ген/локус Локализация гена Наследуемость Мутации 2.4.2. #144650) Подтип 1а Подтип 1b Дефицит ЛПЛ Дефицит аполипопротеина C-II Гиперлипопротеинемия тип 1а; гиперхиломикронемия; гиперлипемия семейная; идиопатическия тип Бюргера Крутца; дефицит липазы D; дефицит LIPD; семейная хиломикронемия Гиперлипопротеинемия тип Ib; анаполипопротеинемия C-II; дефицит APOC2 238600 207750 609708 608083 8p22 19q13.2 Аутосомно-рецессивная (АР) Подтип 1с Хиломикронемия семейная, обусловленная наличием ингибитора ЛПЛ Гиперлипопротеинемия, тип 1C; очень низкая активность постгепариновой липолитической липазы 118830 колик. Возможно развитие гепатоспленомегалии, панкреатита. На фоне атеросклеротического поражения коронарных артерий отмечаются клинические проявления острой коронарной недостаточности. При облитерирующем атеросклерозе артерий нижних конечностей у больных со смешанной гиперхиломикронемией наблюдается перемежающаяся хромота. Типичными кожными проявлениями являются эруптивные ксантомы. При смешанной гиперхиломикронемии понижена толерантность к жирам и углеводам. Заболевание часто сочетается с сахарным диабетом, гиперурикемией. Генетика. Наследственность смешанной гиперхиломикронемии аутосомно-доминантная. Предположительно связана с мутацией гена APOA5 (MIM 606368), липазы I (MIM 609252), гена RP1 (MIM 603937.0005) (табл. 2.7). Таблица 2.7 Связь фенотипа с генотипом смешанной гиперхиломикронемии (МКБ 9/10 – 272.3/ E78.3) Аутосомнодоминантная Название фенотипа Множественные аллели и структуры Смешанная гиперхиломикронемия (ОМIМ Синонимы: гиперлипидемия тип 5 (по классификации Фредриксона); гиперхиломикронемия позднего развития; семейная гиперхиломикронемия с гипетриглицеридемией; гиперлипемия смешанная; комбинированная индуцированная жирами и углеводами. Клиника. Клиническая симптоматика напоминает таковую при чистой ГХМ. Заболевание проявляется на 2–3-м десятилетии жизни. Больные обычно имеют избыточную массу тела. Характерно наличие в анамнезе абдоминальных 57 Номер Позифенотипа ция OMIM локуса Ген/ локус OMIM Дефект Гиперхиломикронемия позднего развития 144650 11q23.3 606368 Мутация гена аполипопротеина A5 (APOA5) Предрасположенность к гиперхиломикронемии 145750 15q11.2q13.1 609252 Мутация гена, липазы I (LIPI) 8q12.1 603937.0005 Мутация гена RP1 Среди родственников больных со смешанной ГХМ в равных отношениях встречается чистая гипертриглицеридемия и смешанная гиперхиломикронемия, а также нормальные липопротеиновые фенотипы. Существует предположение, что аллельные сочетания семейной недостаточности ЛПЛ и чистой гипертриглицеридемии могут фенотипически 58 проявляться в виде смешанной ГХМ. Не исключено мультифакториальное наследование смешанной гиперхиломикронемии. То есть заболевание является следствием взаимодействия генотипа и специфических средовых факторов: контрацептивных эстрогенов, неумеренного потребления алкоголя, большого количества углеводов в др. Глава 3 ОСНОВНЫЕ ПРИНЦИПЫ ПРОФИЛАКТИЧЕСКИ И ЛЕЧЕНИЯ СЕМЕЙНЫХ ГИПЕРЛИПОПРОТЕИДЕМИЙ Основным принципом профилактики и лечения СГЛП является нормокопирование, т.е. достижение нормального фенотипа при патологическом генотипе. Достижение нормального липопротеинового фенотипа снижает риск атеросклеротического поражения сердечно-сосудистой системы и является одним из подходов к первичной и вторичной профилактике ИБС. Лечебно-профилактические мероприятия следует проводить всем больным СГЛП – даже при отсутствии жалоб и клинической симптоматики, с учетом характера наследования (моногенное, полигенное, комбинированное). Наиболее сложными для профилактики и лечения являются моногенные формы СГЛП. Это связано с тем, что при моногенных дефектах, например, это имеет место при семейной ГХС, семейной недостаточности ЛПЛ, наличие патологического гена обязательно реализуется изменениями метаболизма ЛП и обычно в детском возрасте, и профилактику и лечение необходимо проводить с детства. При полигенных мультифакториальных формах СГЛП как заболевание может проявиться под воздействием неблагоприятных средовых факторов риска. 59 60 Таким образом, выявление моногенных типов СГЛП являются абсолютным показанием для проведения специфических лечебных мероприятий (диетологических, медикаментозных и др.). Семейные ГЛП с полигенным наследованием могут быть в определенной степени скоррегированы устранением патогенных воздействий и диетической и медикаментозной терапией. Главные направления профилактики и терапии СГЛП определяются характером биохимических механизмов развития заболевания. Наиболее целесообразными считаются следующие этапы лечебно-профилактических мероприятий. 1. Прежде чем приступить к специфической терапии СГЛП, больного следует нацелить на необходимость устранения вредных привычек – курение, потребление алкоголя; рекомендовать повысить физическую активность при наличии гиподинамии, исключить психоэмоциональные перегрузки. 2. В большинстве случаев необходимым условием является лечение ожирения и достижение идеальной массы тела с учетом возраста, пола, конституциональных особенностей. Наиболее простой способ определения идеальной массы тела – по формуле Брока. По этой формуле нормальная масса тела в килограммах равна росту в сантиметрах минус сто. Достижение идеальной массы тела является ведущим принципом терапии гипертриглицеридами, смешанной гиперлипедемии, гиперхиломикронемии. В отдельных случаях снижение массы тела и устранение влияния неблагоприятных средовых факторов может быть достаточным для нормализации липидного спектра плазмы крови. 3. Основным лечебным подходом при СГЛП служит диетотерапия. Диетологические мероприятия преследуют несколько целей: а) устранение избыточной массы тела; б) проведение дифференциальной днагностики полигенных нарушений метаболизма липопротеидов, индуцируемых избыточной калорийностью питания, углеводами, жирами, алкоголем; в) специфическую коррекцию отдельных фенотипов СГЛП. Коррегирующие диетологические мероприятия – качественные, количественные, модифицированные с учетом липопротеинового фенотипа следует проводить под контролем общеклинического состояния больного, динамики массы тела и лабораторных данных. Больной должен детально усвоить индивидуальные особенности своего рациона питания и придерживаться его в течение всей жизни. 4. Медикаментозная терапия используется в случаях, когда длительное (не менее 3-х месяцев) применение диеты малоэффективно. Медикаментозная терапия применяется после 3–6 месяцев диетотерапии или сразу в комбинации с диетотерапией. При назначении медикаментозной терапии учитывают пол, возраст, степень повышения липидов, наличие атеросклеротических повреждений сосудов (расчетные таблицы риска фатального ИМ или риск заболеть ИБС). Применение медикаментозной терапии, как и диетотерапии, должно быть постоянно и под контролем клинического состояния и лабораторных биохимических параметров. Медикаментозная терапия используется особенно при генетических семейных нарушениях липидного обмена и соответствует фенотипам ДЛП. Определенным фенотипам по механизму действия соответствуют разные классы медикаментов. Группы гиполипидемических препаратов Статины – ингибиторы редуктазы ГМГ-коензима А Препараты этой группы подавляют (блокируют) активность образования ХС в печени. Это наиболее широко представленная группа гипохолестеринового действия, включает препараты химической модификации: ловастатин, симвастатин, правастатин, флювастатин, аторвастатин, розувастатин. Эти препараты имеют 61 62 оригинальные формы и дженерики, общим количеством около 30 фирменных названий. Ингибитор абсорбции ХС в кишечнике – эзетимиб (эзетрол) Производные фиброевой кислоты (фибраты) Возможно применение комбинации этих препаратов. Гиполипидемические препараты различаются преимущественным действием на липопротеины, богатые триглицеридами (фибраты), или на липопротеины, богатые холестерином (ингибиторы редуктазы ГМГ-коензима А). При разных типах дислипопротеидемий отмечаются эти различия, что и определяет применение препаратов. В соответствии с рекомендациями ВНОК эффективность гиполипидемических средств при различных типах СГЛП располагается в следующем порядке. Семейная гиперхиломикронемия типа I. Хорошо реагирует нормокопированием при правильной диетотерапии. При семейной чистой гиперхолестеринемии статины > никотиновая кислота > (препараты группы фибратов или фиброевой кислоты) > препараты ненасыщенных жирных кислот > липотропные вещества. При семейной ГЛП смешанной клофибрат > никотиновая кислота >эстрогены > препараты ненасыщенных жирных кислот. При семейной чистой гипертриглицеридемии (ГЛП типа IV): клофибрат > никотиновая кислота> эстрогены > препараты ненасыщенных жирных кислот. При гиперхиломикронемии – никотиновая кислота. Медикаментозные средства имеют существенные недостатки. В ряде случаев их применение неэффективно, часто наблюдаются побочные явления, индивидуальная непереносимость. 5. В последние годы для коррекции биохимических нарушений при СГЛП используют методы быстрого выведения из организма продуктов аномального обмена. С этой целью применяют гемосорбцню, плазмаферез, липидэкстракцию. Однако опыт такого рода лечения пока еще ограничен. Для поддержания длительного терапевтического эффекта обычно оказывается необходимым использование комплексной терапии. 6. Хирургическое лечение может быть использовано в наиболее тяжелых случаях при гомозиготной семейной ГXC. Разработаны методы радикальной хирургической коррекции: выключение из системы пищеварения части подвздошной кишки – илеошунтнрованне, наложение портакавального анастомоза; наложение фистулы желчного пузыря с перевязкой желчного протока. Оперированные больные для поддержания эффекта радикального лечения нуждаются в постоянном соблюдении специфической диеты. В настоящее время в медицинской практике СГЛП диагностируются, как правило, уже при наличии клинически выраженных осложнений, связанных с атеросклерозом. Проведение диагностики современными методами пока еще сложно и требует комплексного медико-генетического, клинического и биохимического обследования не только больного, но и его родственников. Вместе о тем использование такого подхода позволяет проводить дифференциальную диагностику и фенотипирование СГЛП, а также способствует активному выявлению ранних признаков заболевания в этих семьях. Своевременная диагностика и типоспецифическая коррекция СГЛП является одним из направлений профилактики ИБС и других атеро-склеротических заболеваний сердечно-сосудистой системы. 63 64 Глава 4 ОСНОВЫ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ ПРИ СЕМЕЙНЫХ ГИПЕРЛИПОПРОТЕИДЕМИЯХ Благодаря завершению картирования генома человека, выявлению сотен генетических маркеров, имеющих отношение к гиперлипидемиям, доступности многих генетических тестов, коммерциализации генетической информации сформировались условия использования генетических подходов в медицинской практике. Растет потребность среди членов семей больных с сердечно-сосудистыми заболеваниями (ССЗ) в информации относительно наследования указанных заболеваний среди родственников. Расчетные данные распространенности различных мутаций, детерминирующих нарушения метаболизма липидов и липопротеидов, указывают на то, что распространенность гетерозигот только по гену LDLR составляет 1 на 200–500 человек, что делает эту мутацию наиболее распространенной среди наследственных заболеваний. Полигенная гиперхолестеринемия распространена, по расчетам, у каждого 10-го взрослого, находящегося на европейской диете, семейная чистая гипертриглицеридемия составляет 1% среди общего населения. Конкретные генетические дефекты, приводящие к дислипидемиям, составляют небольшую долю от их общего числа. Основной вклад в заболевания атеросклеротического 65 профиля вносят дислипидемии, обусловленные мультифакториально, т.е. взаимодействием многочисленных генетических факторов, каждый из которых имеет небольшой эффект, и факторов среды, таких как диета, содержащая избыточное количество жиров животного происхождения, малоподвижный образ жизни, злоупотребление алкоголем, сопутствующие заболевания и др. Вместе с тем генетическая детерминация мультифакториальности гиперлипидемий до конца не изучена. Имеется множество косвенных данных, свидетельствующих о том, что мультифакториальность есть степень незнания всего разнообразия генетических факторов, детерминирующих нарушения метаболизма липидов. Гиперлипидемии, обусловленные как конкретными генетическими мутациями, так и дислипидемии с мультифакториальным генезом, являются предметом медико-генетического консультирования. Генетическое консультирование состоит из специфических медицинских функций, с помощью которых пациенты или родственники, имеющие риск наследственного заболевания, получают консультацию относительно последствий этого заболевания, вероятности развития его, способов профилактики или снижения риска. 4.1. Общий алгоритм медико-генетического консультирования (МГК) Проведение медико-генетического консультирования состоит из нескольких составляющих: 1) первичного обследования; 2) генетического тестирования; 3) консультации/совета. Первичное обследование Задачами первичного обследования являются: – определение соответствия обращения консультируемого (по нозологии); 66 – ургентность состояния обратившегося; мотивы его обращения за консультацией; – опасения, ожидания, представление пациента о проведении исследования. Изучение медицинской истории случая включает в себя: – изучение специфической проблемы для пациента относительно его здоровья; – изучение индивидуальной медицинской информации, случаев госпитализации и хирургического лечения по теме консультации, в том числе на основе конкретных медицинских документов; – наличие врожденных аномалий/дефектов при рождении; – наличие приема лекарственных препаратов (тип, показания, дозировка, длительность приема). Изучение семейного анамнеза консультируемого: – составление родословной (3–4 поколения); – изучение доступных медицинских документов родственников и регистрация их в родословной. Физикальное обследование Оценка риска: – оценка семейного риска развития заболевания (причина консультации); – оценка роли наследственности и среды. Информирование (обучение) консультируемого относительно: – базовых сведений о генетических и медицинских концепциях; – наличия соответствующего наследственного синдрома и риска заболевания; – характера наследования; – степени риска для родственников; – выгоды, ограничения и риска генетического тестирования; – методов лечения заболевания и снижения его риска. Все выявленные факторы должны фиксироваться в родословной. Составление родословной является обязательным элементом медико-генетического консультирования. При составлении родословной используются стандартные обозначении (рис. 4.1). Если в конкретном случае имеется соответствующий генетический тест и возможность выполнения генетического тестирования и/или проведения генетического исследования, необходимо: – ознакомить пациента с основными генетическими понятиями; – ознакомить пациента с предполагаемым характером наследственности; – информировать пациента относительно генетических рисков (предиктивность) и возможности модификации риска заболевания; – оценить ответную реакцию пациента на оценку риска. При этом важно обеспечить правильное понимание пациентом рисков его состояния, поскольку от этого зависит согласие консультируемого с предложенным порядком и сроками проведения генетического исследования. Самые ранние проявления ССЗ по возрасту происходят в семьях с положительной историей этого заболевания. Если врач при сборе семейного анамнеза больного обнаруживает наличие сходных случаев в семье, то это служит прямым указанием на возможную наследственную природу этих случаев. Показания для медико-генетического консультирования на основании данных анализа родословной: – 2 или 3 родственника, имеющих ИБС в возрасте до 60 лет; 67 68 4.2. Обоснование составления родословной – наличие случаев инфаркта миокарда, инсультов, внезапной смерти среди ближайших родственников (родители, братья и сестры) в возрасте до 50 лет. Накопление ИБС среди членов одной семьи является показанием для проведения клинико-биохимического обследования как пробанда (лицо, с которого начинается генетическое обследование), так и его ближайших родственников. Показатели липидов, свидетельствующие о дислипидемии Содержание холестерина Общий холестерин Концентрации ХС ЛПВП («хороший» холестерин) Концентрации 6,3 ммоль/л и выше Менее чем 1 ммоль/л (для мужчин) Менее чем 1,3 ммоль/л (для женщин) Более 4,1 ммоль/л >5,65 ммоль/л (500 мг/дл и выше) ХС ЛПНП («плохой» холестерин) Концентрации ТГ в плазме крови И сто чник : http://emedicine.medscape.com/article/126568-clinical#a0218 Обычный риск Умеренный генетический риск Критерии уровней триглицеридов Содержание триглицеридов Нормальное Погранично высокое Высокое Очень высокое Высокий генетический риск Концентрации (мг/дл) < 150 150-199 200-499 >500 Классификация, основанная на содержании ХС в ЛПНП и не-ЛПВП Показатели ИБС* и эквиваленты риска ИБС: сахарный диабет и т.д. ХС ЛПНП (мг/дл) ХС не-ЛПВП (мг/дл) <100 <130 <130 <160 <160 <190 Два и более факторов риска Рис.4.1. Демонстрация случаев ИБС при составлении родословной как важного фактора риска развития заболевания у обследуемого Отсутствие факторов риска * Ишемическая болезнь сердца 69 70 Риск ИБС 10-летний риск для ИБС >20% 10-летний риск для ИБС <20% 0-1 фактор риска 4.3. Анализ родословной и семейной истории При клинико-генеалогическом изучении болезней с наследственной предрасположенностью следует особенно тщательно анализировать родословные. Наряду с получением обычной для моногенных форм информации необходимо обращать особое внимание на следующие вопросы методического характера. По мере развития геномной медицины, с открытием и применением в практику новых генетических тестов и конкретных мутаций значение анализа родословной не снижается, а, наоборот, увеличивается. Это обусловлено тем, что одинаковая мутация в каждой семье проявляется в разной генотипической среде. При анализе родословных необходимо в полной мере обеспечить точность диагностики заболевания у всех явно пораженных или предположительно пораженных членов семьи. При оценке выраженности болезни у разных членов семьи следует отмечать сходство фенотипического проявления среди различных пораженных родственников (форма течения болезни, возраст в момент начала заболевания, степень тяжести, реакция на лекарства и т.д.). Большое сходство клинической картины обычно указывает на существенное значение генетической компоненты и, следовательно, на повышенный риск развития заболевания у близких кровных родственников. Степень кровного родства среди больных членов семьи должна быть установлена достаточно точно. При полигенном характере болезни это важно для оценки риска, потому что степень риска у близких родственников резко возрастает. Распределение случаев ИБС и факторов риска в семейном анамнезе дает достаточную информацию для оценки степени связи с конкретными факторами риска заболевания. Обнаружение в определенной семье каких-либо мутаций 71 можно оценить с помощью сопоставления случаев патогенетически связанных с ними заболеваний в родословной. Например, наличие гиперлипидемии у консультируемого в сочетании с накоплением в семейном анамнезе случаев инфаркта миокарда, стенокардии, инсультов, внезапных смертей дает ориентацию относительно значения данной гиперлипидемии в отношении риска ССЗ. В данном случае тип гиперлипидемии очевидно связан с риском ССЗ. При медико-генетической консультации пациентов с дислипидемиями указанный метод применяется во всех случаях без исключения. Задачей анализа родословной является построение полной родословной с помощью стандартных обозначений. При этом фиксируются все заболевания среди родственников и признаки, имеющие отношение к данному заболеванию. Для этого необходимо выполнить следующие этапы: – сформулировать вопросы конкретно данному пациенту; – установить историю болезни/состояния; – зафиксировать по воспоминаниям (специфические симптомы, диагнозы, курсы лечения); – документировать этничность и близкородственность; – выявить диагнозы, требующие подтверждения. 4.4. Практические рекомендации составления родословной Составление родословной начинается с консультирующегося или с пробанда. Консультирующимся называется лицо, обратившееся к врачу. Пробанд – больной или носитель изучаемого признака, с него начинается обследование членов семьи. Часто консультирующийся и пробанд являются одним и тем же лицом. Дети одной родительской пары называются сибсами (братья и сестры). Семьей называют супружескую пару и их 72 детей (ядерная семья). Обычно родословная собирается по одному или по нескольким признакам. Символ Описание Лицо мужского пола Лицо женского пола Умерли Брачная пара (муж/жена) Сибсы (братья/сестры) Пробанд № 1_demo Рис. 4.2. Пример построения родословной ИМ – инфаркт миокарда; АГ – артериальная гипертония; ГХС – гиперхолестеринемия 73 Составление родословной сопровождают краткой записью о каждом члене родословной с точной характеристикой его родства по отношению к пробанду (легенда родословной) (рис. 4.2). 4.5. Показания для проведения медико-генетического тестирования Генетическое тестирование начинается с члена семьи (пробанда), у которого уже имеются проявления семейного заболевания, с помощью установления генетической мутации, которая может быть причиной основного заболевания. Как только мутация найдена, есть основания проводить генетическое тестирование у кровных родственников с целью выявления, кто из них может быть носителем данной мутации, приводящей к развитию ССЗ. Результаты выявленных данных генетического тестирования способствуют: – уточнению и подтверждению диагноза у консультируемого; – идентификации родственников, имеющих генетический риск. Задачи генетического тестирования: – подтверждение диагноза; – получение информации относительно прогноза заболевания в будущем; – носительство специфического гена, который увеличивает риск заболевания у детей. Алгоритм генетического тестирования Для проведения генетического тестирования необходимо: 1) выбрать генетический тест; 2) выявить наиболее информативных лиц для тестирования; 74 3) объяснить возможные исходы тестирования и способы их использования; 4) обсудить потенциальную стоимость, риски, выгоды и ограничения тестирования; 5) определить субстрат тестирования; 6) получить информированное согласие на проведение генетического тестирования. 4.6. Семейная чистая гиперхолестеринемия (МКБ-10:Е78.0) как пример использования медикогенетического консультирования Семейная ГХС (СГХС) имеет все показания для генетического тестирования в семье. Характеристика семейной чистой гиперхолестеринемии IIа типа: – распространенность гетерозиготной формы в общей популяции составляет 1/500; – характеризуется преждевременным развитием атеросклероза; – причиной заболевания являются мутации в генах LDLR, АPOВ и PCSK9 (см. табл. 2.2); Гетерозиготы: увеличение содержания ХС ЛПНП плазмы в 2-3 раза выше нормы, имеют риск ксантоматоза, липоидной дуги роговицы, ИБС (включая ИМ) в возрасте 30–60 лет. Гомозиготы: повышение уровня ХС ЛПНП плазмы в 8 раз выше нормы, риск развития ИБС (включая ИМ) в возрасте до 10–20 лет. В семье с СГХС медико-генетическое консультирование решает следующие задачи. 1. Идентификация конкретной мутации у пробанда, которая может быть причиной основного заболевания. 75 2. Как только мутация найдена, проводится генетическое тестирование у кровных родственников с целью установления, кто из них может быть носителем данной мутации. Диагноз «определенная» СГХС ставится, если: (a) уровень общего ХС >6,7 ммоль/л или уровень ХС ЛПНП >4 ммоль/л у ребенка младше 16 лет или уровень общего ХС >7,5 ммоль/л или уровень ХС-ЛПНП >4,9 ммоль/л у взрослого, в сочетании; (б) наличие сухожильного ксантоматоза у родственников 1-й степени родства (родители, дети) или у родственников 2-й степени родства (дедушки, бабушки, дяди или тети); или (в) положительный тест ДНК-диагностики, подтверждающий мутации в генах LDLR, АPOВ и PCSK9 (см. табл. 2.2); Диагноз «возможная» СГХС ставится, если: а) уровень общего ХС >6,7 ммоль/л или уровень ХС ЛПНП >4 ммоль/л у ребенка младше 16 лет или общий ХС >7,5 ммоль/л или уровень ХС ЛПНП >4,9 ммоль/л у взрослого (исходный уровень липидов или самый высокий уровень на терапии), плюс (б) одно из нижеперечисленного: – отягощенный семейный анамнез (ИМ) до 50 лет у родственника 2-й степени родства, до 60 лет у родственника 1-й степени родства; – уровень общего ХС >7,5 ммоль/л у взрослого 1-й или 2-й степени родства; – повышение уровня общего ХС >6,7 ммоль/л у ребенка или родственника 1-й степени родства. 4.7. Алгоритм диагностики гипертриглицеридемий Повышенные триглицериды определяются прямым лабораторным анализом сыворотки или плазмы крови после 10–12 часов отстаивания в холодильнике. После определения 76 общего уровня ТГ необходимо определить, за счет каких фракций липопротеидов ТГ повышены. Когда уровни ТГ <1000 мг/дл, ХМ обычно отсутствуют. Если уровни ТГ более этого показателя, обычно присутствуют ХМ. Если ТГ повышены, но менее 1000 мг/дл, а общий ХС тоже повышен, нарушение метаболизма липопротеидов может быть обусловлено: (1) одновременным повышением концентраций ЛПНП и ЛПОНП, что указывает на смешанную (комбинированную) гиперлипидемию (МКБ-10:Е78.2); 2) повышенным содержанием в плазме крови ремнантов ЛПОНП или ЛППП, что указывает на ГЛП или дисбеталипопротеидемию (МКБ-10: Е78.2). Эти два нарушения метаболизма липопротеидов могут различаться с помощью прямого определения ХС ЛПНП, который является доступным в коммерческих лабораториях. Если прямой ХС ЛПНП значительно ниже, более вероятен диагноз ГЛП дисбеталипопротеидемии. Единственная процедура, которая надежно отличает смешанную ГЛП (повышенное содержание ХС ЛПНП и ТГ) от дисбеталипопротеидемии (повышенное содержание ЛППП), – определение количества бета-ЛП на электрофорезе. Этот относительно дорогой анализ требует ультрацентрифугирования, а затем электрофореза. На практике определение различий между этими типами ГЛП редко необходимо, потому что лечение обоих состояний по существу одинаковое. Модификация диеты, физическая нагрузка, соответствующее снижение веса улучшают оба типа ГЛП. Типы IIb и III также хорошо реагируют на назначения никотиновой кислоты (ниацина) и/или производных фиброевой кислоты (фибратов). Отсюда, независимо от того, какой установлен диагноз, лечение – одинаковое. 77 4.8. Алгоритм диагностики гиперхиломикронемий (МКБ-10:Е78.3) Если уровни ТГ больше, чем 1000 мг/дл, должно быть подтверждено наличие ХМ. Самым простым и дешевым тестом определения ХМ в плазме крови является помещение пробирки образца плазмы или сыворотки на 10–12 часов в холодильник. После отстаивания в течение указанного времени наличие верхнего сливкообразного слоя свидетельствует о наличии ХМ, при этом: – если инфранатант (остаточная фракция) мутный, а уровни ЛПОНП высокие, можно предполагать смешанную гиперхиломикронемию (гиперхиломикронемия с гипетриглицеридемией); – если инфранатант прозрачный, содержание ЛПОНП в норме, следует подозревать чистую (гиперхиломикронемию). После получения данных клинического, лабораторного и генетического обследования следует: – провести дифференциальный диагноз; – определить количественную оценку риска; – рассчитать индивидуальный риск проявления, осложнения и т.д. 4.9. Обсуждение диагноза и генетического состояния с пациентом Перед представлением информации о заболевании или признаке необходимо: – сформулировать программу консультирования; – скомпоновать полученные результаты обследования в рабочий диагноз; – разработать план. 78 Информация, представленная консультируемому, должна содержать: диагноз заболевания; его этиологию; историю состояния; изменчивость экспрессии; пенетрантность; прогноз; методы профилактики и лечения. При проведении медико-генетической консультации следует учитывать степень понимания пациента и его реакцию. При необходимости приспособить план работы в соответствии с условиями, в которых находится пациент, ознакомить пациента с основными генетическими понятиями и характером наследственности данного случая. Необходимо оценить ответную реакцию пациента на оценку риска. Следует обращать внимание на правильное понимание пациентом рисков, связанных с результатами тестирования, и корректировать консультацию в зависимости от понимания и ответной реакции пациента. 4.10. Этико-правовые вопросы обследования медикогенетического консультирования При проведении медико-генетического консультирования следует придерживаться нижеследующих положений. 1. Получить информированное согласие (согласие конкретного лица на проведение медицинского тестирования, лечения и иных медицинских процедур, основанное на полном понимании всей информации, необходимой при оценке пользы и риска предлагаемого тестирования и принятия осознанного решения). 2. Относительно личной информации пациента должны использоваться принципы приватности и конфиденциальности. 3. Пациентов необходимо информировать относительно вероятности реального или потенциального дискриминационного риска в связи с установленным генетическим диагнозом. 4. Должны соблюдаться правовые требования при подготовке документации. 79 ПРИЛОЖЕНИЕ ПЕРВИЧНАЯ ГЕНЕТИЧЕСКАЯ КОНСУЛЬТАЦИЯ ЛИПИДОЛОГА Дата ______________ Ф.И.О. ___________________________ ________________________________________________________ Дата рождения ________________ Телефон, адрес ____________________________________________ Направительный диагноз ГЛП _______________________________ _________________________________________________________ Дата ан крови._______ХС_______ТГ_______ХС ЛПНП_______ ХС ЛПВП_______ глюкоза_______ другие показатели _________________________________________ Были или имеются у Вас следующие заболевания Заб-я ГБ Стен Возраст Заб-я Др. заб. Панпечени креатит ИМ Изм ЭКГ СД Глюкозурия Холе- Пода- Порок Ревмасердца тизм цис- гра тит Мик- Гиперсе- тиреоидема дизм Нефроз/ нефрит Билл. Опер. на Ч. мозг. цироз сердце травмы АКШ, ЭКС, др. Возраст Примечание: Х – по словам обследуемого; Д – документально подтверждено; Н – нет. 80 Принимали ли Вы в течение последних 2-х недель следующие препараты Гипо- Диуретенз. тики Нитроглицерин Антикоагулянты Диги- Нар.серд. талис ритма 1239 1239 1239 1239 1239 1239 Лекар- Снижаюства щие моч. Друсниж. кислоту, от гое подгары вес 1239 1239 1239 Примечение: 1 – нет; 2 – рецепт; 3 – интервью; 9 – неизвестно Прием статинов: Да/Нет. Если Да: доза____________ Дительность приема_________________________________ Факторы риска: Курит да /нет / ранее___лет назад. Если курит в наст. вр.__ сигарет в день. Прием алкоголя – не принимает. Принимает (ежедневно, 1 раз в нед, 1 раз в мес, редко). Уровень физической активности: низкая/умеренная/ высокая. Проф. вредность: есть/нет. Хронический (острый) эмоциональный стресс есть / нет. Объективно. АД ____/_____мм рт ст, Пульс_____Рост______Вес_____ ИМТ______ Наличие ксантом: (ксантелазмы, липоидные дуги роговиц, кожные эруптивные, тубероэруптивные, ахилловых сухожилий). Возраст появления ксантом___ Семейный анамнез, наличие у родителей следующих заболеваний У матери Стенокардия ___ лет У отца Стенокардия ___ лет Среди Стенокардия ___ сибсов лет Среди Стенокардия детей АГ Инсульт ГХС АГ Инсульт ГХС АГ Инсульт ГХС АГ Инсульт ГХС Примечание: выделить наличие. Сахарный диабет Сахарный диабет Сахарный диабет Сахарный диабет Заключение Д-з: Гиперлипопротеидемия тип ____________ Первичная (моногенная: гомозигота/гетерозигота, полигенная) / вторичная Атеросклеротические заболевания: ИБС, ИМ, инсульт, периферические сосуды, сахарный диабет Избыточный вес составляет __________ кг Степень отягощенности семейного анамнеза: есть/нет, не выраженная / выраженная Оценка 10-летнего риска развития сердечно-сосудистых заболеваний атеросклеротического генеза составляет % (с учетом пола, возраста, курения, уровней артериального давления, концентраций липидов плазмы крови. Рекомендовано 1. Рекомендуется выполнить следующие инструментальные обследования (подчеркнуть): ЭКГ, УЗДГ БЦА, ЭХОКГ, ХМ ЭКГ, тредмил-тест. 2. Обеспечить достижение целевых уровней липидов плазмы крови: ХС ЛПНП < _________ммоль/л; ХС ЛПВП>_______ммоль/л; массы тела_______кг. 3. Для модификации диеты выдана инструкция, соответствующая типу ____ ГЛП. Цель диетической модификации: 3.1. Снизить концентрации холестерина плазмы крови до целевого значения ____ 3.2. Снизить избыточный вес до целевых значений за счет низкокалорийной диеты и увеличения двигательной активности (см. табл. калорий пищи). 4. Дополнительные рекомендации ____________________________ _________________________________________________________ _________________________________________________________ _________________________________________________________ _________________________________________________________ _________________________________________________________ 5. Контроль через _1 / 3 / 6_ _ месяцев (подчеркнуть). 81 82 ЛИТЕРАТУРА 1. Кошечкин В.А. Биохимическая генетика нарушений метаболизма липидов плазмы крови // Итоги науки и техники. Генетика человека. ВИНИТИ АМН СССР. – М., 1980. – Т. 5. 8. Мешков А.Н., Малышев П.П., Кухарчук В.В. Семейная гиперхолестеринемия в России: генетическая и фенотипическая характеристика // Терапевтический архив. – 2009. – № 9. – С. 34-38. 9. Диагностика и коррекция нарушений липидного обмена с целью профилактики и лечения атеросклероза. Российские рекомендации IV пересмотр. Разработан Комитетом экспертов Всероссийского научного общества кардиологов (ВНОК). – М., 2009. – С. 80. 10. Титов В.Н., Алиджанова Х.Г., Малышев П.П. Семейная гиперхолестеринемия. Этиология, патогенез, диагностика и лечение. – М.: БИНОМ, 2011. – 624 с. 2. Кошечкин В.А., Перова Н.В., Сидоренко Б.А. и др. Диагностика семейных гиперлипопротеидемий (Методические указания). Утв. Министерством здравоохранения СССР, № 10/11-61 от 28.05. 1985 г. – М., 1985. – С. 22. 11.Fredrickson, D.S; Lees, R.S (1965). "A system for phenotyping hyperlipoproteinemia" (PDF). Circulation 31 (3): 321–7. PMID 14262568. URL: http://circ.ahajournals.org/cgi/reprint/31/3/321. 3. Кошечкин В.А., Репин В.С., Чепурненко Н.В., Никифоров А.Н., Фуки И.В., Пыж М.В. Опыт диагностики семейной гиперхолестеринемии методом определения количества ЛПНП-рецепторов на фибробластах // Кардиология. – 1989. – № 5. – С. 108-110. 12. Fredrickson, D.S., Levy, R.I. Familial hyperlipoproteinemia. In: Stanbury, J.B.; Wyngaarden, J.B.; Fredrickson, D.S.: The Metabolic Basis of Inherited Disease. New York: McGraw-Hill (pub.) (3rd ed.) : 1972. Pp. 545-614. 4. Кошечкин В.А. Болезни сердца и сосудов // Наследственная патология человека / Ред. Ю.Е. Вельтищев, Н.П. Бочков. – М., 1992. – Т. 1. – С. 202-231. 13. OMIM, Online Mendelian Inheritance in Man, a database of human genes and genetic disorders developed. URL: www.ncbi.nlm.nih.gov/omim. 5. Крапивнер С.Р., Малышев П.П., Рожкова Т.А., Полтараус А.Б., Кухарчук В.В., Бочков В.Н. Дифференциальная диагностика с помощью анализа ДНК семейной гиперхолестеринемии и семейного дефекта апо В-100 // Терапевтический архив. – 2000. – № 4. – С. 9-12. 14 The International Statistical Classification of Diseases and Related Health Problems, 10th Revision (known as "ICD-10"). 15. NCBI – The National Center for Biotechnology Information. 6. Малышев П.П., Рожкова Т.А., Соловьева Е.Ю., Мешков А.Н., Кухарчук В.В. Развитие ишемической болезни сердца при гетерозиготной форме семейной гиперхолестеринемии // Кардиоваскулярная терапия и профилактика. – 2006. – № 5. – С. 5-13. 7. Малышев П.П., Рожкова Т.А., Соловьева Е.Ю., Мешков А.Н., Каминная В.И., Кухарчук В.В. Фенотипические особенности гетерозиготной формы семейной гиперхолестеринемии // Терапевтический архив. – 2007. – № 9. – С. 34-38. 83 84 СПИСОК ТЕРМИНОВ Аллель – одна из двух (или нескольких) форм гена. Аллели одного признака занимают в хромосоме один и тот же локус. В клетке могут одновременно находиться два аллеля одного признака, если она содержит две гомологичные хромосомы. Аллели возникают в результате мутаций. Потенциальное число аллелей для каждого гена практически не ограничено. Аллельная гетерогенность (гетерозиготность) – наличие в рамках единой нозологической формы различных вариантов заболевания, обусловленных различными мутантными аллелями одного гена. Аллельные заболевания – фенотипически различные заболевания, обусловленные разными мутациями в одном и том же локусе (гене). Амплификация – увеличение числа копий определенного фрагмента ДНК. Антиципация – более раннее появление и более тяжелое течение заболевания в каждом последующем поколении. Аполипопротеины (апо) – белки, входящие в состав липопротеидов. Апо А (семейства апо-A) – апо АI и AII – основные белковые компоненты ЛПВП-плазмы. Апо АI кодируется геном APOA1, который локализуется в области длинного плеча 11 хромосомы в районе 11q23q24. Мутации APOA1 связаны с дефицитом ЛПВП, включая 85 болезнь Танжера и системный не-нейропатический амилоидоз. Апо АII-ген (APOA2) локализован на хромосоме 1. Мутации APOA2 могут быть причиной дефицита аполипопротеина AII или гиперхолестеринемии. Aпо AIV – плазменный белок, синтезирующийся в печени, располагается на поверхности первоначально синтезированных ХМ. Aпо AIV-ген (APOA4) локализован на хромосоме 11, сцеплен с генами APOA1 и APOC3. Апо AV – белок, являющийся составной частью нескольких фракций липопротеидов, включая ЛПОНП, ЛПВП, ХМ. Aпо AV кодируется геном APOA5. Данный ген локализован в проксимальном отделе кластера аполипопротеиновых генов на хромосоме 11q23. Апо В – ключевой белок, вовлеченный в метаболизм ЛП и поддержание нормального гомеостаза уровней ХС плазмы крови. Апо В-48 синтезируется исключительно в тонком кишечнике, является структурным компонентом хиломикронов. Апо В-100 синтезируется в печени. Повышенные концентрации апо B в плазме крови связаны с сердечными заболеваниями. Aпо B-генетика. Апо В-48 и апо В-100 кодируются одним геном APOB, однако апо В-48 образуется в результате редактирования мРНК апо В-100, что и приводит к синтезу более короткого полипептида. Мутации APOB могут детерминировать гипобеталипопротеидемию, гипобеталипопротеидемию в сочетании с нормальными уровнями ТГ в плазме 86 крови, гиперхолестеринемию, обусловленную дефектом связывающих функций апо B. Aпо C (семейство аполипопротеинов CI, СII, CIII, CIV). Апо С плазмы человека относят к так называемым регуляторным белкам. Они составляют 40–80% от общего содержания белка хиломикронов и ЛПОНП и содержатся также в ЛПВП. Апо СI является составной частью ЛП, богатых ТГ. Aпо CI кодируется геном APOC1, который расположен в хромосоме 19, внутри кластера аполипопротеиновых генов. Этот ген экспрессируется, главным образом, в печени. Апо СII является активатором ЛПЛ-фермента, ответственного за удаление ТГ из ЛПОНП. Апо СII кодируется геном APOC2. Апо СIII является белком ЛПОНП, который ингибирует ЛПЛ и печеночную липазу. Повышение уровня апо СIII сопровождается гипертриглицеридемией. Апо CIV – апобелок, который кодируется APOC4 [1][2] . Ген APOC4 является членом семейства генов апоgene липопротеинов С. Экспрессия гена проявляется в клетках печени; белковая структура характерна для семейства апо С[1]. В геноме человека гены APOA1, APOC3 и APOA4 тесно сцеплены. Апо Е вовлечен во многие стадии гомеостаза ЛП. Он является лигандом с высокой степенью сродства к ЛПНПрецептору и другим членам этого семейства, таким как LRP1, ЛПОНП-рецептор и апо Е2-рецептор (LРR8). Полиморфизм апо Е обусловлен неоднородностью локуса гена AРОЕ, который может присутствовать в одной из трех форм (аллелей): ε2, ε3 («дикий» тип), ε4. Таким образом, в популяции возможно существование шесть фенотипов апо Е, отличающихся как по своей распространенности, так и функциональной значимости. Аутосома – любая неполовая хромосома. У человека имеются 22 пары аутосом. Аутосомно-доминантные заболевания – наследственные заболевания, которые передаются из поколения в поколение. У больного ребенка обязательно болен один из родителей. Аутосомно-доминантное наследование – тип наследования, при котором одного мутантного аллеля, локализованного в аутосоме, достаточно, чтобы болезнь (или признак) могла проявиться. Аутосомно-рецессивное наследование – тип наследования признака или болезни, при котором мутантный аллель, локализованный в аутосоме, должен быть унаследован от обоих родителей. Ацил-КоА-холестерин-ацилтрансфераза (АХАТ) – фермент, катализирующий внутриклеточный синтез эфиров ХС. Белок, переносящий эфиры холестерина (БПЭХ) – секретируется в печени и переносит эфиры ХС от ЛПВП к ЛП, богатым ТГ, и к ЛПНП, а также ТГ от ЛП, богатых ТГ, к ЛПВП. БПЭХ модулирует уровни ЛП плазмы, транспортируя неполярные липиды между разными классами ЛП. Ген и белок БПЭХ хорошо изучены. Белки, связывающие жирные кислоты (БСЖК) – играют важную роль в: а) контролировании процессов поглощения ЖК клетками и их последующего метаболизма, б) распределении/хранении ЖК внутри клеток, в) модуляции внутриклеточной сигнальной трансдукции, г) переносе сигнальных ЖК к ядерным рецепторам, таким как PPAR. Белок микросомальный триглицерид-переносящий (МТПБ) – является внутриклеточным липид-переносящим белком, отвечающим за перенос липидных молекул (в частности, эфиров ХС, ТГ и ФЛ) из цитозоля и/или мембраны 87 88 эндоплазматического ретикулума к насцентным апо В для образования апо В-содержащих ЛП. Белок, переносящий фосфолипиды (БПФЛ) – обеспечивает ускорение переноса ФЛ между митохондриями, микросомами, клеточными мембранами и ЛП плазмы. БПФЛ усиливает связывание на поверхности клетки и ремоделирование ЛПВП-частиц, что повышает их способность усиливать отток ХС и ФЛ. Гаплотип – комбинация конкретных аллелей сцепленных локусов (генов) на одной хромосоме. Ген – субъединица ДНК, содержащая генетическую информацию конкретного продукта клетки (белка, фермента). В геноме человека содержится примерно 25 000 генов. Генетическая ассоциация – достоверная взаимосвязь между заболеванием и определенным аллелем изучаемого гена. Генетический – проявление или состояние, детерминированное одним или большим количеством генов. Генетический риск – вероятность появления определенного наследственного заболевания. Генетическое консультирование – процесс представления персональной информации относительно генетического дефекта. Генетическое скринирование – процесс поиска изменения конкретного гена, способного детерминировать развитие заболевания или наследование. Генная инженерия – совокупность приемов, методов и технологий получения рекомбинантных РНК и ДНК, выделения генов из организма (клеток), осуществления манипуляций с генами и введения их в другие организмы. Генная терапия (генотерапия) – введение генетического материала (ДНК или РНК) в клетку, функцию которой (или функцию организма) он изменяет. Генные болезни – болезни, вызываемые генными мутациями. Генная инженерия – совокупность приемов, методов и технологий получения рекомбинантных РНК и ДНК, выделения генов из организма (клеток), осуществления манипуляций с генами и введения их в другие организмы. Генодиагностика (или ДНК-диагностика) – совокупность методов по выявлению мутаций, приводящих к наследственной патологии. Генокопия – клинический синдром, манифестирующий под маской известного наследственного заболевания с установленной генетической природой, но обусловленный мутацией в другом гене (локусе). Генотип – сумма всех генов организма. Геном – полный состав ДНК клетки, т.е. совокупность всех генов и межгенных участков конкретного организма. Геном содержит полный набор инструкций для формирования и функционирования индивида. Геномика – наука, изучающая общие принципы построения геномов и их структурно-функциональную организацию с помощью секвенирования, картирования и идентификации функций генов и внегенных элементов. Геномика человека является основой молекулярной медицины и имеет важнейшее значение для разработки методов диагностики, лечения и профилактики наследственных и ненаследственных болезней. Методы геномики направлены на расшифровку новых закономерностей биологических систем и процессов. Геномика медицинская изучает наследственные заболевания на основе знания генома человека. Геномика функциональная – изучение геномов для определения биологической функции всех генов и их продуктов, а также взаимодействий между генами. 89 90 Генотип – набор генов индивида или вся генетическая информация организма. Может относиться к конкретной паре аллелей, которые индивид имеет в данном участке генома. Гетерозигота – целый организм или диплоидная клетка, несущие два различных аллеля одного или нескольких рассматриваемых генов. Гетерозиготный организм – организм, имеющий две различные формы данного гена (разные аллели) в гомологичных хромосомах. Гибридизация (ренатурация) – взаимодействие комплементарных цепей ДНК (или ДНК и РНК), приводящее к образованию двухцепочечной молекулы. Гибридизация in situ – гибридизация между денатурированной ДНК клеток на предметном стекле и меченной радиоактивными изотопами или иммунофлюоресцентными соединениями одноцепочечной РНК или ДНК. Гиперлипидемии – метаболические заболевания, характеризующиеся повышением содержания липидов и/или липопротеидов в плазме крови. Гиперлипидемии являются одной из главных причин развития сердечно-сосудистых заболеваний. Гиперлипопротеинемии, гиперлипопротеидемии (общее название – дислипидемии/дислипопротеидемии) разделяют на семейные (первичные) и приобретенные (вторичные) [МКБ-10 Е78, МКБ – 9 272.0-272.4], представляющие собой повышенные уровни одного или нескольких классов липопротеидов в плазме крови, в составе которых содержится холестерин и/или триглицериды. Гиперлипопротеидемии семейные – наследственные заболевания, обусловленные конкретными генетическими дефектами. Гомозигота – клетка (или организм), содержащая два одинаковых аллеля в конкретном локусе гомологичных хромосом. Гомозиготный – организм, имеющий две идентичные копии данного гена в гомологичных хромосомах. Гомологичные хромосомы – хромосомы, одинаковые по набору составляющих их генов. Гомеостаз – генетически обусловленный компонент генотипа. Норма реакции. Группа сцепления – все гены, локализованные в одной хромосоме. Делеция – тип хромосомной мутации, при которой утрачивается участок хромосомы; тип генной мутации, при которой выпадает участок молекулы ДНК. Диагностическое тестирование – тест, выявляющий наличие или отсутствие специфического заболевания или состояния. Диплоидный – содержащий двойной набор хромосом. ДНК (дезоксирибонуклеиновая кислота) – крупная молекула, содержащая всю генетическую информацию, необходимую для контроля жизнедеятельности клетки. ДНК кодирующая – последовательности, транскрибирующиеся в белковую структуру (син. – экзоны). ДНК комплементарная (кДНК) – последовательность ДНК, полученная с помощью обратной транскриптазы с информационной РНК. ДНК повторяющаяся – последовательности разной длины, существующие в геноме в виде многократно повторяющихся копий; составляет большую часть генома. ДНК-полимераза – фермент, осуществляющий комплементарный синтез (репликацию) ДНК. 91 92 Домен – участок аминокислотной последовательности белка, связанный с определенной функцией. Доминантный – признак или соответствующий аллель, проявляющийся у гетерозигот. Доминирование – явление, при котором один из двух аллелей гетерозиготы (доминантный аллель) отчетливо подавляет проявление другого (рецессивного) аллеля. При доминантном наследовании заболевание проявляется, если хотя бы одна из гомологичных хромосом несет патологический аллель. Жиры – см. триглицериды. Инбредные браки – браки между кровными родственниками 2-й или далее степени родства. Инверсия – тип хромосомной мутации, при которой последовательность генов в участке хромосом изменена на обратную. Инсерция – тип генной мутации, при которой имеется вставка отрезка ДНК в структуру гена. Интрон – сегмент ДНК (гена), не содержащий информации о структуре белкового продукта. Информативная семья – семья с наследственным заболеванием, в которой имеется достаточное число больных и здоровых родственников из разных поколений, что позволяет оценивать расхождение признаков в изучаемой родословной при анализе генетического сцепления. Информированное согласие – согласие конкретного лица на проведение медицинского тестирования, лечения и иных медицинских процедур, основанное на полном понимании всей информации, необходимой при оценке пользы и риска предлагаемого тестирования и принятии осознанного решения. 93 Картирование – определение локализации гена на хромосоме. Генетическое – определение расположения изучаемого гена по отношению к другим генам в определенной хромосомной области. Клонирование гена – получение необходимого числа (миллионов) идентичных копий определенного участка ДНК с использованием микроорганизмов. Кодоминантные аллели – аллели, каждый из которых проявляется в гетерозиготе (например, группа крови АВ). Кодон – три рядом находящихся основания, обеспечивающих включение одного аминокислотного остатка в полипептидную цепь, либо сигнал начала или завершения транскрипции. Лецитин-холестерин-ацилтрансфераза (ЛХАТ) катализирует синтез эфиров ХС в ЛПВП, в которых главным активатором является апо АI. ЛХАТ-реакция играет важную роль в опосредуемом ЛПВП транспорте ХС от периферических тканей в печень. Лигáнд (от лат. ligare – связывать) – атом, ион или молекула, связанные с неким центром (акцептором). Понятие применяется в биохимии для обозначения агентов, соединяющихся с биологическими акцепторами (рецепторами, иммуноглобулинами), а также в химии комплексных соединений, обозначая присоединенные к одному или нескольким центральным (комплексообразующим) атомам металла частицы. Лизосомные болезни – группа наследственных болезней, характеризующихся унаследованной недостаточной продукцией лизосомных ферментов. Липидология – наука о нарушениях обмена липидов и липопротеидов в плазме крови. 94 Липиды – одни из основных компонентов биологических мембран, влияя на их проницаемость, участвуют в передаче нервного импульса, создании межклеточных контактов. Липиды также входят в состав липопротеидов, которые участвуют в транспорте липидов в организме. Липиды, классификация. К липидам относят: жирные кислоты; нейтральные жиры (триглицериды); фосфолипиды; стероиды (стерины или стеролы (холестерин и эфиры холестерина)). Липопротеидлипаза печеночная (EC 3.1.1.3.) – фермент, действующий на ремнанты ЛПОНП (бета-ЛПОНП и ЛППП ). Липопротеиды (липопротеины) представляют собой комплексы, состоящие из белков (аполипопротеинов; сокращенно – апо) и липидов, связь между которыми осуществляется посредством гидрофобных и электростатических взаимодействий. Липопротеиды очень низкой плотности (ЛПОНП) формируются в печени, транспортируют ХС и ТГ эндогенного происхождения, в отличие от ХМ, которые транспортируют ХС и ТГ экзогенного (диетического) происхождения. Липопротеиды промежуточной плотности (ЛППП) – отличие ЛППП от ЛПОНП состоит в том, что ЛППП содержат небольшое количество ТГ, при этом сохраняют в своем составе эфиры ХС. Липопротеиды низкой плотности (ЛПНП) являются главным представителем группы ЛП, богатых ХС. Их размер позволяет им пересекать эндотелий сосуда и проникать в тканевую жидкость, снабжая ткани ХС. ЛПНП имеют липидное ядро, почти целиком состоящее из эфиров ХС (приблизительно 1500 молекул на одну ЛПНП-частицу). На поверхности этих ЛП содержится единственный апобелок – апо В-100. Липопротеиды высокой плотности (ЛПВП) переносят эфиры ХС. Основными апобелками ЛПВП являются aпo AI и aпo AII. ЛПВП синтезируются печенью в виде комплексов, содержащих аполипопротеины и фосфолипиды. ЛПВП – гетерогенный класс ЛП, включающий несколько подклассов, различающихся по размеру и заряду при электрофорезе. Липопротеид (а), ЛП(a) – особая липопротеидная частица, характеризующаяся наличием уникального гликопротеина (а), связанного с апо В-100 дисульфидными связями. Плотность ЛП(а) занимает промежуточное положение между ЛПНП и ЛПВП. Концентрации ЛП(a) в плазме наследуются в высокой степени и контролируются геном LPA. Локус – область локализации определенного генетического элемента на хромосоме. Локусная гетерогенность – наличие в рамках единой нозологической формы различных генетических вариантов заболевания, вызываемых мутациями самостоятельных генов в разных хромосомных локусах. Маркер – аллель (или признак), наследование которого прослеживается в потомстве. 95 96 Маркер генетический – полиморфный участок ДНК строго определенной локализации, разные аллели которого позволяют дифференцировать различные по происхождению хромосомы и анализировать их сегрегацию в родословной. Медико-генетическое консультирование – специализированный вид медицинской помощи, при которой пациенты или родственники, имеющие риск наследственного заболевания, получают информацию о возможных последствиях и о природе этого заболевания, возможности его развития или наследования. Миссенс-мутации – см. мутация. Митохоидриальное наследование – наследование признаков, передаваемых через ДНК митохондрий. Мицеллы – частицы в коллоидных растворах. В клетках слизистой тонкого кишечника содержимое мицелл (желчные кислоты, гидрофильные и гидрофобные липиды) используется для синтеза хиломикронов. Множественные аллели – наличие в популяции (или у вида) более двух аллелей одного и того же локуса. Моногенное наследование – признак, кодируемый одним геном, который наследуется в соответствии с законами Менделя и называется менделирующим. Наследование моногенных болезней – аутосомное или сцепленное с X-хромосомой – можно определить при изучении родословной. Мультифакториальные болезни – те, которые развиваются в результате взаимодействия определенных комбинаций аллелей разных локусов и специфических воздействий факторов окружающей среды. Мутант – организм, несущий мутантный аллель. Мутация – изменение в наследственных структурах (ДНК, ген, хромосома, геном): – генная – изменение последовательности нуклеотидов в определенном участке молекулы ДНК; – геномная – изменение числа хромосом (кратное гаплоидному – полиплоидия, некратное – анеуплоидия); – динамическая – мутация по типу экспансии тандемных тринуклеотидных повторов; – мажорная – встречающаяся с высокой частотой в определенной популяции; – миссенс – замена нуклеотида, сопровождающаяся изменением аминокислотного шифра кодона и ведущая к замене аминокислоты в составе белка; нейтральная (молчащая) – не сопровождающаяся изменением фенотипа; – нонсенс – замена нуклеотида, приводящая к замещению информационно значимого кодона на стоп-кодон и сопровождающаяся преждевременным обрывом трансляции; – нулевая – приводящая к отсутствию синтеза скольконибудь функционально значимого продукта; – регуляторная – затрагивающая регуляторные последовательности гена и нарушающая его экспрессию; – со сдвигом рамки – приводящая к нарушению нормального отсчета кодирующих триплетов (делеции или вставки участков молекулы ДНК, размеры которых не кратны трем основаниям); – сплайсинговая – затрагивающая сайт сплайсинга и приводящая к неправильному вырезанию интрона либо к удалению из молекулы РНК информационно значимой экзонной последовательности; – структурная – приводящая к протяженному (мультинуклеотидному) дефекту гена; – точковая (точечная) – затрагивающая один нуклеотид, либо 1–2 соседних нуклеотида; – хромосомная – любое нарушение структуры хромосом (деления, дупликация, инверсия, транслокация). Наследование полигенное – характер наследования, которое не подчиняется законам Менделя и не соответствует классическим типам аутосомно-доминантного, аутосомнорецессивного наследования и наследования, сцепленного с X-хромосомой. Наследственная болезнь – болезнь, для которой этиологическим фактором является генная, хромосомная или геномная мутация. Наследуемость – часть общей фенотипической изменчивости, обусловленной генетическими факторами. 97 98 Нонсенс-мутации – см. мутация. Норма реакции – границы выраженности фенотипов при одном и том же генотипе в различных условиях среды. Носительство – статус носительства определяется генетическим тестированием. Означает, что данный индивид имеет ген, способный вызвать заболевание и который может передаваться потомкам. Однонуклеотидный полиморфизм (англ. Single nucleotide polymorphism, SNP) – отличия последовательности ДНК размером в один нуклеотид (A, T, G или C) в геноме (или в другой сравниваемой последовательности) представителей одного вида или между гомологичными участками гомологичных хромосом индивида. Две последовательности ДНК – AAGCCTA и AAGCTTA – отличаются на один нуклеотид. В таком случае говорят о существовании двух аллелей: C и T. SNP возникают в результате точечных мутаций. SNP используются в качестве молекулярно-генетических маркеров (наряду с ПДРФ (RFLP) и ПДАФ (AFLP)). Изучение SNP широко используется в генной систематике – построении биологической систематики на основе дивергенции (расхождения) гомологичных участков ДНК в филогенезе. В данной области наиболее часто используются спейсеры генов рибосомальной РНК. Ввиду того, что мутации в данных спейсерах не сказываются на структуре конечных продуктов гена (теоретически они не влияют на жизнеспособность), в первом приближении постулируется прямая зависимость между степенью полиморфизма и филогенетическим расстоянием между организмами. Пенетрантность – частота или вероятность проявления аномального гена или генной комбинации в признаках носителя в зависимости от условий внешней среды и генотипа. Если у части лиц, несущих данный ген, он фенотипически не проявляется, то говорят о неполной пенетрантности. Плейотропность – влияние одного гена на развитие двух или более фенотипических признаков. Полигенность (полигенный) – признак (заболевание), которые контролируются сразу несколькими генами. Проявление признака во многом зависит от экзогенных факторов. Генетический риск полигенных болезней в большой степени зависит от семейной предрасположенности и от тяжести заболевания у родителей. Генетический риск значительно снижается с уменьшением степени родства. Полигенные признаки – признаки, обусловленные многими генами, каждый из которых оказывает лишь небольшое влияние на степень экспрессии данного признака. Полимеразная цепная реакция (ПЦР) – метод циклического синтеза in vitro огромного числа копий строго определенного участка ДНК длиной от десятков до нескольких тысяч пар нуклеотидов: – количественная – реакция, по результатам которой оценивается количественный выход продуктов амплификации с помощью соответствующих сканирующих устройств; – мультиплексная (мультипраймерная) – одновременная амплификация в одной реакции нескольких участков исследуемого гена; – обратно-транскриптазная – реакция, в которой матрицей служат молекулы кДНК. Полиморфизм длин рестрикционных фрагментов (ПДРФ) – наличие участков ДНК разной длины после обработки определенной рестриктазой. Полиморфизм – варианты последовательностей ДНК, распространенные в общей популяции с частотой не менее 1%. Половые хромосомы – хромосомы, определяющие пол индивида (у человека – Х- и Y-хромосомы). 99 100 Предрасположенность генетическая – комбинация аллелей разных локусов, предрасполагающих к более раннему возникновению заболеваний под влиянием факторов окружающей среды и к более тяжелому их течению. Пробанд – лицо, с которого началось исследование родословной. Прогностическое (предсказательное) ДНК-тестирование – проведение ДНК-анализа у клинически здорового человека с целью установить его генетический статус, который может привести к развитию наследственной болезни или болезни с наследственной предрасположенностью. Просеивающие программы – см. скрининг. Протеом – полный набор белков, кодируемых геномом. Регуляторная область – сегмент ДНК, контролирующий наличие и степень экспрессии гена. Рестриктаза (рестрикционная эндонуклеаза) – фермент бактериального происхождения, распознающий специфическую нуклеотидную последовательность длиной от 4 до 10 пар нуклеотидов и «разрезающий» двунитевую молекулу ДНК в этом месте. Рестрикционный анализ – метод анализа ДНК с использованием рестрикционных эндонуклеаз. Рецепторы ЛПВП – скэвенджеры (очистители) входят в состав клеточных мембран и находятся во многих типах клеток и тканей, включая печень и адренальные органы. Они обеспечивают перенос ХС из периферических тканей в печень для дальнейшей их экскреции. Этот процесс переноса ХС известен как обратный транспорт ХС и служит в качестве защитного механизма от развития атеросклероза. Рецепторы ЛППП и ЛПНП – объединяют в группу белков под общим названием LRP. Эти белки выполняют функции рецептора в плазменных мембранах клеток, тем самым обеспечивают рецепторопосредованный эндоцитоз. У человека белок LRP1 кодируется геном LRP1. Рецессивность – при рецессивном наследовании заболевание проявляется только в том случае, когда обе гомологичные хромосомы несут патологический аллель. РНК-полимераза – фермент, осуществляющий синтез РНК на ДНК-матрице. Родословная – схема, показывающая родство между членами одной семьи в ряду поколений. Сайт – определенное место (позиция) в молекуле ДНК. Секвенирование – определение последовательности нуклеотидов в молекуле ДНК или последовательности аминокислот в молекуле белка. Семейные болезни – болезни, наблюдающиеся у нескольких членов семьи в одном или нескольких поколениях. Семейные заболевания – заболевания, имеющие тенденцию к накоплению в отдельных семьях (болезни с наследственным предрасположением). Семейные гиперлипопротеидемии (СГЛП) – наиболее распространенные нарушения обмена веществ вследствие генетических дефектов в метаболизме липопротеидов (ЛП), характеризующиеся повышением концентраций холестерина (ХC) и триглицеридов (ТГ) в плазме крови. Сердечно-сосудистые заболевания – в данной книге этот термин ограничивается описанием сердечно-сосудистых заболеваний атеросклеротического генеза. Сибсы – братья/сестры пробанда (родные, двоюродные и т.д.). Скрининг (син. – просеивание) – обследование больших групп людей на выявление каких-либо состояний (болезней или носительства) с целью активной профилактики 101 102 тяжелых форм болезней; предположительное выявление не диагностированной ранее болезни с помощью простых методов, дающих быстрый ответ. Сцепление генов – совместная передача генов (признаков). Тестирование диагностическое – тест, выявляющий наличие или отсутствие специфического заболевания или состояния. Тестирование (предиктивное, предупредительное) – генетический тест, проводимый с целью определить, имеет ли тестируемый изменения в одном или более генах, способных увеличить риск развития определенного заболевания или состояния в будущем. Тестирование предсимптоматическое – генетический тест, выполняемый до возникновения любых симптомов, с целью выявить, имеет ли тестируемый изменение в гене в конечном счете, способное детерминировать развитие заболевания или состояния. Трансляция – передача наследственной информации; синтез белковой молекулы или перевод последовательности оснований мРНК в последовательность аминокислот в полипептидной цепи. Триацилглицероллипаза печеночная (LIPC) – см. липопротеидлипаза печеночная. Триглицериды (жиры) – природные органические соединения, входят в состав липидов. Фенокопия – клинический синдром, сходный по своим проявлениям с наследственным заболеванием, но имеющий негенетическую природу возникновения. Фенотип – совокупность наблюдаемых признаков организма. Фенотип не всегда служит прямым и полным выражением генотипа, а представляет собой результат взаимодействия между генотипом и окружающей средой. Фенотипическая классификация семейных гиперлипопротеидемий Фредриксона основана на электрофоретических свойствах и по плотности при ультрацентрифугировании, принята Всемирной организацией здравоохранения. В соответствии с классификацией семейные гиперлипопротеидемии делят на пять типов: – семейная гиперлипопротеидемия, тип I (гиперхиломикронемия) – повышенное содержание хиломикронов в плазме крови; – семейная гиперлипопротеидемия, тип II. Данный тип делят на два подтипа: IIа (чистая гиперхолестеринемия) и IIб (гиперхолестеринемия с гипертриглицеридемией); – семейная гиперлипопротеидемия, тип III (болезнь «широких бета», дисбеталипопротеидемия); Фактор транскрипции – белок, связывающийся с регуляторной областью гена и регулирующий его экспрессию. Фармакогеномика – создание новых типов лекарств на основе геномной информации. – семейная гиперлипопротеидемия, тип IV (гипертриглицеридемия); – семейная гиперлипопротеидемия, тип V (хиломикронемия с гипертриглицеридемией). Хиломикроны (ХМ) – самые крупные липопротеидные комплексы, они содержат 85–92% ТГ, 6–12% фосфолипидов, 1–3% белков; основной белок ХМ – апо В-48. Насцентные (т.е. вновь синтезируемые) ХМ формируются в энтероцитах тонкого кишечника. 103 104 Холестерин (холестерол) – органическое соединение, природный жирный (липофильный) спирт, содержащийся в клеточных мембранах живых организмов. Хромосомная мутация (или аберрация) – изменение в структуре хромосомы. Хромосомный набор – совокупность хромосом в ядре гаметы, зиготы или соматической клетки. Х-сцепленное наследование – тип наследования признаков, гены которых локализованы в Х-хромосоме. Экзон – отдельный фрагмент прерывистого гена, сохраняющийся в зрелой РНК. Экспрессивность – степень фенотипического проявления одного и того же гена у разных лиц. Разная экспрессивность встречается при большинстве моногенных болезней. Экспрессия гена – активизация транскрипции гена, в процессе которой на смысловой нити ДНК синтезируется мРНК. Владимир Анатольевич Кошечкин Павел Прокопьевич Малышев Татьяна Алексеевна Рожкова ПРАКТИЧЕСКАЯ ЛИПИДОЛОГИЯ С МЕТОДАМИ МЕДИЦИНСКОЙ ГЕНЕТИКИ Учебное пособие Редактор Т.В. Анисимова Технический редактор Н.А. Ясько Компьютерная верстка М.Н. Заикина Дизайн обложки М.В. Рогова Тематический план 2011 г., № 27а Этерификация (от др.-греч. αἰθήρ – эфир и лат. facio – делаю) – реакция образования сложных эфиров при взаимодействии кислот и спиртов. Подписано в печать 0.0.12 г. Формат 60×84/16. Печать офсетная. Усл. печ. л. 7,9. Тираж 100 экз. Заказ 1320 Российский университет дружбы народов 115419, ГСП-1, г. Москва, ул. Орджоникидзе, д. 3 Типография РУДН 115419, ГСП-1, г. Москва, ул. Орджоникидзе, д. 3, тел. 952-04-41 105 106